
Development of In Vitro Assays for Advancing Radioimmunotherapy against Brain Tumors

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culture Methods, and Cell Preparation for Experiments
2.2. Drug Dosage and In Vitro Treatment of Cells
2.3. Irradiation of Cells: Faxitron CellRad
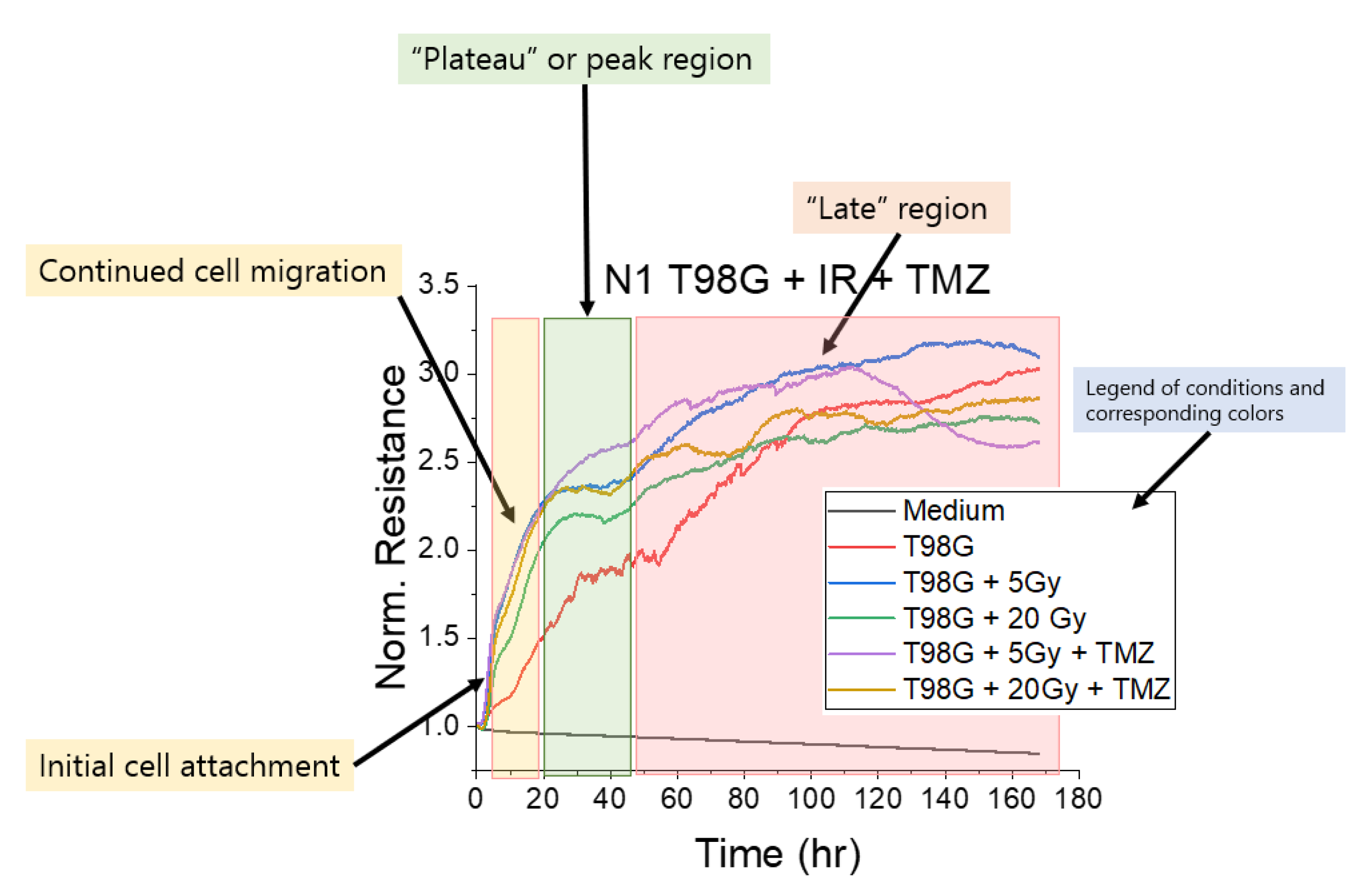
2.4. Electric Cell Impedance Sensing (ECIS) for Migration Measurement
