
Insulin Resistance in Peripheral Tissues and the Brain: A Tale of Two Sites



Abstract
:1. Introduction
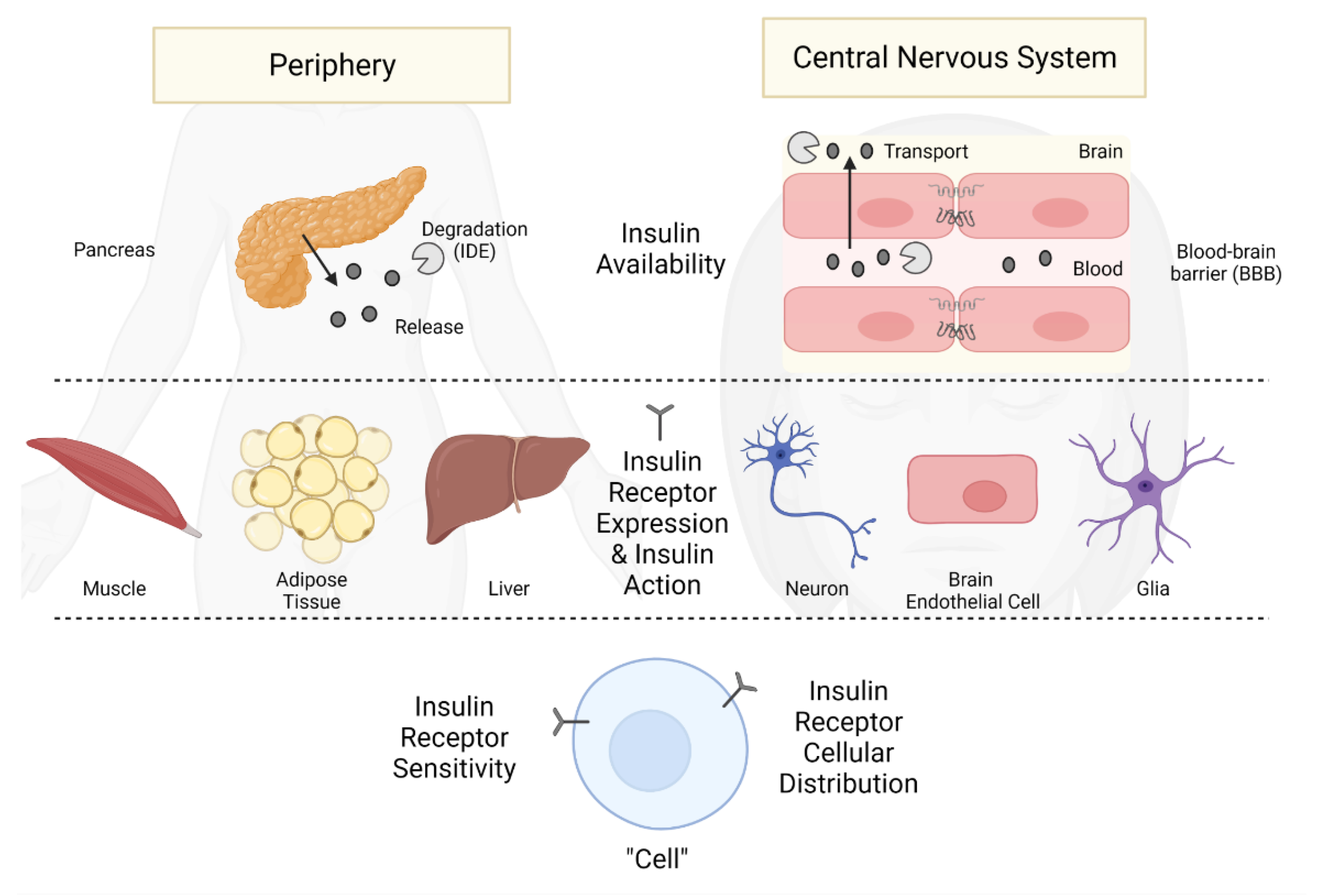
1.1. Periphery versus CNS: Separate Entities Connected by a Regulated Interface
1.2. Peripheral versus CNS apoE Pools
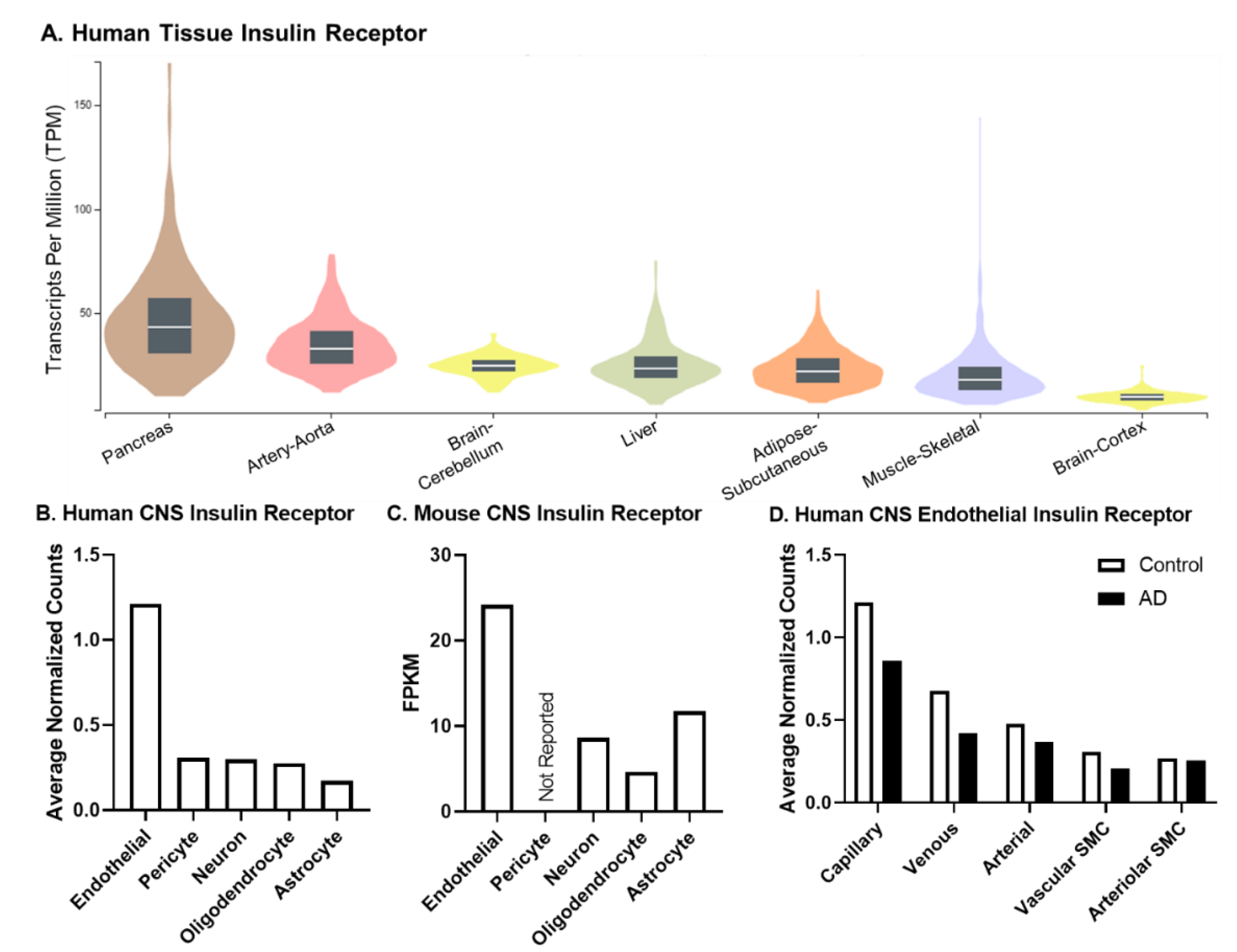
2. Insulin Receptor and Signaling
3. Peripheral Insulin Resistance
3.1. Peripheral Insulin Resistance and COVID-19
3.2. Role of apoE Status on Peripheral Insulin Resistance
3.3. Role of Ethnicity in Central and Peripheral apoE Effects
4. CNS Insulin Resistance
Impact of ApoE Status on CNS Insulin Resistance
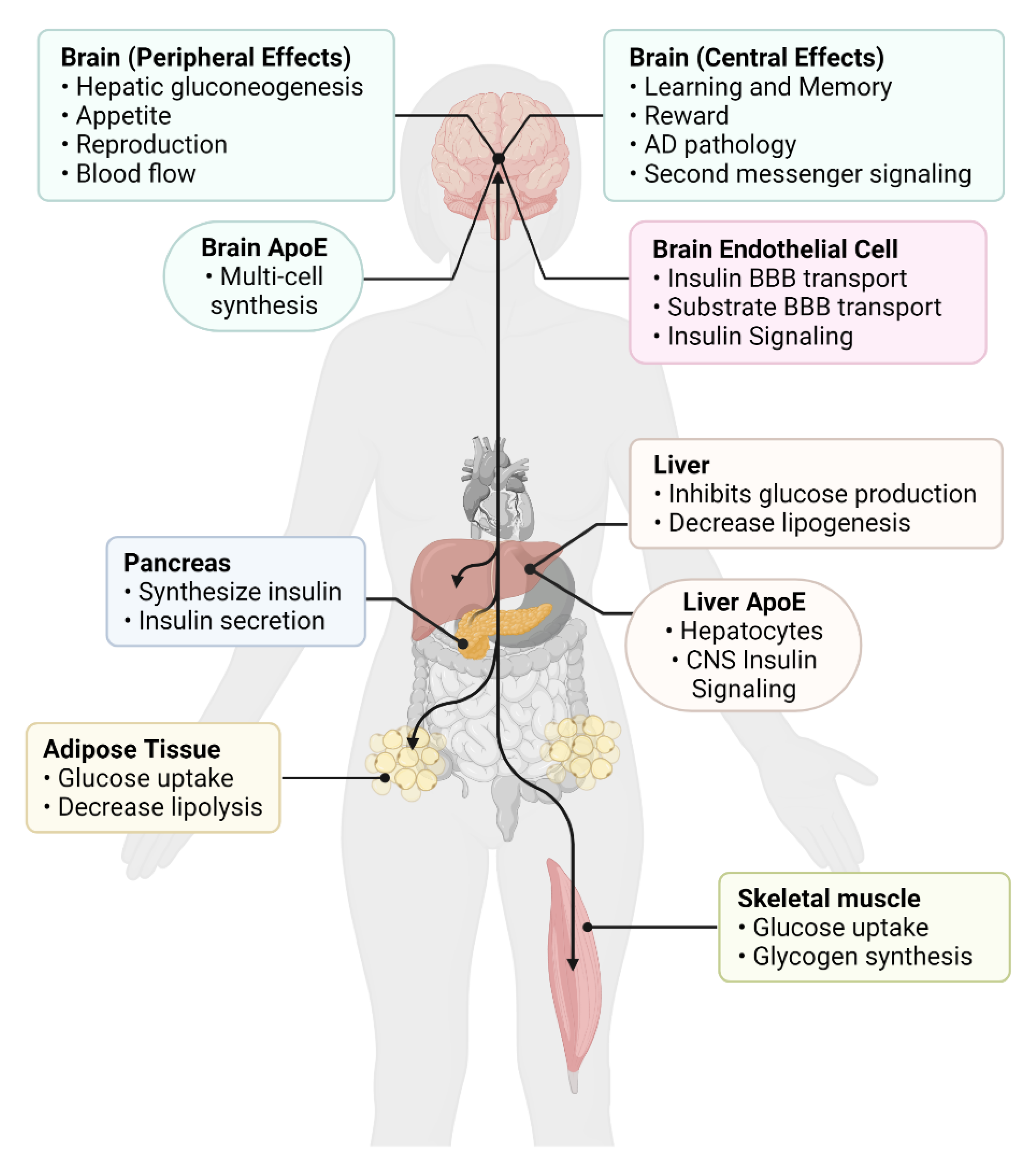
5. Contributions of the Periphery to CNS Insulin Actions
ApoE-Related Serum Factors Contributing to CNS Insulin Action
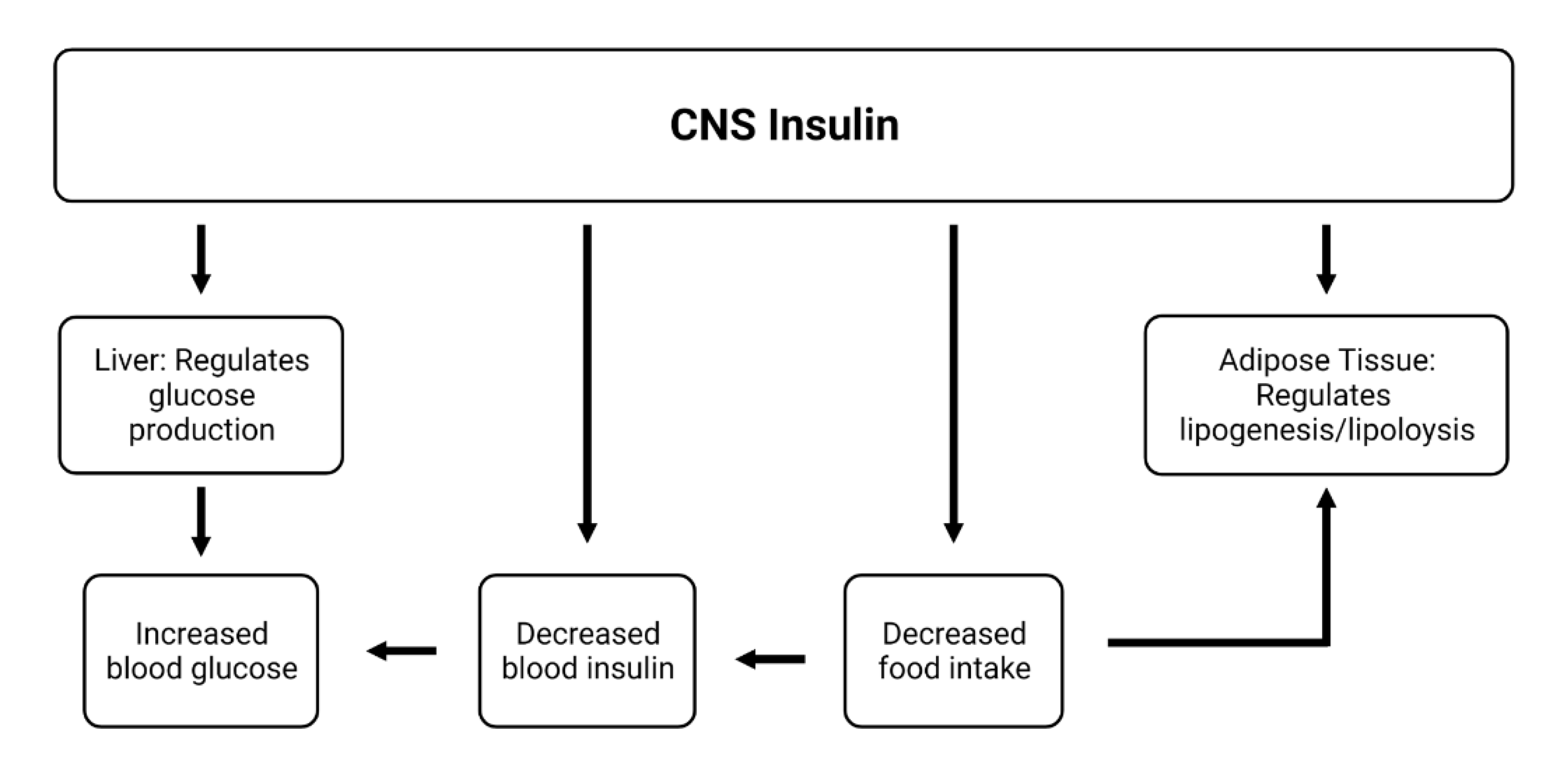
6. Influences of CNS Insulin on Peripheral Action
Impact of CNS ApoE on the Periphery
7. Conclusions and Remaining Questions
Funding
Data Availability Statement
Conflicts of Interest
References
- Insulin Resistance and Sensitivity. Br. Med. J. 1940, 1, 734. [CrossRef] [Green Version]
- Root, H.F.; Carpenter, T.M. Studies of Carbohydrate Metabolism in Cases of Insulin Resistance. Trans. Am. Clin. Climatol. Assoc. 1940, 56, 1–11. [Google Scholar] [PubMed]
- Lozinski, E.; Frohlich, L.I. Resistance to Insulin. Can Med. Assoc. J. 1942, 46, 62–65. [Google Scholar] [PubMed]
- Albright, F.; Burnett, C.H.; Smith, P.H.; Parson, W. Pseudohypoparathyroidism—An example of “Seabright-Bantam syndrome”. Endocrinology 1942, 30, 922–932. [Google Scholar]
- Verhoeven, G.F.; Wilson, J.D. The syndromes of primary hormone resistance. N. Engl. J. Med. 1979, 28, 253–289. [Google Scholar] [CrossRef]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Losada-Barragan, M. Physiological effects of nutrients on insulin release by pancreatic beta cells. Mol. Cell Biochem. 2021, 476, 3127–3139. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Batista, T.M.; Haider, N.; Kahn, C.R. Defining the underlying defect in insulin action in type 2 diabetes. Diabetologia 2021, 64, 994–1006. [Google Scholar] [CrossRef]
- Freeman, A.M.; Pennings, N. Insulin Resistance. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Terrie, Y. Insulin Resistance: Recognizing the Hidden Danger. Available online: https://www.pharmacytimes.com/view/insulin-resistance-recognizing-the-hidden-danger (accessed on 14 April 2022).
