
Heterocycle-Based Multicomponent Reactions in Drug Discovery: From Hit Finding to Rational Design



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Enzymes
2.1. Acetyl-Cholinesterase (AChE)
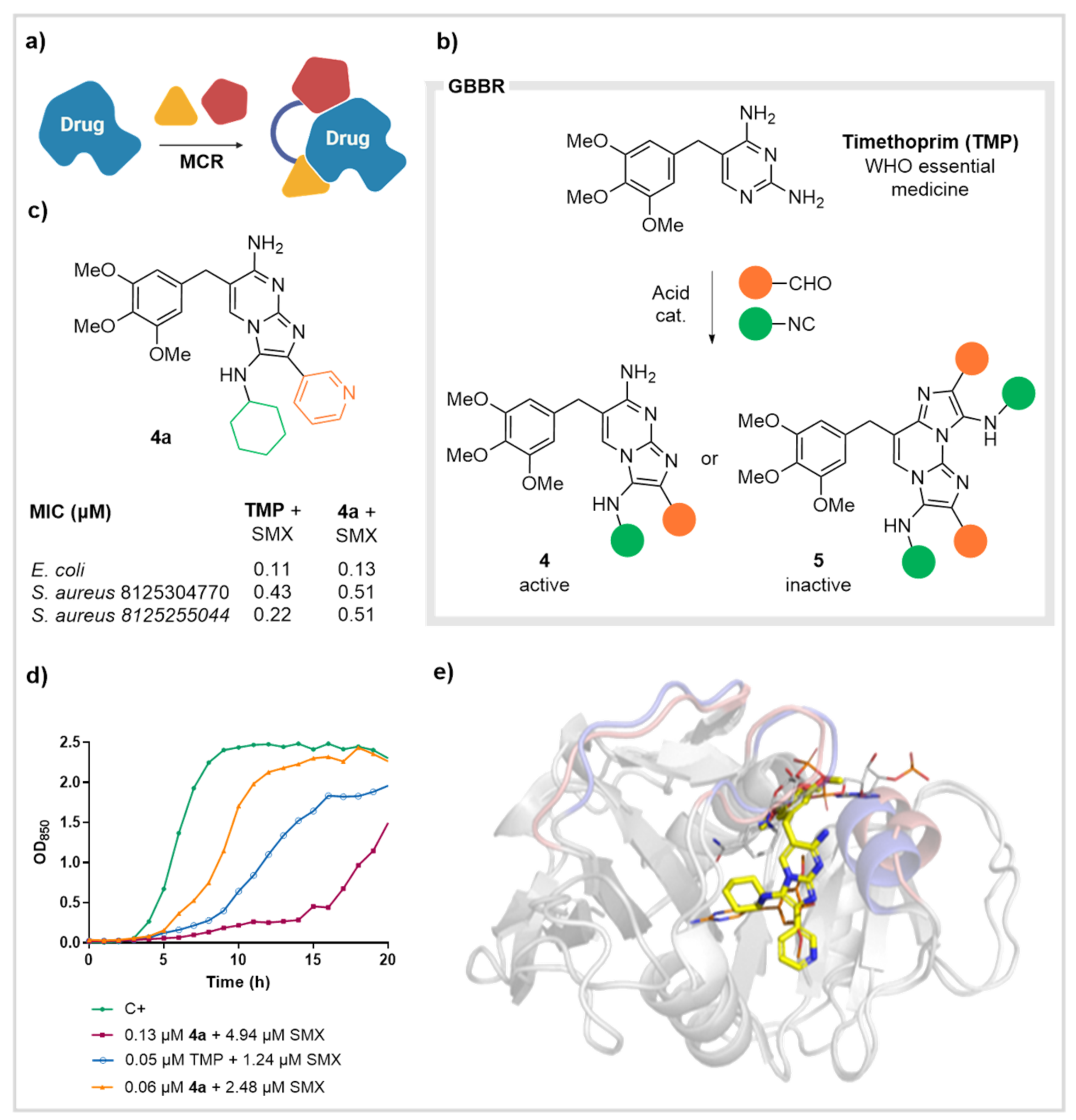
2.2. Dihydrofolate Reductase (DHFR)
3. Transcription Factors
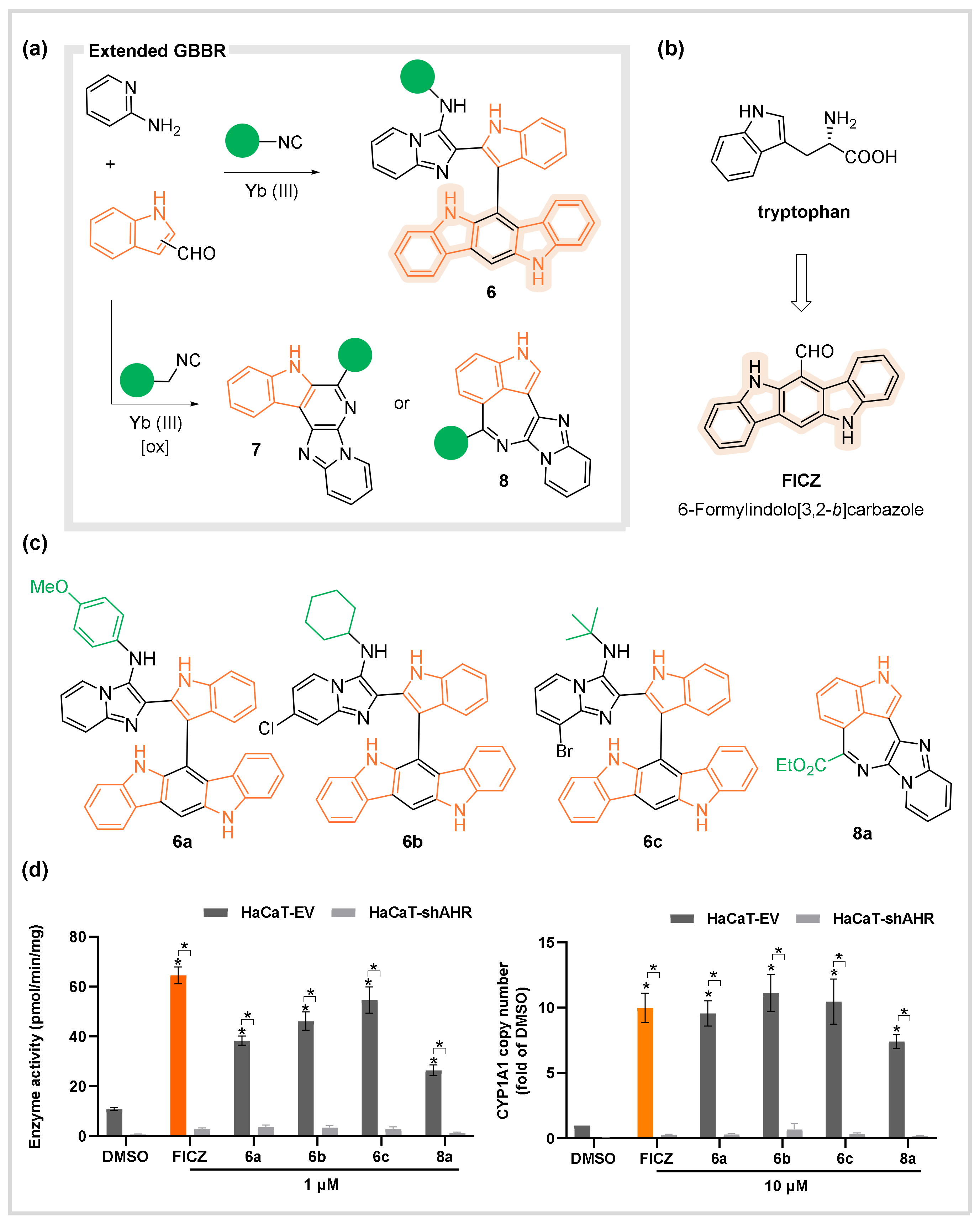
Aryl Hydrocarbon Receptor (AhR)
4. Chemotherapeutic Agents
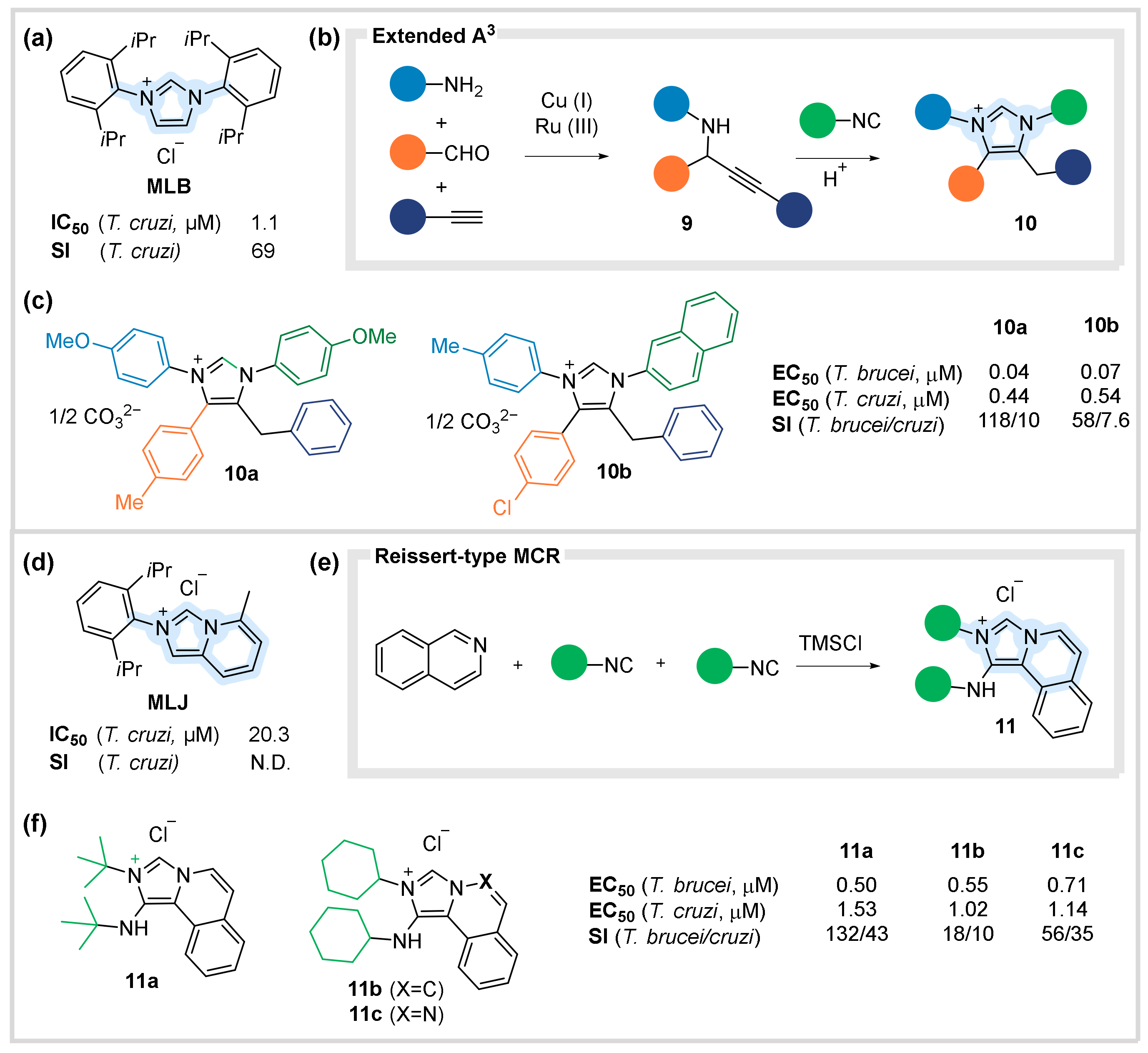
4.1. Antiparacitic Agents against Trypanosoma
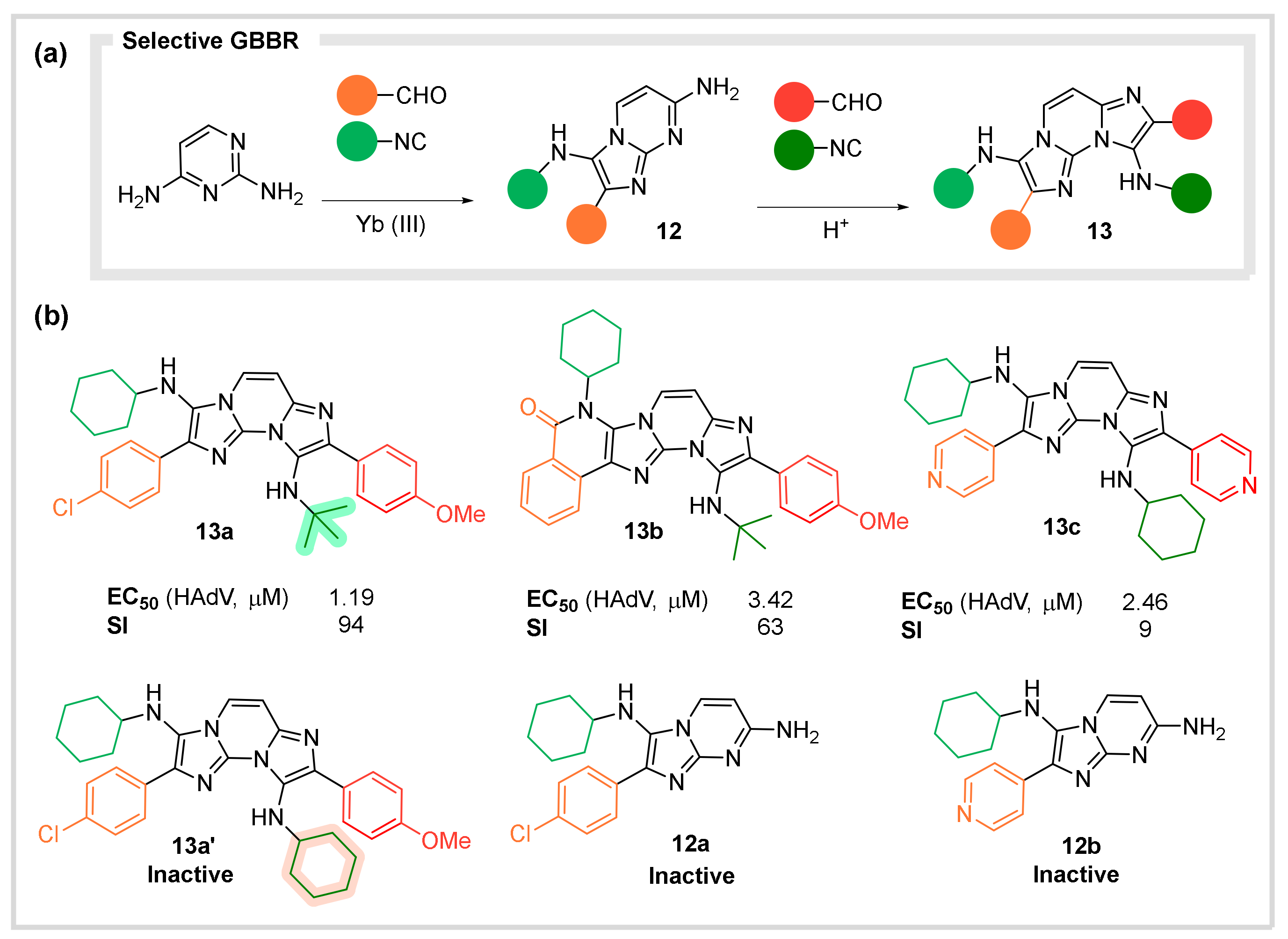
4.2. Antiviral Agents against Human Adenovirous
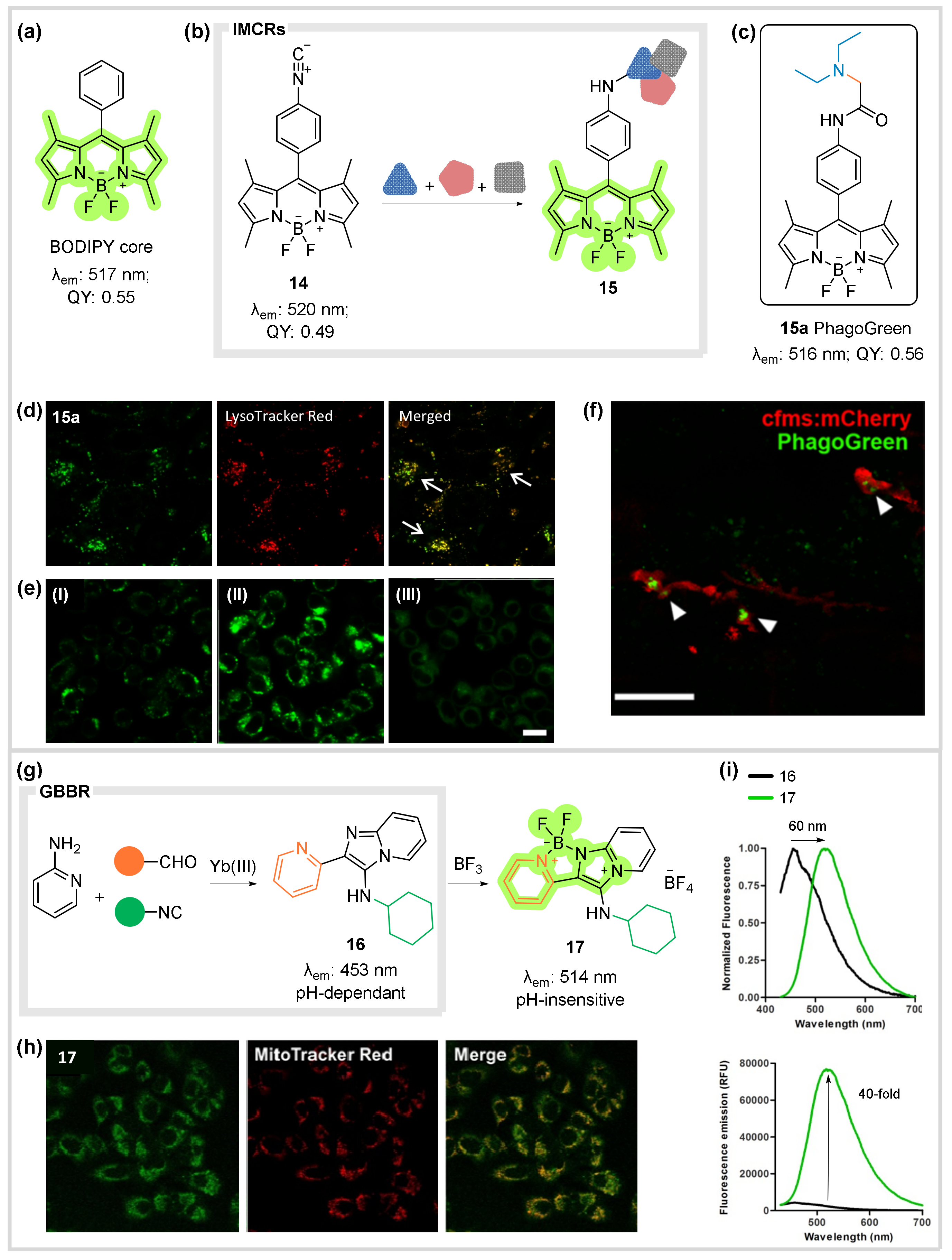
5. Fluorescent Bioprobes
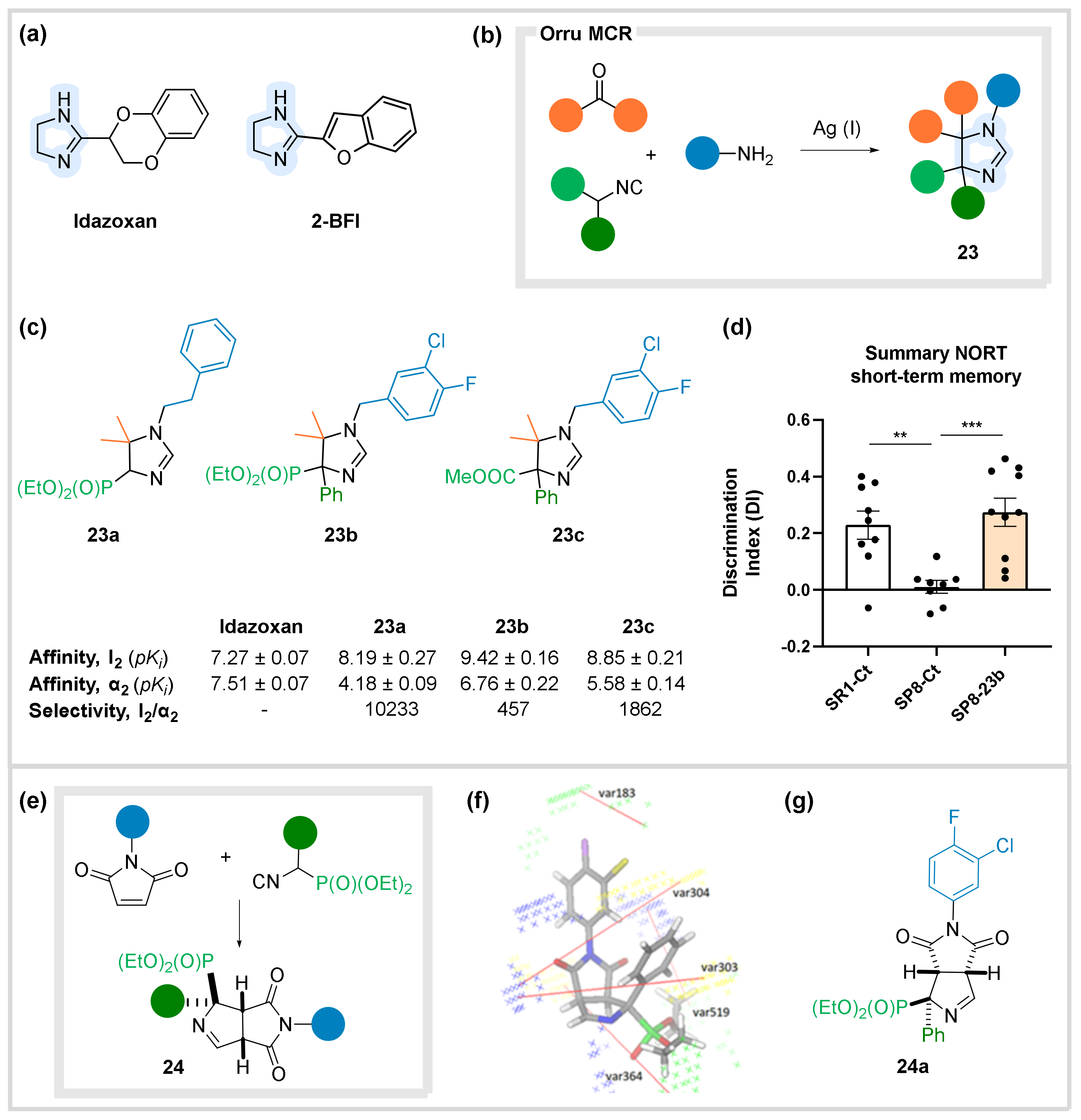
6. Receptors
7. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AChE | Acetylcholinesterase |
| AD | Alzheimer disease |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| BACE | Beta secretase |
| GBBR | Groebke-Blackburn-Bienaymé MCR |
| TMP | Trimethoprim |
| WHO | World Health Organization |
| MIC | Minimum inhibition concentration |
| PDB | Protein data bank |
| DHFR | Dihydrofolate reductase |
| SMX | Sulfamethoxazole |
| PaβN | Phenylalanine-arginine β-naphthylamide |
| NADPH | Reduced nicotinamide adenine dinucleotide phosphate |
| AhR | Hydrocarbon Receptor |
| FICZ | 6-Formylindolo [3,2-b]carbazole |
| SAR | Structure-Activity Relationship |
| Trp | Tryptophan |
| TMSCl | Trimethylsilyl chloride |
| PBS | Phosphate buffer saline |
| AR | α2-Adrenoreceptors |
| PhosMic | Diethyl isocyanomethylphosphonate |
References
- Gioiello, A.; Piccinno, A.; Lozza, A.M.; Cerra, B. The Medicinal Chemistry in the Era of Machines and Automation: Recent Advances in Continuous Flow Technology. J. Med. Chem. 2020, 63, 6624–6647. [Google Scholar] [CrossRef]
- Blakemore, D.C.; Castro, L.; Churcher, I.; Rees, D.C.; Thomas, A.W.; Wilson, D.M.; Wood, A. Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nat. Chem. 2018, 10, 383–394. [Google Scholar] [CrossRef]
- Cernak, T.; Dykstra, K.D.; Tyagarajan, S.; Vachal, P.; Krska, S.W. The Medicinal Chemist’s Toolbox for Late Stage Functionalization of Drug-like Molecules. Chem. Soc. Rev. 2016, 45, 546–576. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Burke, M.D.; Schreiber, S.L.; Burke, M.D. A Planning Strategy for Diversity-Oriented Synthesis. Angew. Chem. Int. Ed. 2004, 43, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, T.; Baran, P.S.; Hoffmann, R.W. The Economies of Synthesis. Chem. Soc. Rev. 2009, 38, 3010–3021. [Google Scholar] [CrossRef]