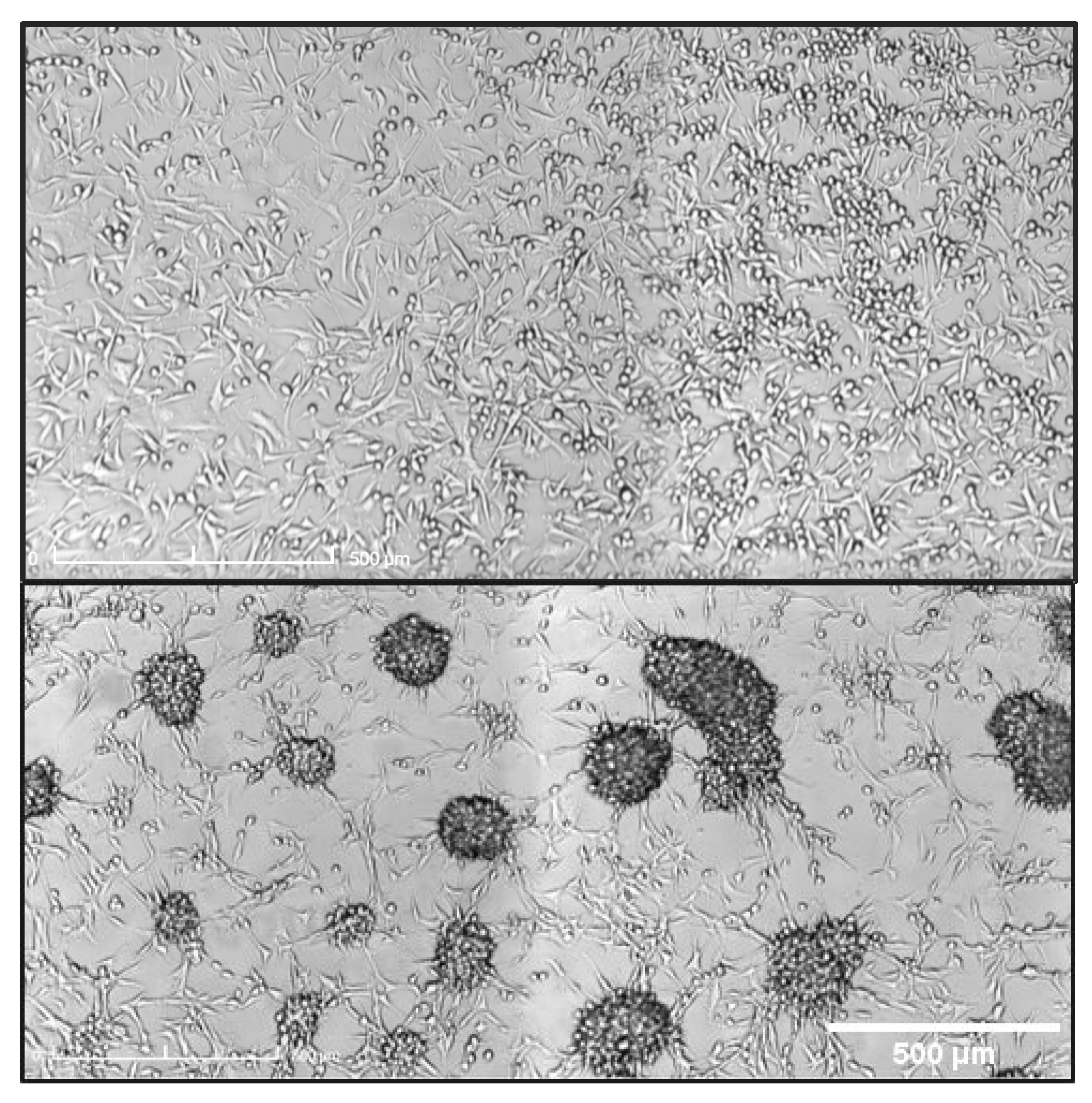
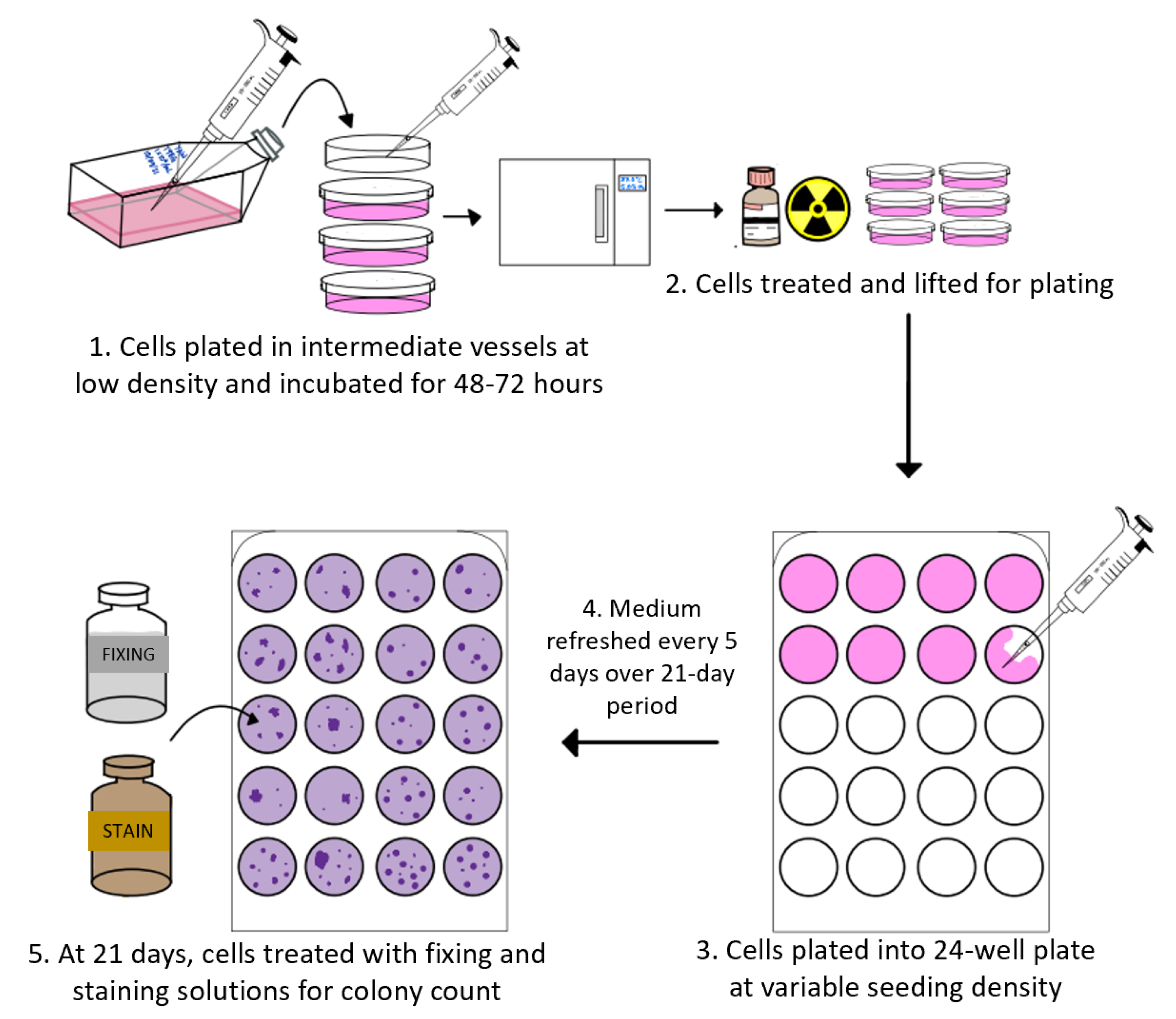
2.5. Clonogenic Assays
2.6. Statistical and Error Analyses
3. Results
3.1. Results Follow from Hypotheses