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Rhea, E.M.; Raber, J.; Banks, W.A. ApoE and cerebral insulin: Trafficking, receptors, and resistance. Neurobiol. Dis. 2020, 137, 104755. [Google Scholar] [CrossRef]
- Kuusisto, J.; Koivisto, K.; Mykkanen, L.; Helkala, E.L.; Vanhanen, M.; Hanninen, T.; Kervinen, K.; Kesaniemi, Y.A.; Riekkinen, P.J.; Laakso, M. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype: Cross sectional population based study. Br. Med. J. 1997, 315, 1045–1049. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Refetoff, S. Syndromes of thyroid hormone resistance. Am. J. Physiol. 1982, 243, E88–E98. [Google Scholar] [CrossRef]
- Sarret, C.; Oliver Petit, I.; Tonduti, D. Allan-Herndon-Dudley Syndrome. In GeneReview; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef]
- Linton, M.F.; Gish, R.; Hubl, S.T.; Bütler, E.; Esquivel, C.; Bry, W.I.; Boyles, J.K.; Wardell, M.R.; Young, S.G. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J. Clin. Investig. 1991, 88, 270–281. [Google Scholar] [CrossRef] [Green Version]
- Roses, A.D. Apolipoprotein E affects the rate of Alzheimer disease expression: B-amyloid burden is a secondary consequence dependent on APOE genotype and duration of disease. J. Neuropathol. Exp. Neurol. 1994, 53, 429–437. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Tangirala, R.; Chun, S.; Usher, D.; Pure, E.; Rader, D.J. Hepatic expression of apolipoprotein E inhibits progression of atherosclerosis without reducing cholesterol levels in LDL receptor-deficient mice. Mol. Ther. 2000, 1, 189–194. [Google Scholar] [CrossRef]
- Nascimento, J.; Matos, G.; Perreira, L.; Mourao, A.; Sampaio, A.; Oria, R.; Toniutto, P. Impact of apolipoprotein E genetic polymorphisms on liver disease: An essential review. Ann. Heptol. 2020, 19, 24–30. [Google Scholar] [CrossRef]
- Beaupere, C.; Liboz, A.; Feve, B.; Blondeau, B.; Guilemain, G. Molecular Mechanisms of Glucocorticoid-Induced Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 623. [Google Scholar] [CrossRef]
- Prack, M.M.; Nicosia, M.; Williams, D.L.; Gwynne, J. Relationship between apolipoprotein E mRNA expression and tissue cholestorol content in rat adrenal gland. J. Lipid Res. 1991, 32, 1611–1618. [Google Scholar] [CrossRef]
- Nicosia, M.; Prack, M.M.; Williams, D.L. Differential regulation of apolipoprotein-E messenger RNA in zona fasciculata cells of rat adrenal gland determined by in situ hybridization. Mol. Endocrinol. 1992, 6, 288–298. [Google Scholar]
- Raber, J.; Akana, S.F.; Bhatnager, S.; Dallman, M.F.; Wong, D.; Mucke, L. Hypothalamic-pituitary-adrenal dysfunction in ApoE−/− mice: Possible role in behavioral and metabolic alterations. J. Neurosci. 2000, 20, 2064–2071. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, D.; Spellicy, C.; Harding, M.; Graham, C. Apolipoprotein E DNA methylation and posttraumatic stress disorder are associated with plasma ApoE level: A preliminary study. Behav. Brain Res. 2019, 356, 415–422. [Google Scholar] [CrossRef]
- Buttini, M.; Orth, M.; Bellosta, S.; Akeefe, H.; Pitas, R.E.; Wyss-Coray, T.; Mucke, L.; Mahley, R.W. Expression of human apolipoprotein E3 or E4 in the brains of Apoe–/– mice: Isoform-specific effects on neurodegeneration. J. Neurosci. 1999, 19, 4867–4880. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Hyejin, Y.; Jungsu, K. Apolipoprotein E metabolism and functions in brain and its role in Alzheimer’s disease. Curr. Opin. Lipidol. 2017, 28, 60–67. [Google Scholar]
- Torres, E.; Luo, J.; Boehnlein, J.; Towns, D.; Kinzie, D.; DeBarber, A.; Raber, J. Apolipoprotein E Isoform-specific changes related to stress and trauma exposure. Transl. Psychiatry 2022, 12, 125. [Google Scholar] [CrossRef]
- Johnson, L.; Olsen, R.; Merkens, L.; DeBarber, A.; Steiner, R.; Sullivan, P.; Maeda, N.; Raber, J. Apolipoprotein E–low density lipoprotein receptor interaction affects spatial memory retention and brain ApoE levels in an isoform-dependent manner. Neurobiol. Dis. 2014, 64, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, T.-P.V.; Wang, C.; Tran, A.; Tabor, T.; Mahan, T.; Francis, C.; Finn, M.; Spellman, R.; Manis, M.; Tanzi, R.E.; et al. Lack of hepatic apoE does not influence early Aβ deposition: Observations from a new APOE knock-in model. Mol. Neurodegener. 2019, 14, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernick, D.; Ortiz-Valle, S.; Jeong, A.; Li, L. Peripheral Versus Central Nervous System APOE in Alzheimer’s Disease: Interplay across the Blood-Brain Barrier. Neurosci. Lett. 2019, 708, 134306. [Google Scholar] [CrossRef] [PubMed]
- Giannisis, A.; Patra, K.; Edlund, A.; Nieto, L.; Benedicto-Gras, J.; Moussaud, S.; de la Rosa, A.; Twohig, D.; Bengtsson, T.; Fu, Y.; et al. Brain integrity is altered by hepatic APOE ε4 in humanized-liver mice. Mol. Psychiatry, 2022; Epub ahead of print. [Google Scholar] [CrossRef]
- Patra, K.; Giannisis, A.; Edlund, A.; Sando, S.; Lauridsen, C.; Berge, G.; GR, F.; Brathen, G.; White, L.; Nielsen, H. Plasma Apolipoprotein E Monomer and Dimer Profile and Relevance to Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 71, 1217–1231. [Google Scholar] [CrossRef]
- Rasmussen, K.L.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Plasma apolipoprotein E levels and risk of dementia: A Mendelian randomization study of 106,562 individuals. Alzheimers Dement. 2018, 14, 71–80. [Google Scholar] [CrossRef]
- Nielsen, H.M.; Chen, K.; Lee, W.; Chen, Y.; Bauer, R.J., 3rd; Reiman, E.; Caselli, R.; Bu, G. Peripheral apoE isoform levels in cognitively normal APOE epsilon3/epsilon4 individuals are associated with regional gray matter volume and cerebral glucose metabolism. Alzheimers Res. Ther. 2017, 9, 5. [Google Scholar] [CrossRef]
- Martinez-Morillo, E.; Hansson, O.; Atagi, Y.; Bu, G.; Minthon, L.; Diamandis, E.P.; Nielsen, H.M. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol. 2014, 127, 633–643. [Google Scholar] [CrossRef]
- Baker-Nigh, A.T.; Mawuenyega, K.G.; Bollinger, J.G.; Ovod, V.; Kasten, T.; Franklin, E.E.; Liao, F.; Jiang, H.; Holtzman, D.; Cairns, N.J.; et al. Human Central Nervous System (CNS) ApoE Isoforms Are Increased by Age, Differentially Altered by Amyloidosis, and Relative Amounts Reversed in the CNS Compared with Plasma. J. Biol. Chem. 2016, 291, 27204–27218. [Google Scholar] [CrossRef] [Green Version]
- Sideris-Mapretsas, G.; Malcangio, M. Pain-resolving microglia. Science 2022, 376, 33–34. [Google Scholar] [CrossRef]
- Kohno, K.; Shirasaka, K.; TYoshihara, K.; Mikuriya, S.; Tanaka, K.; Takanami, K.; Inoue, K.; Sakamoto, H.; Ohkawa, Y.; Masuda, T.; et al. A spinal microglia population involved in remitting and relapsing neuropathic pain. Science 2022, 376, 86–90. [Google Scholar] [CrossRef]
- Kasuga, M.; Fujita-Yamaguchi, Y.; Blithe, D.L.; White, M.F.; Kahn, C.R. Characterization of the insulin receptor kinase purified from human placental membranes. J. Biol. Chem. 1983, 258, 10973–10980. [Google Scholar] [CrossRef]
- Nakamura, S.; Mori, K.; Okuma, H.; Sekine, T.; Miyazaki, A.; Tsuchiya, K. Age-associated decline of monocyte insulin sensitivity in diabetic and healthy individuals. Diabetes Vasc. Dis. Res. 2021, 18, 1479164121989281. [Google Scholar] [CrossRef]
- Yang, A.C.; Vest, R.T.; Kern, F.; Lee, D.P.; Agam, M.; Maat, C.A.; Losada, P.M.; Chen, M.B.; Schaum, N.; Khoury, N.; et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 2022, 603, 885–892. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Slaaby, R. Specific insulin/IGF1 hybrid receptor activation assay reveals IGF1 as a more potent ligand than insulin. Sci. Rep. 2015, 5, 7911. [Google Scholar] [CrossRef] [Green Version]