- Hayashi, Y. Time Economy in Total Synthesis. J. Org. Chem. 2021, 86, 1–23. [Google Scholar] [CrossRef]
- Lipinski, C.; Hopkins, A. Navigating Chemical Space for Biology and Medicine. Nature 2004, 432, 855–861. [Google Scholar] [CrossRef]
- Dobson, C.M. Chemical Space and Biology. Nature 2004, 432, 824–828. [Google Scholar] [CrossRef]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [Green Version]
- Grygorenko, O.O.; Radchenko, D.S.; Dziuba, I.; Chuprina, A.; Gubina, K.E.; Moroz, Y.S. Generating Multibillion Chemical Space of Readily Accessible Screening Compounds. iScience 2020, 23, 101681. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Boström, J. Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem. 2016, 59, 4443–4458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Akritopoulou-Zanze, I. When Analoging Is Not Enough: Scaffold Discovery in Medicinal Chemistry. Expert Opin. Drug Discov. 2010, 5, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Bienaymé, H. Multicomponent Reactions; Zhu, J., Bienaymé, H., Eds.; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Zhu, J.; Wang, Q.; Wang, M.-X. Multicomponent Reactions in Organic Synthesis; Zhu, J., Wang, Q., Wang, M.-X., Eds.; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Hulme, C. Applications of Multicomponent Reactions in Drug Discovery—Lead Generation to Process Development. In Multicomponent Reactions; Zhu, J., Bienayme, H., Eds.; Wiley-VCH: Weinheim, Germany, 2005; pp. 311–341. [Google Scholar]
- Wang, Z.; Domling, A. Multicomponent Reactions in Medicinal Chemistry. In Multicomponent Reactions towards Heterocycles; van der Eycken, E., Sharma, U.K., Eds.; Wiley-VCH: Weinhiem, Germany, 2022; pp. 91–137. [Google Scholar]
- Slobbe, P.; Ruijter, E.; Orru, R.V.A. Recent Applications of Multicomponent Reactions in Medicinal Chemistry. Medchemcomm 2012, 3, 1189–1218. [Google Scholar] [CrossRef]
- Isambert, N.; Lavilla, R. Heterocycles as Key Substrates in Multicomponent Reactions: The Fast Lane towards Molecular Complexity. Chem. Eur. J. 2008, 14, 8444–8454. [Google Scholar] [CrossRef] [PubMed]