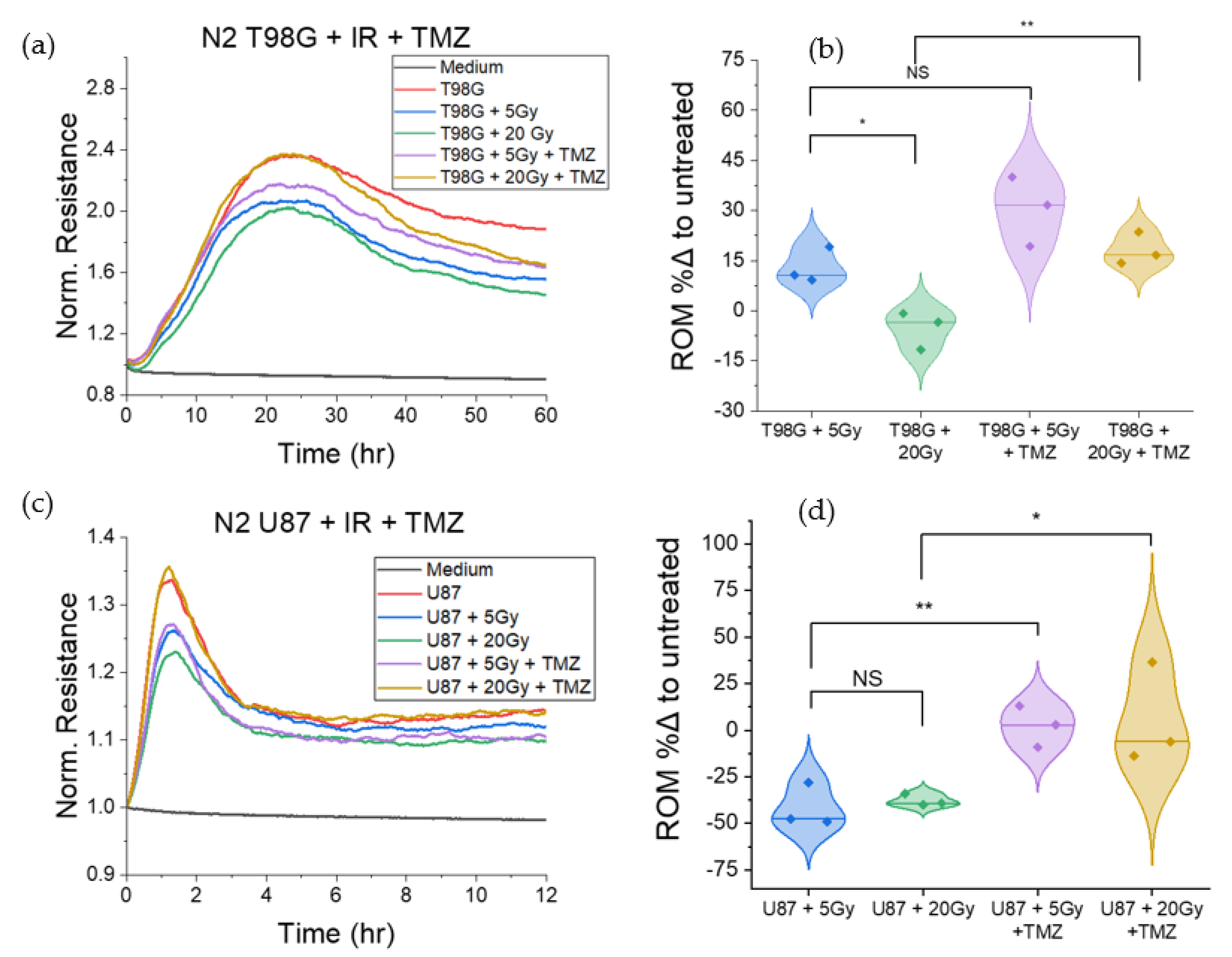
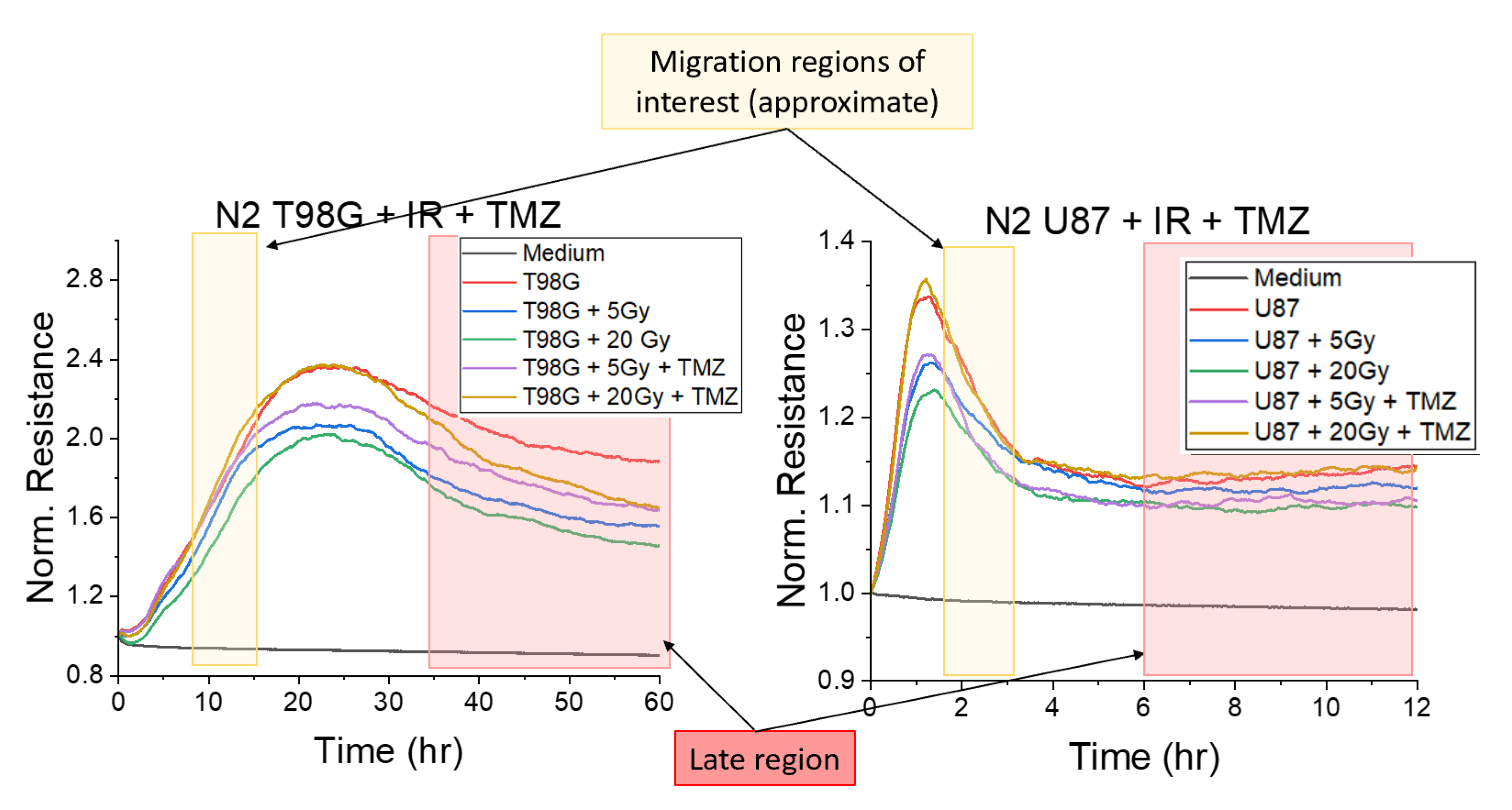
3.1.1. Treatment with Radiation and Concurrent Temozolomide Increases Cell Migration Relative to Radiation Alone