- Leclerc, M.; Bourassa, P.; Tremblay, C.; Caron, V.; Sugère, C.; Emond, V.; Bennett, D.A.; Calon, F. Cerebrovascular insulin receptors are defective in Alzheimer’s disease. bioRxiv 2021. [Google Scholar] [CrossRef]
- Banks, W.A.; Farr, S.A.; Morley, J.E. Permeability of the blood-brain barrier to albumin and insulin in the young and aged SAMP8 mouse. J. Gerontol. A Biol. Sci. Med. Sci. 2000, 55, B601–B606. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.L.; Zheng, B.; Hu, Z.; Price, S.R.; Mitch, W.E. Chronic kidney disease causes defects in signaling through the insulin receptor substrate/phosphatidylinositol 3-kinase/Akt pathway: Implications for muscle atrophy. J. Am. Soc. Nephrol. 2006, 17, 1388–1394. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Sabbagh, F.; Muhamad, I.I.; Niazmand, R.; Dikshit, P.K.; Kim, B.S. Recent progress in polymeric non-invasive insulin delivery. Int. J. Biol. Macromol. 2022, 203, 222–243. [Google Scholar] [CrossRef]
- De Meyts, P. The Insulin Receptor and Its Signal Transduction Network. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Bergeron, J.J.; Di Guglielmo, G.M.; Dahan, S.; Dominguez, M.; Posner, B.I. Spatial and Temporal Regulation of Receptor Tyrosine Kinase Activation and Intracellular Signal Transduction. Annu. Rev. Biochem. 2016, 85, 573–597. [Google Scholar] [CrossRef]
- Accili, D.; Drago, J.; Lee, E.J.; Johnson, M.D.; Cool, M.H.; Salvatore, P.; Asico, L.D.; Jose, P.A.; Taylor, S.I.; Westphal, H. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat. Genet. 1996, 12, 106–109. [Google Scholar] [CrossRef]
- Gonzalez-Casimiro, C.M.; Merino, B.; Casanueva-Alvarez, E.; Postigo-Casado, T.; Camara-Torres, P.; Fernandez-Diaz, C.M.; Leissring, M.A.; Cozar-Castellano, I.; Perdomo, G. Modulation of Insulin Sensitivity by Insulin-Degrading Enzyme. Biomedicines 2021, 9, 86. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Krycer, J.R.; Kearney, A.L.; Hocking, S.L.; James, D.E. Muscle and adipose tissue insulin resistance: Malady without mechanism? J. Lipid Res. 2019, 60, 1720–1732. [Google Scholar] [CrossRef]
- Nesto, R. C-reactive protein, its role in inflammation, Type 2 diabetes and cardiovascular disease, and the effects of insulin-sensitizing treatment with thiazolidinediones. Diabet. Med. 2004, 21, 810–817. [Google Scholar] [CrossRef]
- Kubaszek, A.; Pihlajamaki, J.; Komarovski, V.; Lindi, V.; Lindstrom, J.; Eriksson, J.; Valle, T.T.; Hamalainen, H.; Ilanne-Parikka, P.; Keinanen-Kiukaanniemi, S.; et al. Promoter polymorphisms of the TNF-alpha (G-308A) and IL-6 (C-174G) genes predict the conversion from impaired glucose tolerance to type 2 diabetes: The Finnish Diabetes Prevention Study. Diabetes 2003, 52, 1872–1876. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. J. Biomed. Biotechnol. 2010, 2010, 476279. [Google Scholar] [CrossRef] [Green Version]
- McGarry, J.D. Banting lecture 2001: Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002, 51, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Nono Nankam, P.A.; Bluher, M. Retinol-binding protein 4 in obesity and metabolic dysfunctions. Mol. Cell Endocrinol. 2021, 531, 111312. [Google Scholar] [CrossRef]
- Rhea, E.M.; Banks, W.A. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front. Neurosci. 2019, 13, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A. The blood-brain barrier as an endocrine tissue. Nat. Rev. Endocrinol. 2019, 15, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Rhea, E.M. The Blood-Brain Barrier, Oxidative Stress, and Insulin Resistance. Antioxidants 2021, 10, 1695. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Banks, W.A. Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions. Pharmacol. Rev. 2018, 70, 278–314. [Google Scholar] [CrossRef]
- Schaum, N.; Lehallier, B.; Hahn, O.; Palovics, R.; Hosseinzadeh, S.; Lee, S.E.; Sit, R.; Lee, D.P.; Losada, P.M.; Zardeneta, M.E.; et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 2020, 583, 596–602. [Google Scholar] [CrossRef]
- DeFronzo, R.; Deibert, D.; Hendler, R.; Felig, P.; Soman, V. Insulin sensitivity and insulin binding to monocytes in maturity-onset diabetes. J. Clin. Investig. 1979, 63, 939–946. [Google Scholar] [CrossRef]
- Serrano, R.; Villar, M.; Gallardo, N.; Carrascosa, J.M.; Martinez, C.; Andres, A. The effect of aging on insulin signalling pathway is tissue dependent: Central role of adipose tissue in the insulin resistance of aging. Mech. Ageing Dev. 2009, 130, 189–197. [Google Scholar] [CrossRef]
- Carvalho, C.R.; Brenelli, S.L.; Silva, A.C.; Nunes, A.L.; Velloso, L.A.; Saad, M.J. Effect of aging on insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of rats. Endocrinology 1996, 137, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Mykkanen, L.; Kuusisto, J.; Pyorala, K.; Laakso, M. Cardiovascular disease risk factors as predictors of type 2 (non-insulin-dependent) diabetes mellitus in elderly subjects. Diabetologia 1993, 36, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Stolk, R.P.; Breteler, M.M.; Ott, A.; Pols, H.A.; Lamberts, S.W.; Grobbee, D.E.; Hofman, A. Insulin and cognitive function in an elderly population. The Rotterdam Study. Diabetes Care 1997, 20, 792–795. [Google Scholar] [CrossRef]
- Craft, S.; Newcomer, J.; Kanne, S.; Dagogo-Jack, S.; Cryer, P.; Sheline, Y.; Luby, J.; Dagogo-Jack, A.; Alderson, A. Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiol. Aging 1996, 17, 123–130. [Google Scholar] [CrossRef]
- Salameh, T.S.; Shah, G.N.; Price, T.O.; Hayden, M.R.; Banks, W.A. Blood-Brain Barrier Disruption and Neurovascular Unit Dysfunction in Diabetic Mice: Protection with the Mitochondrial Carbonic Anhydrase Inhibitor Topiramate. J. Pharmacol. Exp. Ther. 2016, 359, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Rhea, E.M.; Salameh, T.S.; Banks, W.A. Routes for the delivery of insulin to the central nervous system: A comparative review. Exp. Neurol. 2019, 313, 10–15. [Google Scholar] [CrossRef]
- Frolich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Turk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. 1998, 105, 423–438. [Google Scholar] [CrossRef]
- Hoyer, S. Is sporadic Alzheimer disease the brain type of non-insulin dependent diabetes mellitus? A challenging hypothesis. J. Neural Transm. 1998, 105, 415–422. [Google Scholar] [CrossRef]
- Denson, J.L.; Gillet, A.S.; Zu, Y.; Brown, M.; Pham, T.; Yoshida, Y.; Mauvais-Jarvis, F.; Douglas, I.S.; Moore, M.; Tea, K.; et al. Metabolic Syndrome and Acute Respiratory Distress Syndrome in Hospitalized Patients With COVID-19. JAMA Netw. Open 2021, 4, e2140568. [Google Scholar] [CrossRef]
- Ren, H.; Yang, Y.; Wang, F.; Yan, Y.; Shi, X.; Dong, K.; Yu, X.; Zhang, S. Association of the insulin resistance marker TyG index with the severity and mortality of COVID-19. Cardiovasc. Diabetol. 2020, 19, 58. [Google Scholar] [CrossRef]
- Cromer, S.J.; Colling, C.; Schatoff, D.; Leary, M.; Stamou, M.I.; Selen, D.J.; Putman, M.S.; Wexler, D.J. Newly diagnosed diabetes vs. pre-existing diabetes upon admission for COVID-19: Associated factors, short-term outcomes, and long-term glycemic phenotypes. J. Diabetes Complicat. 2022, 36, 108145. [Google Scholar] [CrossRef]