- Vicente-García, E.; Kielland, N.; Lavilla, R. Functionalization of Heterocycles by MCRs. In Multicomponent Reactions in Organic Synthesis; Zhu, J., Wang, Q., Wang, M., Eds.; Wiley-VCH: Weinheim, Germany, 2015; pp. 159–182. [Google Scholar]
- Ghashghaei, O.; Pedrola, M.; Escolano, C.; Lavilla, R. Heterocycles as Inputs in MCRs: An Update. In Multicomponent Reactions towards Heterocycles; van der Eycken, E., Sharma, U.K., Eds.; Wiley-VCH: Weinheim, Germany, 2022; pp. 1–43. [Google Scholar]
- Ghashghaei, O.; Nadal Rodríguez, P.; Lavilla, R. Development of Heterocyclic Multicomponent Reactions through Guided Exploration: Direct, Reasonable and Unpredictable Processes. Synlett 2022, 33, 822–835. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alvarez, A.; Pérez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase Accelerates Assembly of Amyloid-Peptides into Alzheimer’s Fibrils: Possible Role of the Peripheral Site of the Enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Torrero, D.; Camps, P. Dimeric and Hybrid Anti-Alzheimer Drug Candidates. Curr. Med. Chem. 2006, 13, 399–422. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Capsoni, S.; Andrisano, V.; Bartolini, M.; Margotti, E.; Cattaneo, A.; Recanatini, M.; Melchiorre, C. A Small Molecule Targeting the Multifactorial Nature of Alzheimer’s Disease. Angew. Chem. Int. Ed. 2007, 46, 3689–3692. [Google Scholar] [CrossRef]
- Kouznetsov, V. Recent Synthetic Developments in a Powerful Imino Diels-Alder Reaction (Povarov Reaction): Application to the Synthesis of N-Polyheterocycles and Related Alkaloids. Tetrahedron 2009, 65, 2721–2750. [Google Scholar] [CrossRef]
- Ghashghaei, O.; Masdeu, C.; Alonso, C.; Palacios, F.; Lavilla, R. Recent Advances of the Povarov Reaction in Medicinal Chemistry. Drug Discov. Today Technol. 2018, 29, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Camps, P.; Formosa, X.; Galdeano, C.; Muñoz-Torrero, D.; Ramírez, L.; Gómez, E.; Isambert, N.; Lavilla, R.; Badia, A.; Clos, M.V.; et al. Pyrano[3,2-c]Quinoline-6-Chlorotacrine Hybrids as a Novel Family of Acetylcholinesterase-and β-Amyloid-Directed Anti-Alzheimer Compounds. J. Med. Chem. 2009, 52, 5365–5379. [Google Scholar] [CrossRef]