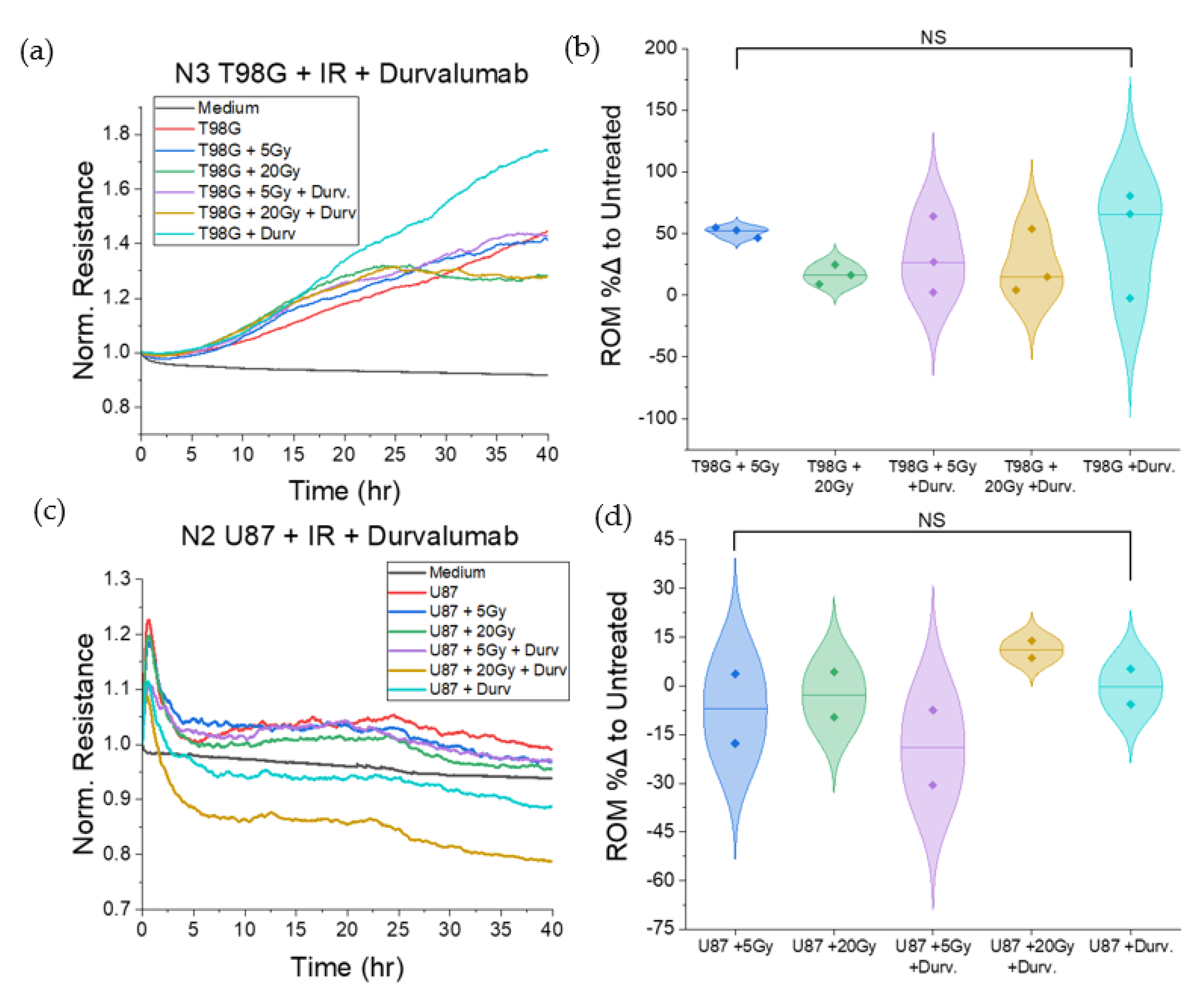
3.1.2. Treatment with Durvalumab Does Not Significantly Alter T98G or U87 Cell Migration
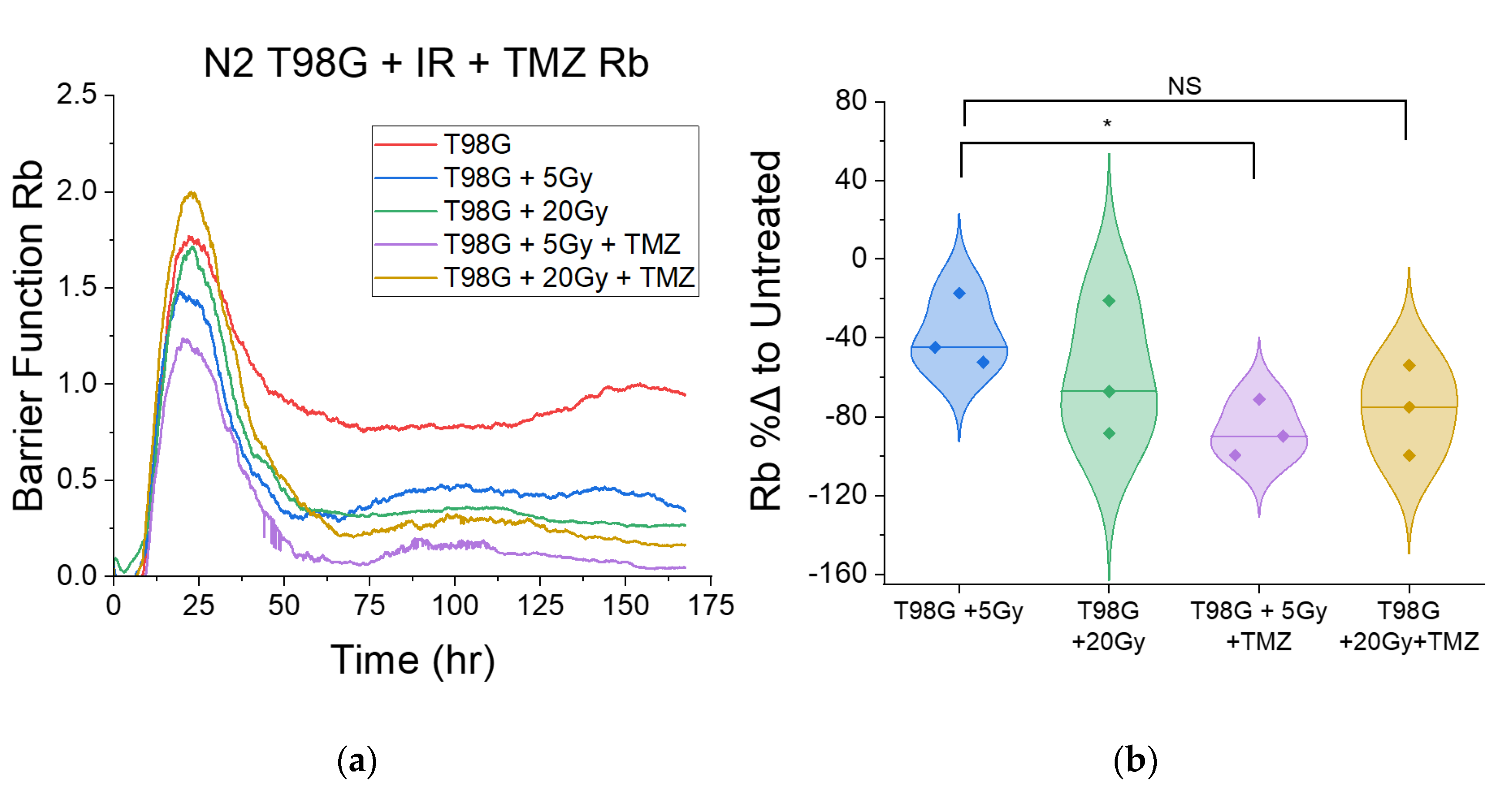