- Montefusco, L.; Ben Nasr, M.; D’Addio, F.; Loretelli, C.; Rossi, A.; Pastore, I.; Daniele, G.; Abdelsalam, A.; Maestroni, A.; Dell’Acqua, M.; et al. Acute and long-term disruption of glycometabolic control after SARS-CoV-2 infection. Nat. Metab. 2021, 3, 774–785. [Google Scholar] [CrossRef]
- He, X.; Liu, C.; Peng, J.; Li, Z.; Li, F.; Wang, J.; Hu, A.; Peng, M.; Huang, K.; Fan, D.; et al. COVID-19 induces new-onset insulin resistance and lipid metabolic dysregulation via regulation of secreted metabolic factors. Signal Transduct. Target. Ther. 2021, 6, 427. [Google Scholar] [CrossRef]
- Chen, M.; Zhu, B.; Chen, D.; Hu, X.; Xu, X.; Shen, W.J.; Hu, C.; Li, J.; Qu, S. COVID-19 May Increase the Risk of Insulin Resistance in Adult Patients Without Diabetes: A 6-Month Prospective Study. Endocr. Pract. 2021, 27, 834–841. [Google Scholar] [CrossRef]
- Srivastava, A.; Rockman-Greenberg, C.; Sareen, N.; Lionetti, V.; Dhingra, S. An insight into the mechanisms of COVID-19, SARS-CoV2 infection severity concerning β-cell survival and cardiovascular conditions in diabetic patients. Mol. Cell Biochem. 2022, 477, 1681–1695. [Google Scholar] [CrossRef]
- Govender, N.; Khaliq, O.P.; Moodley, J.; Naicker, T. Insulin resistance in COVID-19 and diabetes. Prim. Care Diabetes 2021, 15, 629–634. [Google Scholar] [CrossRef]
- Zangiabadian, M.; Nejadghaderi, S.A.; Zahmatkesh, M.M.; Hajikhani, B.; Mirsaeidi, M.; Nasiri, M.J. The Efficacy and Potential Mechanisms of Metformin in the Treatment of COVID-19 in the Diabetics: A Systematic Review. Front. Endocrinol. 2021, 12, 645194. [Google Scholar] [CrossRef]
- Liu, Y.H.; Chen, Y.; Wang, Q.H.; Wang, L.R.; Jiang, L.; Yang, Y.; Chen, X.; Li, Y.; Cen, Y.; Xu, C.; et al. One-Year Trajectory of Cognitive Changes in Older Survivors of COVID-19 in Wuhan, China: A Longitudinal Cohort Study. JAMA Neurol. 2022, 79, 509–517. [Google Scholar] [CrossRef]
- Reiken, S.; Sittenfeld, L.; Dridi, H.; Liu, Y.; Liu, X.; Marks, A.R. Alzheimer’s-like signaling in brains of COVID-19 patients. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2022, 18, 955–965. [Google Scholar] [CrossRef]
- Starks, E.J.; O’Grady, J.; Hoscheidt, S.; Racine, A.; Carlsson, C.; Zetterberg, H.; Blennow, K.; Okonkwo, A.; Puglielli, L.; Asthana, S.; et al. Insulin Resistance is Associated with Higher Cerebrospinal Fluid Tau Levels in Asymptomatic APOEɛ4 Carriers. J. Alzheimer’s Dis. 2015, 46, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Ekblad, L.; Johansson, J.; Helin, S.; Viitanen, M.; Laine, H.; Puuka, P.; Jula, A.; Rinne, J. Midlife insulin resistance, APOE genotype, and late-life brain amyloid accumulation. Neurology 2018, 90, e1150–e1157. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Ski, J.; Li, Y.; Yang, Y.; Ren, S. Association between ApoE Polymorphism and Type 2 Diabetes: A Meta-Analysis of 59 Studies. Biomed. Environ. Sci. 2019, 32, 823–838. [Google Scholar]
- Johnson, L.A.; Torres, E.R.S.; Impey, S.; Stevens, J.F.; Raber, J. Apolipoprotein E4 and insulin resistance interact to impair cognition and alter the epigenome and metabolome. Sci. Rep. 2017, 7, srep43701. [Google Scholar] [CrossRef] [Green Version]
- Raygor, V.; Abbasi, F.; Lazzeroni, L.; Kim, S.; Ingelsson, E.; Reaven, G.; Knowles, J. Impact of race/ethnicity on insulin resistance and hypertriglyceridaemia. Diabetes Vasc. Dis. Res. 2019, 16, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Laws, A.; Jeppesen, J.; Maheux, P. Resistance to insulin-stimulated glucose uptake and dyslipidemia in Asian Indians. Arter. Thromb 1994, 14, 917–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanaya, A.M.; Herrington, D.; Vittinghoff, E.; Ewing, S.K.; Liu, K.; Blaha, M.J.; Dave, S.S.; Qureshi, F.; Kandula, N.R. Understanding the high prevalence of diabetes in U.S. South Asians compared with four racial/ethnic groups: The MASALA and MESA studies. Diabetes Care 2014, 37, 1621–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raji, A.; Seely, E.; Arky, R.; Simonson, D. Body fat distribution and insulin resistance in healthy Asian Indians and Caucasians. J. Clin. Biochem. Nutr. 2001, 86, 5366–5371. [Google Scholar] [CrossRef]
- do Vale Moreira, N.; Ceriello, A.; Basit, A.; Balde, N.; Mohan, V.; Gupta, R.; Misra, A.; Bhowmik, B.; Lee, M.; Zuo, H.-Z.; et al. Race/ethnicity and challenges for optimal insulin therapy. Diabetes Res. Clin. Pract. 2021, 175, 108823. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. J. Am. Med. Assoc. 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Turney, I.; Chesebro, A.; Renteria, M.; Lao, P.; Beato, J.; Schupf, N.; Mayeux, R.; Manly, J.; Brickman, A.M. APOE ε4 and resting-state functional connectivity in racially/ethnically diverse older adults. Alzheimer Dement. 2020, 12, e12094. [Google Scholar]
- Hyman, B.; Tanzi, R. Effects of Species-Specific Genetics on Alzheimer’s Mouse Models. Neuron 2019, 101, 351–352. [Google Scholar] [CrossRef] [Green Version]
- Neuner, S.; Heuer, S.; Huentelman, M.; O’Connell, K.; Kaczorowski, C. Harnessing genetic complexity to enhance translatability of Alzheimer’s disease mouse models: A path toward precision medicine. Neuron 2019, 101, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Grillo, C.A.; Piroli, G.G.; Hendry, R.M.; Reagan, L.P. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res. 2009, 1296, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Thibault, O.; Anderson, K.L.; DeMoll, C.; Brewer, L.D.; Landfield, P.W.; Porter, N.M. Hippocampal calcium dysregulation at the nexus of diabetes and brain aging. Eur. J. Pharmacol. 2013, 719, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Spanswick, D.; Smith, M.A.; Mirshamsi, S.; Routh, V.H.; Ashford, M.L. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat. Neurosci. 2000, 3, 757–758. [Google Scholar] [CrossRef]
- Gratuze, M.; Julien, J.; Petry, F.R.; Morin, F.; Planel, E. Insulin deprivation induces PP2A inhibition and tau hyperphosphorylation in hTau mice, a model of Alzheimer’s disease-like tau pathology. Sci. Rep. 2017, 7, 46359. [Google Scholar] [CrossRef] [Green Version]
- Vandal, M.; Bourassa, P.; Calon, F. Can insulin signaling pathways be targeted to transport Abeta out of the brain? Front. Aging Neurosci. 2015, 7, 114. [Google Scholar] [CrossRef]
- Figlewicz, D.P. Insulin, food intake, and reward. Semin. Clin. Neuropsychiatry 2003, 8, 82–93. [Google Scholar] [CrossRef]
- Zhao, W.Q.; Alkon, D.L. Role of insulin and insulin receptor in learning and memory. Mol. Cell. Endocrinol. 2001, 177, 125–134. [Google Scholar] [CrossRef]
- Cabou, C.; Cani, P.D.; Campistron, G.; Knauf, C.; Mathieu, C.; Sartori, C.; Amar, J.; Scherrer, U.; Burcelin, R. Central insulin regulates heart rate and arterial blood flow: An endothelial nitric oxide synthase-dependent mechanism altered during diabetes. Diabetes 2007, 56, 2872–2877. [Google Scholar] [CrossRef] [Green Version]
- Bruning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Muller-Wieland, D.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef]
- Ruud, J.; Steculorum, S.M.; Bruning, J.C. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat. Commun. 2017, 8, 15259. [Google Scholar] [CrossRef] [Green Version]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Haring, H.U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef] [Green Version]