- Di Pietro, O.; Viayna, E.; Vicente-García, E.; Bartolini, M.; Ramón, R.; Juárez-Jiménez, J.; Clos, M.V.; Pérez, B.; Andrisano, V.; Luque, F.J.; et al. 1,2,3,4-Tetrahydrobenzo[h][1,6]Naphthyridines as a New Family of Potent Peripheral-to-Midgorge-Site Inhibitors of Acetylcholinesterase: Synthesis, Pharmacological Evaluation and Mechanistic Studies. Eur. J. Med. Chem. 2014, 73, 141–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pietro, O.; Pérez-Areales, F.J.; Juárez-Jiménez, J.; Espargaró, A.; Clos, M.V.; Pérez, B.; Lavilla, R.; Sabaté, R.; Luque, F.J.; Muñoz-Torrero, D. Tetrahydrobenzo[h][1,6]Naphthyridine-6-Chlorotacrine Hybrids as a New Family of Anti-Alzheimer Agents Targeting β-Amyloid, Tau, and Cholinesterase Pathologies. Eur. J. Med. Chem. 2014, 84, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Di Pietro, O.; Vicente-Garcia, E.; Taylor, M.; Berenguer, D.; Viayna, E.; Lanzoni, A.; Sola, I.; Sayago, H.; Riera, C.; Fisa, R.; et al. Multicomponent Reaction-Based Synthesis and Biological Evaluation of Tricyclic Heterofused Quinolines with Multi-Trypanosomatid Activity. Eur. J. Med. Chem. 2015, 105, 120–137. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaei, O.; Caputo, S.; Sintes, M.; Revés, M.; Kielland, N.; Estarellas, C.; Luque, F.J.; Aviñó, A.; Eritja, R.; Serna-Gallego, A.; et al. Multiple Multicomponent Reactions: Unexplored Substrates, Selective Processes, and Versatile Chemotypes in Biomedicine. Chem. Eur. J. 2018, 24, 14513–14521. [Google Scholar] [CrossRef]
- Boltjes, A.; Dömling, A. The Groebke-Blackburn-Bienaymé Reaction. Eur. J. Org. Chem. 2019, 2019, 7007–7049. [Google Scholar] [CrossRef]
- Pedrola, M.; Jorba, M.; Jardas, E.; Jardi, F.; Ghashghaei, O.; Viñas, M.; Lavilla, R. Multicomponent Reactions Upon the Known Drug Trimethoprim as a Source of Novel Antimicrobial Agents. Front. Chem. 2019, 7. [Google Scholar] [CrossRef]
- Jorba, M.; Pedrola, M.; Ghashghaei, O.; Herráez, R.; Campos-Vicens, L.; Luque, F.J.; Lavilla, R.; Viñas, M. New Trimethoprim-Like Molecules: Bacteriological Evaluation and Insights into Their Action. Antibiotics 2021, 10, 709. [Google Scholar] [CrossRef]
- Ghashghaei, O.; Pedrola, M.; Seghetti, F.; Martin, V.V.; Zavarce, R.; Babiak, M.; Novacek, J.; Hartung, F.; Rolfes, K.M.; Haarmann-Stemmann, T.; et al. Extended Multicomponent Reactions with Indole Aldehydes: Access to Unprecedented Polyheterocyclic Scaffolds, Ligands of the Aryl Hydrocarbon Receptor. Angew. Chem. Int. Ed. 2021, 60, 2603–2608. [Google Scholar] [CrossRef]