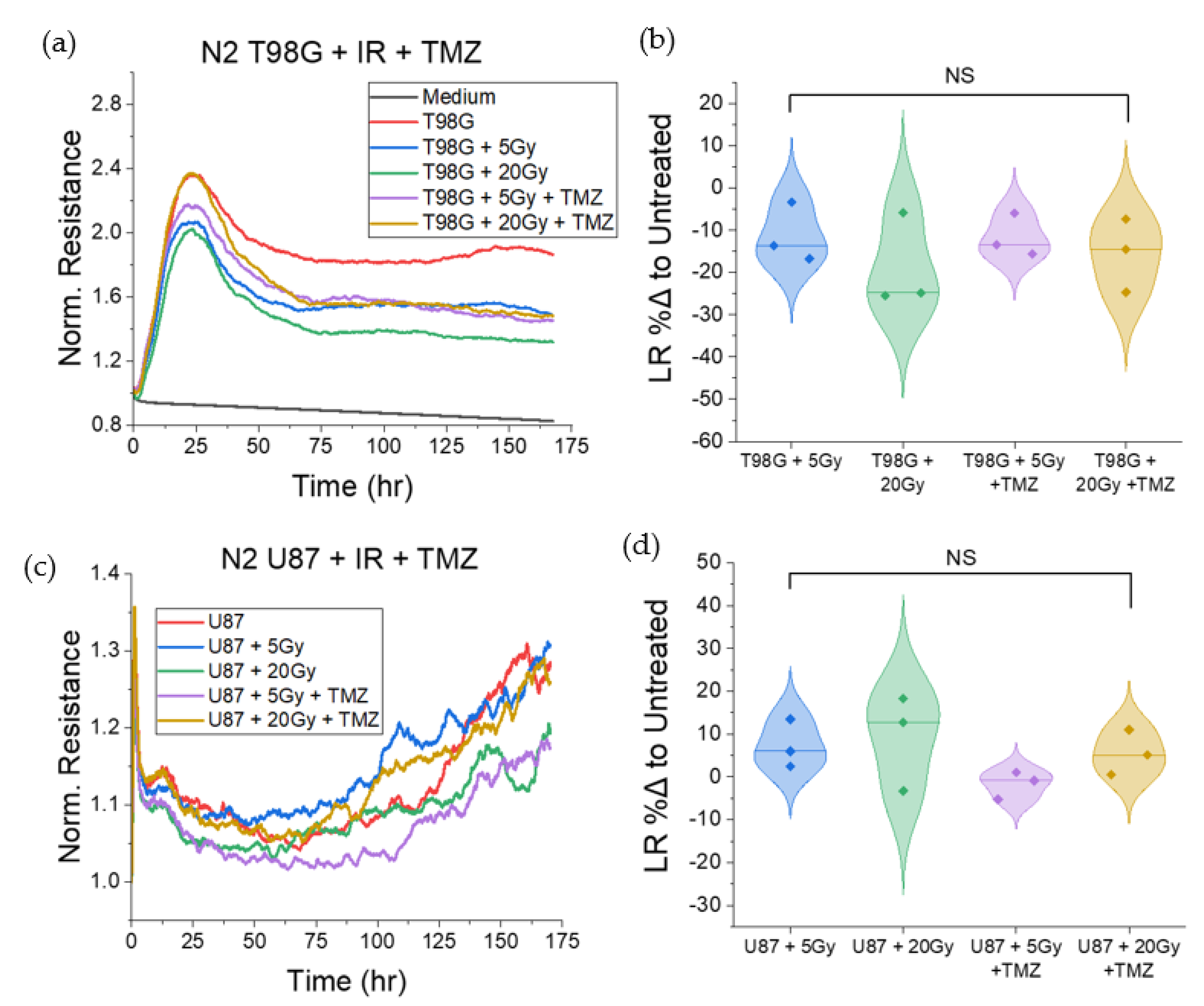
3.1.3. Treatment with Concurrent TMZ Marginally Changes T98G Barrier Function Relative to Radiation Only
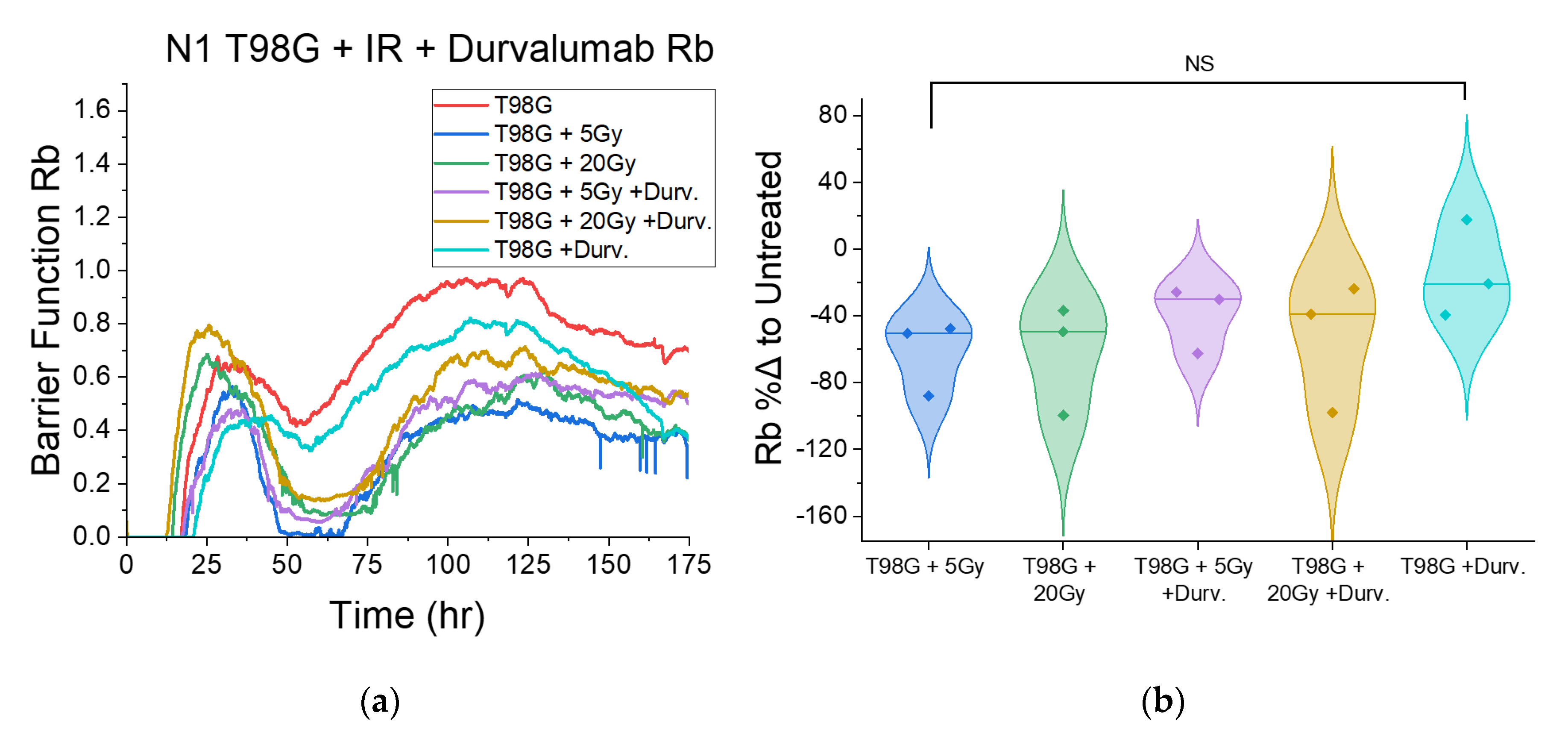