- Tschritter, O.; Preissl, H.; Hennige, A.M.; Stumvoll, M.; Porubska, K.; Frost, R.; Marx, H.; Klosel, B.; Lutzenberger, W.; Birbaumer, N.; et al. The cerebrocortical response to hyperinsulinemia is reduced in overweight humans: A magnetoencephalographic study. Proc. Natl. Acad. Sci. USA 2006, 103, 12103–12108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirvonen, J.; Virtanen, K.A.; Nummenmaa, L.; Hannukainen, J.C.; Honka, M.J.; Bucci, M.; Nesterov, S.V.; Parkkola, R.; Rinne, J.; Iozzo, P.; et al. Effects of insulin on brain glucose metabolism in impaired glucose tolerance. Diabetes 2011, 60, 443–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhea, E.M.; Banks, W.A. A historical perspective on the interactions of insulin at the blood-brain barrier. J. Neuroendocrinol. 2021, 33, e12929. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; Pemberton, S.; Babin, A.; Abdulhameed, N.; Noonan, C.; Brown, M.B.; Banks, W.A.; Rhea, E.M. Insulin blood-brain barrier transport and interactions are greater following exercise in mice. J. Appl. Physiol. 2022, 132, 824–834. [Google Scholar] [CrossRef]
- Pocai, A.; Lam, T.K.; Gutierrez-Juarez, R.; Obici, S.; Schwartz, G.J.; Bryan, J.; Aguilar-Bryan, L.; Rossetti, L. Hypothalamic K(ATP) channels control hepatic glucose production. Nature 2005, 434, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Goetzl, E.J.; Kapogiannis, D.; Lista, S.; Vergallo, A. Biomarker-Drug and Liquid Biopsy Co-development for Disease Staging and Targeted Therapy: Cornerstones for Alzheimer’s Precision Medicine and Pharmacology. Front. Pharmacol. 2019, 10, 310. [Google Scholar] [CrossRef]
- Mullins, R.J.; Mustapic, M.; Goetzl, E.J.; Kapogiannis, D. Exosomal biomarkers of brain insulin resistance associated with regional atrophy in Alzheimer’s disease. Hum. Brain Mapp. 2017, 38, 1933–1940. [Google Scholar] [CrossRef] [Green Version]
- Walker, K.A.; Chawla, S.; Nogueras-Ortiz, C.; Coresh, J.; Sharrett, A.R.; Wong, D.F.; Jack, C.R., Jr.; Spychalla, A.J.; Gottesman, R.F.; Kapogiannis, D. Neuronal insulin signaling and brain structure in nondemented older adults: The Atherosclerosis Risk in Communities Study. Neurobiol. Aging 2021, 97, 65–72. [Google Scholar] [CrossRef]
- Craft, S.; Peskind, E.; Schwartz, M.W.; Schellenberg, G.D.; Raskind, M.; Porte, D., Jr. Cerebrosinal fluid and plasma insulin levels in Alzheimer’s disease: Relationship to severity of dementia and apolipoprotein E genotype. Neurology 1998, 50, 164–168. [Google Scholar] [CrossRef]
- Gil-Bea, F.J.; Solas, M.; Solomon, A.; Mugueta, C.; Winblad, B.; Kvipelto, M.; Ramierez, M.J.; Cedazo-Minguez, A. Insulin levels are decreased in the cerebrospinal fluid of women with prodomal Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 22, 405–413. [Google Scholar] [CrossRef]
- Sartorius, T.; Peter, A.; Heni, M.; Maetzler, W.; Fritsche, A.; Haring, H.U.; Hennige, A.M. The brain response to peripheral insulin declines with age: A contribution of the blood-brain barrier? PLoS ONE 2015, 10, e0126804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, S.C.; Porte, D., Jr. Relationship between plasma and cerebrospinal fluid insulin levels of dogs. Am. J. Physiol. 1977, 233, E331–E334. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Jaspan, J.B.; Huang, W.; Kastin, A.J. Transport of insulin across the blood-brain barrier: Saturability at euglycemic doses of insulin. Peptides 1997, 18, 1423–1429. [Google Scholar] [CrossRef]
- Banks, W.A.; Jaspan, J.B.; Kastin, A.J. Selective, physiological transport of insulin across the blood-brain barrier: Novel demonstration by species-specific enzyme immunoassays. Peptides 1997, 18, 1257–1262. [Google Scholar] [CrossRef]
- Rhea, E.M.; Rask-Madsen, C.; Banks, W.A. Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J. Physiol. 2018, 596, 4753–4765. [Google Scholar] [CrossRef]
- Urayama, A.; Banks, W.A. Starvation and triglycerides reverse the obesity-induced impairment of insulin transport at the blood-brain barrier. Endocrinology 2008, 149, 3592–3597. [Google Scholar] [CrossRef]
- Banks, W.A.; Farr, S.A.; Salameh, T.S.; Niehoff, M.L.; Rhea, E.M.; Morley, J.E.; HAnson, A.J.; Hansen, K.M.; Craft, S. Triglycerides cross the blood-brain barrier and induce central leptin and insulin receptor resistance. Int. J. Obes. 2018, 42, 391–397. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Helmerhorst, E.; Taddel, K.; Plewright, B.; van Bronswijk, W.; Martins, R. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J. Neurosci. 2002, 22, RC221. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.G.; Leverenz, J.B.; McMillan, P.J.; Kulstad, J.J.; Ericksen, S.; Roth, R.A.; Schellenberg, G.D.; Jin, L.W.; Kovacina, K.S.; Craft, S. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer’s disease is associated with apolipoprotein -epsilon4 allele. Am. J. Pathol. 2003, 162, 313–319. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef] [Green Version]
- De Felice, F.G.; Goncalves, R.A.; Ferreira, S.T. Impaired insulin signalling and allostatic load in Alzheimer disease. Nat. Rev. Neurosci. 2022, 23, 215–230. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef]
- Sharma, N.; Tramutola, A.; Lanzillotta, C.; Arena, A.; Blarzino, C.; Cassano, T.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease. Neurobiol. Dis. 2019, 125, 176–189. [Google Scholar] [CrossRef]
- Cimini, F.A.; Barchetta, I.; Zuliani, I.; Pagnotta, S.; Bertoccini, L.; Dule, S.; Zampieri, M.; Reale, A.; Baroni, M.G.; Cavallo, M.G.; et al. Biliverdin reductase-A protein levels are reduced in type 2 diabetes and are associated with poor glycometabolic control. Life Sci. 2021, 284, 119913. [Google Scholar] [CrossRef]
- Cai, W.; Xue, C.; Sakaguchi, M.; Konishi, M.; Shirazian, A.; Ferris, H.A.; Li, M.E.; Yu, R.; Kleinridders, A.; Pothos, E.N.; et al. Insulin regulates astrocyte gliotransmission and modulates behavior. J. Clin. Investig. 2018, 128, 2914–2926. [Google Scholar] [CrossRef]
- Reagan, L.P.; Cowan, H.B.; Woodruff, J.L.; Piroli, G.G.; Erichsen, J.M.; Evans, A.N.; Burzynski, H.E.; Maxwell, N.D.; Loyo-Rosado, F.Z.; Macht, V.A.; et al. Hippocampal-specific insulin resistance elicits behavioral despair and hippocampal dendritic atrophy. Neurobiol. Stress 2021, 15, 100354. [Google Scholar] [CrossRef]
- Kullmann, S.; Kleinridders, A.; Small, D.M.; Fritsche, A.; Haring, H.U.; Preissl, H.; Heni, M. Central nervous pathways of insulin action in the control of metabolism and food intake. Lancet Diabetes Endocrinol. 2020, 8, 524–534. [Google Scholar] [CrossRef]
- Erichsen, J.M.; Fadel, J.R.; Reagan, L.P. Peripheral versus central insulin and leptin resistance: Role in metabolic disorders, cognition, and neuropsychiatric diseases. Neuropharmacology 2022, 203, 108877. [Google Scholar] [CrossRef]
- Hughes, T.M.; Craft, S. The role of insulin in the vascular contributions to age-related dementia. Biochim. Biophys. Acta 2016, 1862, 983–991. [Google Scholar] [CrossRef]
- Kullmann, S.; Heni, M.; Veit, R.; Scheffler, K.; Machann, J.; Haring, H.U.; Fritsche, A.; Preissl, H. Selective insulin resistance in homeostatic and cognitive control brain areas in overweight and obese adults. Diabetes Care 2015, 38, 1044–1050. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.J.; Friston, K.J.; Colebatch, J.G.; Frackowiak, R.S. Decreases in regional cerebral blood flow with normal aging. J. Cereb. Blood Flow Metab. 1991, 11, 684–689. [Google Scholar] [CrossRef] [Green Version]