- Muñoz-Torrero, D.; Lavilla, R.; Pérez-Areales, F.J.; Ghashghaei, O. Multicomponent Reactions: A Mighty Journey Partner for Infectious Tropical Disease Drug Discovery. In Medicinal Chemistry Approaches to Malaria and Other Tropical Diseases; Chibale, K., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 181–217. ISBN 9780128205518. [Google Scholar]
- Faral-Tello, P.; Liang, M.; Mahler, G.; Wipf, P.; Robello, C. Imidazolium Compounds Are Active against All Stages of Trypanosoma Cruzi. Int. J. Antimicrob. Agents 2014, 43, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaei, O.; Revés, M.; Kielland, N.; Lavilla, R. Modular Access to Tetrasubstituted Imidazolium Salts through Acid-Catalyzed Addition of Isocyanides to Propargylamines. Eur. J. Org. Chem. 2015, 15, 4383–4388. [Google Scholar] [CrossRef]
- Ghashghaei, O.; Kielland, N.; Revés, M.; Taylor, M.C.; Kelly, J.M.; Di Pietro, O.; Muñoz-Torrero, D.; Pérez, B.; Lavilla, R. Tetrasubstituted Imidazolium Salts as Potent Antiparasitic Agents against African and American Trypanosomiases. Molecules 2018, 23, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishore, K.G.; Ghashghaei, O.; Estarellas, C.; Mestre, M.M.; Monturiol, C.; Kielland, N.; Kelly, J.M.; Francisco, A.F.; Jayawardhana, S.; Muñoz-Torrero, D.; et al. Insertion of Isocyanides into N–Si Bonds: Multicomponent Reactions with Azines Leading to Potent Antiparasitic Compounds. Angew. Chem. Int. Ed. 2016, 55, 8994–8998. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Campbell, R.E.; Ting, A.Y.; Tsien, R.Y. Creating New Fluorescent Probes for Cell Biology. Nat. Rev. Mol. Cell Biol. 2002, 3, 906–918. [Google Scholar] [CrossRef]
- Kikuchi, K. Design, Synthesis and Biological Application of Chemical Probes for Bio-Imaging. Chem. Soc. Rev. 2010, 39, 2048–2053. [Google Scholar] [CrossRef]
- Fu, Y.; Finney, N.S. Small-Molecule Fluorescent Probes and Their Design. RSC Adv. 2018, 8, 29051–29061. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Singh, K. Recent Advances in the Application of BODIPY in Bioimaging and Chemosensing. J. Mater. Chem. C. 2019, 7, 11361–11405. [Google Scholar] [CrossRef]
- Loudet, A.; Burgess, K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef]
- Vázquez-Romero, A.; Kielland, N.; Arévalo, M.J.; Preciado, S.; Mellanby, R.J.; Feng, Y.; Lavilla, R.; Vendrell, M. Multicomponent Reactions for de Novo Synthesis of Bodipy Probes: In Vivo Imaging of Phagocytic Macrophages. J. Am. Chem. Soc. 2013, 135, 16018–16021. [Google Scholar] [CrossRef]
- Kielland, N.; Vendrell, M.; Lavilla, R.; Chang, Y.T. Imaging Histamine in Live Basophils and Macrophages with a Fluorescent Mesoionic Acid Fluoride. ChemComm 2012, 48, 7401–7403. [Google Scholar] [CrossRef] [PubMed]
- De Moliner, F.; Kielland, N.; Lavilla, R.; Vendrell, M. Modern Synthetic Avenues for the Preparation of Functional Fluorophores. Angew. Chem. Int. Ed. 2017, 56, 3758–3769. [Google Scholar] [CrossRef] [PubMed]
- Sintes, M.; de Moliner, F.; Caballero-Lima, D.; Denning, D.W.; Read, N.D.; Kielland, N.; Vendrell, M.; Lavilla, R. Electrophilic, Activation-Free Fluorogenic Reagent for Labeling Bioactive Amines. Bioconjug. Chem. 2016, 27, 1430–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggiero, D.A.; Regunathan, S.; Wang, H.; Milner, T.A.; Reis, D.J. Immunocytochemical Localization of an Imidazoline Receptor Protein in the Central Nervous System. Brain Res. 1998, 780, 270–293. [Google Scholar] [CrossRef]
- Li, J.X. Imidazoline I2 Receptors: An Update. Pharmacol. Ther. 2017, 178, 48–56. [Google Scholar] [CrossRef]
- Bousquet, P.; Hudson, A.; García-Sevilla, J.A.; Li, J.X. Imidazoline Receptor System: The Past, the Present, and the Future. Pharmacol. Rev. 2020, 72, 50–79. [Google Scholar] [CrossRef]
- Rovati, L.C.; Brambilla, N.; Blicharski, T.; Connell, J.; Vitalini, C.; Bonazzi, A.; Giacovelli, G.; Girolami, F.; D’Amato, M. Efficacy and Safety of the First-in-Class Imidazoline-2 Receptor Ligand CR4056 in Pain from Knee Osteoarthritis and Disease Phenotypes: A Randomized, Double-Blind, Placebo-Controlled Phase 2 Trial. Osteoarthr. Cartil. 2020, 28, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.L.; Jessop, D.S.; Finn, D.P. Modulation of Stress by Imidazoline Binding Sites: Implications for Psychiatric Disorders. Stress 2009, 12, 97–114. [Google Scholar] [CrossRef] [Green Version]
- Callado, L.F.; Martín-Gómez, J.I.; Ruiz, J.; Garibi, J.M.; Meana, J.J. Imidazoline I2 Receptor Density Increases with the Malignancy of Human Gliomas. J. Neurol. Neurosurg. Psychiatry 2004, 75, 785–787. [Google Scholar] [CrossRef] [Green Version]