- Sano, M.; Zhu, C.W.; Grossman, H.; Schimming, C. Longitudinal Cognitive Profiles in Diabetes: Results from the National Alzheimer’s Coordinating Center’s Uniform Data. J. Am. Geriatr. Soc. 2017, 65, 2198–2204. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Yamaguchi, K.; Matsui, K.; Sano, T.; Kubota, T.; Hashimoto, T.; Mano, A.; Yamada, K.; Matsuo, Y.; Kubota, N.; et al. Differential effects of diet- and genetically-induced brain insulin resistance on amyloid pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 15. [Google Scholar] [CrossRef]
- Peila, R.; Rodriguez, B.; Launer, L. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia aging study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef] [Green Version]
- Claxton, A.; Baker, L.; Hanson, A.L.; Trittschuh, E.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimer’s Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Claxton, A.; Baker, L.; Wilkinson, C.; Trittschuh, E.; Chapman, D.; Watson, G.; Cholerton, B.; Plymate, S.; Arbuckle, M.; Craft, S. Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 35, 789–797. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Hu, J.-C.; Tsai, C.; Yue, M.; Melrose, H.; Kanekiyo, T.; Bu, G. Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J. Neurosci. 2015, 35, 5851–5859. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G.J. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e5. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.; Torres, E.R.; Weber Boutros, S.; Patel, E.; Akinyeke, T.; Alkayed, N.; Raber, J. Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J. Cereb. Blood Flow Metab. 2017, 39, 770–781. [Google Scholar] [CrossRef] [Green Version]
- Rhea, E.; Torres, E.R.S.; Raber, J.; Banks, W.A. Insulin BBB pharmacokinetics in young apoE male and female transgenic mice. PLoS ONE 2020, 15, e0228455. [Google Scholar] [CrossRef]
- Rhea, E.; Hansen, K.; Pemberton, S.; Torres, E.; Holden, S.; Raber, J.; Banks, W. Effects of apolipoprotein E isoform, sex, and diet on insulin BBB pharmacokinetics in mice. Sci. Rep. 2021, 11, 18636. [Google Scholar] [CrossRef]
- Lacroix, M.C.; Badonnel, K.; Meunier, N.; Tan, F.; Schlegel-Le Poupon, C.; Durieux, D.; Monnerie, R.; Baly, C.; Congar, P.; Salesse, R.; et al. Expression of insulin system in the olfactory epithelium: First approaches to its role and regulation. J. Neuroendocrinol. 2008, 20, 1176–1190. [Google Scholar] [CrossRef]
- Ghosh, S.; Leng, W.; Wilsch-Brauninger, M.; Barrera-Velazquez, M.; Leopold, P.; Eaton, S. A local insulin reservoir in Drosophila alpha cell homologs ensures developmental progression under nutrient shortage. Curr. Biol. 2022, 32, 1788–1797. [Google Scholar] [CrossRef]
- Xaio, H.; Banks, W.A.; Niehoff, M.L.; Morley, J.E. Effect of LPS on the permeability of the blood-brain barrier to insulin. Brain Res. 2001, 896, 36–42. [Google Scholar] [CrossRef]
- Kaiyala, K.J.; Prigeon, R.L.; Kahn, S.E.; Woods, S.C.; Schwartz, M.W. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes 2000, 49, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- May, A.A.; Liu, M.; Woods, S.C.; Begg, D.P. CCK increases the transport of insulin into the brain. Physiol. Behav. 2016, 165, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Dohgu, S.; Nakaoke, R.; Lynch, J.L.; Fleegal-DeMotta, M.A.; Erickson, M.A.; Vo, T.Q. Nitric oxide isoenzymes regulate LPS-enhanced insulin transport across the blood-brain barrier. Endocrinology 2008, 149, 1514–1523. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Hsuchou, H.; He, Y.; Sakharkar, A.; Cain, C.; Yu, C.; Kastin, A.J. Astrocyte leptin receptor (ObR) and leptin transport in adult-onset obese mice. Endocrinology 2008, 149, 2798–2806. [Google Scholar] [CrossRef] [Green Version]
- Konishi, M.; Sakaguchi, M.; Lockhart, S.M.; Cai, W.; Li, M.E.; Homan, E.P.; Rask-Madsen, C.; Kahn, C.R. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc. Natl. Acad. Sci. USA 2017, 114, E8478–E8487. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Caceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Sun, Z. Antiaging Gene Klotho Enhances Glucose-Induced Insulin Secretion by Up-Regulating Plasma Membrane Levels of TRPV2 in MIN6 β-Cells. Endocrinology 2012, 153, 3029–3039. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Fergusson, M.; Castillo, R.; Castilho, R.; Liu, J.; Cao, L.; Chen, J.; Malide, D.; Rovira, I.; Schimel, D.; et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science 2007, 317, 803–806. [Google Scholar] [CrossRef] [Green Version]
- Landry, T.; Li, P.; Shookster, D.; Jiang, Z.; Laing, B.; Bunner, W.; Langton, T.; Tong, Q.; Huang, H. Centrally circulating α-klotho inversely correlates with human obesity and modulates arcuate cell populations in mice. Mol. Metab. 2021, 44, 101136. [Google Scholar] [CrossRef]
- Kundu, P.; Zimmerman, B.; Quinn, J.; Kaye, J.; Mattek, N.; Westaway, S.; Raber, J. Serum Levels of -Klotho Are Correlated with Cerebrospinal Fluid Levels and Predict Measures of Cognitive Function. J. Alzheimer’s Dis. 2022, 86, 1471–1481. [Google Scholar] [CrossRef]
- Zhao, Y.; Zeng, C.-Y.; Li, X.-H.; Yang, T.-T.; Kuang, X.; Du, J.-R. Klotho overexpression improves amyloid-β clearance and cognition in the APP/PS1 mouse model of Alzheimer’s disease. Aging Cell 2020, 19, e13239. [Google Scholar] [CrossRef]
- Mazucanti, C.; Kawamato, E.; Mattson, M.; Scavone, C.; Camandola, S. Activity-dependent neuronal Klotho enhances astrocytic aerobic glycolysis. J. Cereb Blood Flow Metab 2018, 39, 1544–1556. [Google Scholar] [CrossRef]
- Kenyon, C. The plasticity of aging: Insights from long-lived mutants. Cell 2005, 120, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Kurosu, H.; Yamamoto, M.; Clark, J.; Pastor, J.; Nandi, A.; Gurnani, P.; McGuinness, O.; Ch’ikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [Green Version]
- McGowan, M.K.; Andrews, K.M.; Grossman, S.P. Chronic intrahypothalamic infusions of insulin or insulin antibodies alter body weight and food intake in the rat. Physiol. Behav. 1992, 51, 753–766. [Google Scholar] [CrossRef]
- Chowers, I.; Lavy, S.; Halpern, L. Effect of insulin administered intracisternally on the glucose level of the blood and the cerebrospinal fluid in vagotomized dogs. Exp. Neurol. 1966, 14, 383–389. [Google Scholar] [CrossRef]
- Ajaya, B.; Haranath, P.S. Effects of insulin administered into cerebrospinal fluid spaces on blood glucose in unanaesthetized and anaesthestized dogs. Indian, J. Med. Res. 1982, 75, 607–615. [Google Scholar]
- Muntzel, M.S.; Morgan, D.A.; Mark, A.L.; Johnson, A.K. Intracerebroventricular insulin produces nonuniform regional increases in sympathetic nerve activity. Am. J. Physiol. 1994, 267, R1350–R1355. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, D.A.; Obici, S.; Seeley, R.J. Targeting the CNS to treat type 2 diabetes. Nat. Rev. Drug Discov. 2009, 8, 386–398. [Google Scholar] [CrossRef]
- Scherer, T.; O’Hare, J.; Diggs-Andrews, K.; Schweiger, M.; Cheng, B.; Lindtner, C.; Zielinski, E.; Vempati, P.; Su, K.; Dighe, S.; et al. Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab. 2011, 13, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, J.R.; Lyra, E.S.N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; Frazao, R.; et al. Alzheimer-associated Abeta oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol. Med. 2015, 7, 190–210. [Google Scholar] [CrossRef]
- Foster, J.; McVey Neufeld, K.-A. Gut-brain axis: How the microbiome influences anxiety and depression. Trends Neurosci. 2013, 36, 305–312. [Google Scholar] [CrossRef]