- Dardonville, C.; Rozas, I. Imidazoline Binding Sites and Their Ligands: An Overview of the Different Chemical Structures. Med. Res. Rev. 2004, 24, 639–661. [Google Scholar] [CrossRef]
- Rodriguez-Arévalo, S.; Bagán, A.; Griñán-Ferré, C.; Vasilopoulou, F.; Pallàs, M.; Brocos-Mosquera, I.; Callado, L.F.; Loza, M.I.; Martínez, A.L.; Brea, J.; et al. Benzofuranyl-2-Imidazoles as Imidazoline I2 Receptor Ligands for Alzheimer’s Disease. Eur. J. Med. Chem. 2021, 222, 113540. [Google Scholar] [CrossRef] [PubMed]
- Vasilopoulou, F.; Rodríguez-Arévalo, S.; Bagán, A.; Escolano, C.; Griñán-Ferré, C.; Pallàs, M. Disease-Modifying Treatment with I2 Imidazoline Receptor Ligand LSL60101 in an Alzheimer’s Disease Mouse Model: A Comparative Study with Donepezil. Br. J. Pharmacol. 2021, 178, 3017–3033. [Google Scholar] [CrossRef] [PubMed]
- Elders, N.; Schmitz, R.F.; de Kanter, F.J.J.; Ruijter, E.; Groen, M.B.; Orru, R.V.A. A Resource-Efficient and Highly Flexible Procedure for a Three-Component Synthesis of 2-Imidazolines. J. Org. Chem. 2007, 72, 6135–6142. [Google Scholar] [CrossRef] [PubMed]
- Elders, N.; Ruijter, E.; de Kanter, F.J.J.; Groen, M.B.; Orru, R.V.A. Selective Formation of 2-Imidazolines and 2-Substituted Oxazoles by Using a Three-Component Reaction. Chem. Eur. J. 2008, 14, 4961–4973. [Google Scholar] [CrossRef]
- Abás, S.; Estarellas, C.; Luque, F.J.; Escolano, C. Easy Access to (2-Imidazolin-4-Yl)Phosphonates by a Microwave Assisted Multicomponent Reaction. Tetrahedron 2015, 71, 2872–2881. [Google Scholar] [CrossRef]
- Abás, S.; Erdozain, A.M.; Keller, B.; Rodríguez-Arévalo, S.; Callado, L.F.; García-Sevilla, J.A.; Escolano, C. Neuroprotective Effects of a Structurally New Family of High Affinity Imidazoline I2 Receptor Ligands. ACS Chem. Neurosci. 2017, 8, 737–742. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Vasilopoulou, F.; Abás, S.; Rodríguez-Arévalo, S.; Bagán, A.; Sureda, F.X.; Pérez, B.; Callado, L.F.; García-Sevilla, J.A.; García-Fuster, M.J.; et al. Behavioral and Cognitive Improvement Induced by Novel Imidazoline I2 Receptor Ligands in Female SAMP8 Mice. Neurotherapeutics 2019, 16, 416–431. [Google Scholar] [CrossRef] [Green Version]
- Vasilopoulou, F.; Bagan, A.; Rodriguez-Arevalo, S.; Escolano, C.; Griñán-Ferré, C.; Pallàs, M. Amelioration of BPSD-Like Phenotype and Cognitive Decline in SAMP8 Mice Model Accompanied by Molecular Changes after Treatment with I2-Imidazoline Receptor Ligand MCR5. Pharmaceutics 2020, 12, 475. [Google Scholar] [CrossRef]
- Arróniz, C.; Molina, J.; Abás, S.; Molins, E.; Campanera, J.M.; Luque, F.J.; Escolano, C. First Diastereoselective [3 + 2] Cycloaddition Reaction of Diethyl Isocyanomethylphosphonate and Maleimides. Org. Biomol. Chem. 2013, 11, 1640–1649. [Google Scholar] [CrossRef]
- Abás, S.; Rodríguez-Arévalo, S.; Bagán, A.; Griñán-Ferré, C.; Vasilopoulou, F.; Brocos-Mosquera, I.; Muguruza, C.; Pérez, B.; Molins, E.; Luque, F.J.; et al. Bicyclic α-Iminophosphonates as High Affinity Imidazoline I2 Receptor Ligands for Alzheimer’s Disease. J. Med. Chem. 2020, 63, 3610–3633. [Google Scholar] [CrossRef]
- Escolano, C.; Pallás, M.; Griñán-Ferreé, C.; Abás, S.; Callado, L.F.; García-Sevilla, J.A. Synthetic I2 Imidazoline Receptor Ligands for Prevention or Treatment of Human Brain Disorders. Patent No. WO2019121853, 27 June 2019. [Google Scholar]
- Vasilopoulou, F.; Griñán-Ferré, C.; Rodríguez-Arévalo, S.; Bagán, A.; Abás, S.; Escolano, C.; Pallàs, M. I2 Imidazoline Receptor Modulation Protects Aged SAMP8 Mice against Cognitive Decline by Suppressing the Calcineurin Pathway. GeroScience 2020, 43, 965–983. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadal Rodríguez, P.; Ghashghaei, O.; Bagán, A.; Escolano, C.; Lavilla, R. Heterocycle-Based Multicomponent Reactions in Drug Discovery: From Hit Finding to Rational Design. Biomedicines 2022, 10, 1488. https://doi.org/10.3390/biomedicines10071488
Nadal Rodríguez P, Ghashghaei O, Bagán A, Escolano C, Lavilla R. Heterocycle-Based Multicomponent Reactions in Drug Discovery: From Hit Finding to Rational Design. Biomedicines. 2022; 10(7):1488. https://doi.org/10.3390/biomedicines10071488
Chicago/Turabian StyleNadal Rodríguez, Pau, Ouldouz Ghashghaei, Andrea Bagán, Carmen Escolano, and Rodolfo Lavilla. 2022. "Heterocycle-Based Multicomponent Reactions in Drug Discovery: From Hit Finding to Rational Design" Biomedicines 10, no. 7: 1488. https://doi.org/10.3390/biomedicines10071488
APA StyleNadal Rodríguez, P., Ghashghaei, O., Bagán, A., Escolano, C., & Lavilla, R. (2022). Heterocycle-Based Multicomponent Reactions in Drug Discovery: From Hit Finding to Rational Design. Biomedicines, 10(7), 1488. https://doi.org/10.3390/biomedicines10071488