- Allen, A.; Dinan, T.; Clarke, G.; Cryan, J. A psychology of the human brain-gut-microbiome axis. Soc. Pers. Psychol. Compass 2017, 11, e12309. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.; Kennedy, P.; Cryan, J.F.; Dinan, T.; Clarke, G.; Hyland, H. Breaking down the barriers: The gut miocrobiome, intestinal permeability and stress-related psychiatric disorders. Front. Cell Neurosci. 2015, 9, 392. [Google Scholar] [CrossRef] [Green Version]
- Lynch, J.; Hsiao, E. Microbiomes as sources of emergent host phenotypes. Science 2019, 365, 1405–1409. [Google Scholar] [CrossRef]
- Vuong, H.; Yano, J.; Fung, T.; Hsiao, E. The microbbiome and host behavior. Annu. Rev. Neurosci. 2017, 40, 21–49. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.-N.; Kubo, C.; Koga, Y. Postnatal microbbila colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, A.F.; Erickson, M.A.; Rhea, E.M.; Salameh, T.S.; Banks, W.A. Gut reactions: How the blood-brain barrier connects the microbiome and the brain. Exp. Biol. Med. 2018, 243, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Corsini, S.; Kellinggray, L.; Hegarty, C.; Le Gail, C.; Narbad, A.; Muller, M.; Tereja, N.; O’Toole, P.; Minihane, A.-M.; et al. APOE genotype influences the gut microbiome structure and function in humans and mice: Relevance for Alzheimer’s disease pathophysiology. FASEB J. 2019, 33, 8221–8231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, P.; Torres, E.; Stagaman, K.; Kasschau, K.; Okhovat, M.; Holden, S.; Ward, S.; Nevonen, K.; Davis, B.; Saito, T.; et al. Integrated analysis of behavioral, epigenetic, and gut microbiome analyses in AppNL-G-F, AppNL-F, and wild type mice. Sci. Rep. 2021, 11, 4678. [Google Scholar] [CrossRef]
- Kundu, P.; Stagaman, K.; Kasschau, K.; Holden, S.; Shulzhenko, N.; Sharpton, T.; Raber, J. Fecal implants from AppNL-G-F and AppNL-G-F/E4 donor mice sufficient to induce behavioral phenotypes in germ-free mice. Front. Behav. Neurosci. 2022, 16, 791128. [Google Scholar] [CrossRef]
- Wolfe, C.; Fitz, N.; Nam, K.; Lefterov, I.; Koldamova, R. The role of APOE and TREM2 in Alzheimer’s disease—Current understanding and perspectives. Int. J. Mol. Med. 2018, 20, 81. [Google Scholar] [CrossRef] [Green Version]
- Prokop, S.; Miller, K.; Labra, S.; Pitkin, R.; Hoxha, K.; Narasimhan, S.; Changolkar, L.; Rosenbloom, A.; Lee, V.M.-Y.; Trojanowski, J.Q. Impact of TREM2 risk variants on brain region-specific immune activation and plaque microenvironment in Alzheimer’s disease patient brain samples. Acta Neuropahol. 2019, 138, 613–620. [Google Scholar] [CrossRef]
- Lee, C.D.; Daggett, A.; Gu, X.; Jiang, L.-L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. TREM2 maintains microglial metabolic firness in Alzheimer’s disease. Cell 2017, 170, 649–663. [Google Scholar]
- Lee, C.; Daggett, A.; Gu, X.-s.; Jiang, L.; Langfelder, P.; Li, X.; Xang, N.; Zhao, Y. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 2018, 97, 1032–1048. [Google Scholar] [CrossRef] [Green Version]
- Bonstanciklioglu, M. The role of gut microbiota in pathogenesis of Alzheimer’s disease. J. Appl. Microbiol. 2019, 127, 954–967. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relationship | Condition/Treatment | Patient Numbers * | Mean Age | Study Type | Date of Study | Inclusion Criteria | Population % | Main Findings | COVID-19 Mortality | Notes | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Diabetes worsenes COVID-19 outcome | Metabolic Syndrome | 46,441 | 61.2 ± 17.8 years old (SD) | Retrospective | 15 February 2020 to 18 February 2021 | Completed discharge status | 17.5% had metabolic syndrome | Increased risk of ICU admission, invasive mechanical ventilation, ARDS, and mortality; increased ICU and hospital LOS | Increased | MS defined as 3 or more conditions: obesity, prediabetes or diabetes, hypertension, and dyslipidemia) | [80] |
| Triglyceride and Glucose Index (TyG) | 151 | 59.5 ± 15.9 years old (SD) | Retrospective | 12 January 2020 to 13 Febreuary 2020 | Completed medical records and follow-up data | 25.8% had diabetes | TyG index levels were significantly higher in the severe cases and death group | Increased | TyG: marker of insulin resistance | [81] | |
| Diabetes | 1902 | 64 years old | Retrospective | 1 March 2020 to 27 September 2020 | COVID-19 | 31.2% had diabetes | 36% admitted to the ICU | 19% of those with diabetes died | [82] | ||
| COVID-19 increases risk for developing diabetes | Newly Diagnosed Diabetes Mellitus (NDDM) | 594 | 54.1 years old | Retrospective, with follow-up observations | 1 March 2020 to 27 September 2020 | COVID-19 and Diabetes | 13% had NDDM | Younger age in NDDM; NDDM had lower glucose levels but worsened COVID-19 (increased LOS, ICU admission); 56% still classified as DM at mean follow up of 323 days | No effect of NDDM | NDDM defined as fasting blood glucose >125–140 mg/dL or any glucose >140–180 mg/dL during admission | [82] |
| Development of diabetes | 551 | 61 ± 0.7 years old (SEM) | Retrospective | 1 February 2020 to 15 May 2020 | No pre-existing diabetes | 46% hyperglycemic; 27% normoglycemic | 12% had new classification of diabetes; and 18.5% had transient hyperglycemia; DM incread LOS; Glycemic abnormalities persisted for at least 2 months after resolved COVID-19 | DM increased | [83] | ||
| COVID-19 induction of diabetes | 124 | Non-severe COVID: 36.6 ± 15.8 years old; Severe COVID: 59.0 ± 13.9 years old (SEM) | Retrospective | 22 January 2020 to 7 April 2020 | No pre-existing diabetes | 25.8% had metabolic-related diseases | COVID-19 increased blood glucose and insulin levels compared to controls and persisted after virus elimination | Did not investigate | Compared to 30 non-COVID controls; looked into mechanism | [84] | |
| COVID-19 induction of diabetes | 64 | 44.3 ± 13.5 years old (SD) | Prospective | 17 January 2020 to 9 February 2020 (initial cohort) | No pre-existing diabetes | 84% had mild COVID; 15.6% had severe COVID | C-peptide and TyG indices increased with decreased fasting glucose levels up to 6 months post discharge | Followed patients at 3 and 6 months post hospital discharge | [85] | ||
| Treating COVID-19 with Diabetes Drugs | Metformin treatment | 6659 in the 3 observational studies | Systematic Review | Up to 30 July 2020 | English publications selected by 3 independent reviewers | 9 out of 14 articles | Positive benefit of metformin treatment in COVID-19 w/or w/o diabetes | 2/3 studies showed decreased mortality | Keywords used: COVID-19, SARS-CoV-2, 2019-nCoV, metformin, and antidiabetic drug | [88] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rhea, E.M.; Banks, W.A.; Raber, J. Insulin Resistance in Peripheral Tissues and the Brain: A Tale of Two Sites. Biomedicines 2022, 10, 1582. https://doi.org/10.3390/biomedicines10071582
Rhea EM, Banks WA, Raber J. Insulin Resistance in Peripheral Tissues and the Brain: A Tale of Two Sites. Biomedicines. 2022; 10(7):1582. https://doi.org/10.3390/biomedicines10071582
Chicago/Turabian StyleRhea, Elizabeth M., William A. Banks, and Jacob Raber. 2022. "Insulin Resistance in Peripheral Tissues and the Brain: A Tale of Two Sites" Biomedicines 10, no. 7: 1582. https://doi.org/10.3390/biomedicines10071582
APA StyleRhea, E. M., Banks, W. A., & Raber, J. (2022). Insulin Resistance in Peripheral Tissues and the Brain: A Tale of Two Sites. Biomedicines, 10(7), 1582. https://doi.org/10.3390/biomedicines10071582