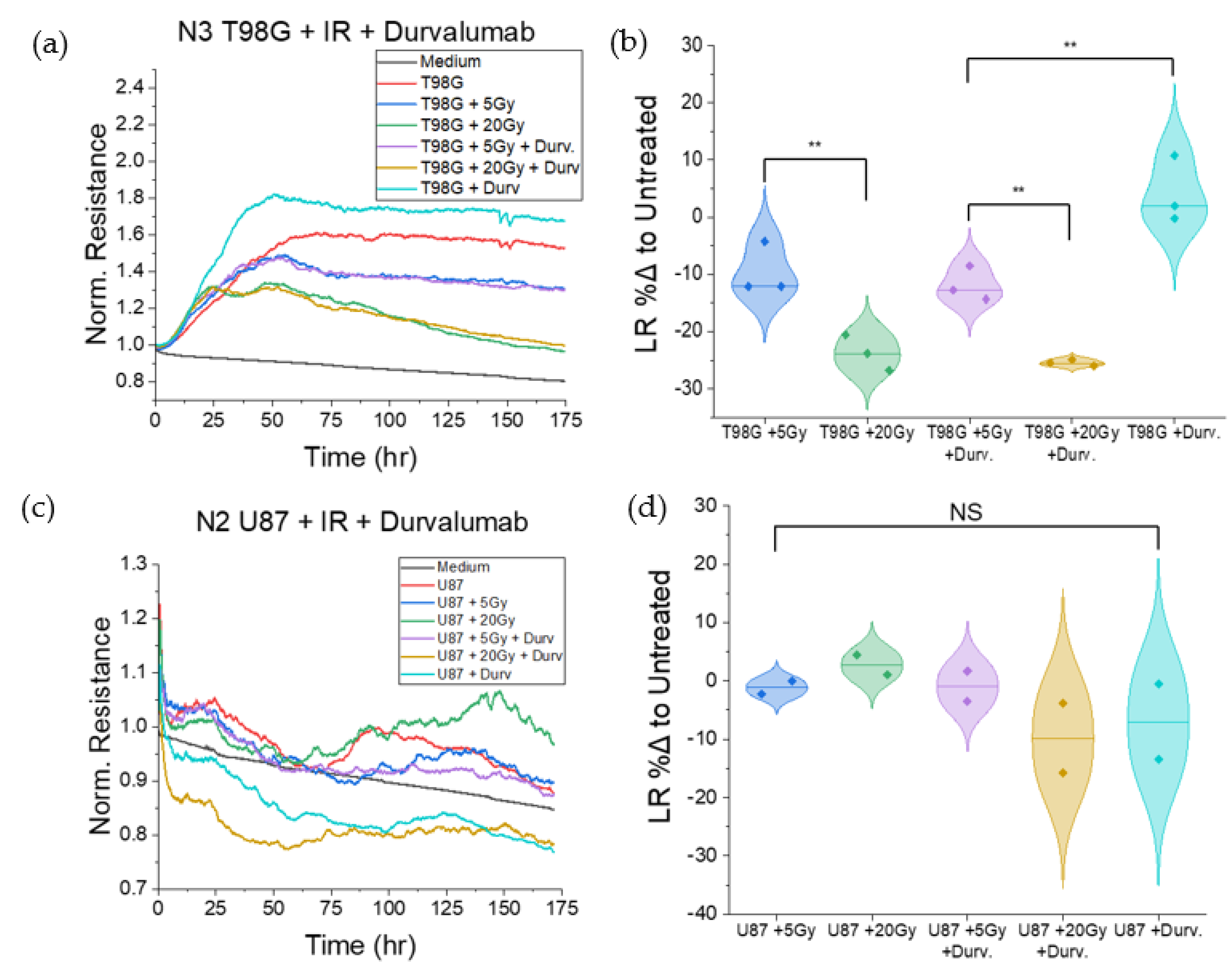
3.1.4. Treatment with Concurrent Durvalumab Does Not Significantly Affect T98G Barrier Function Relative to Radiation Only
3.1.5. T98G and U87 Treated with Radiation and TMZ Show No Significant Change to Late Resistance Relative to Untreated Cells
3.1.6. T98G Treated with Radiation and Durvalumab Show Decreased Late Resistance Compared to Untreated Cells
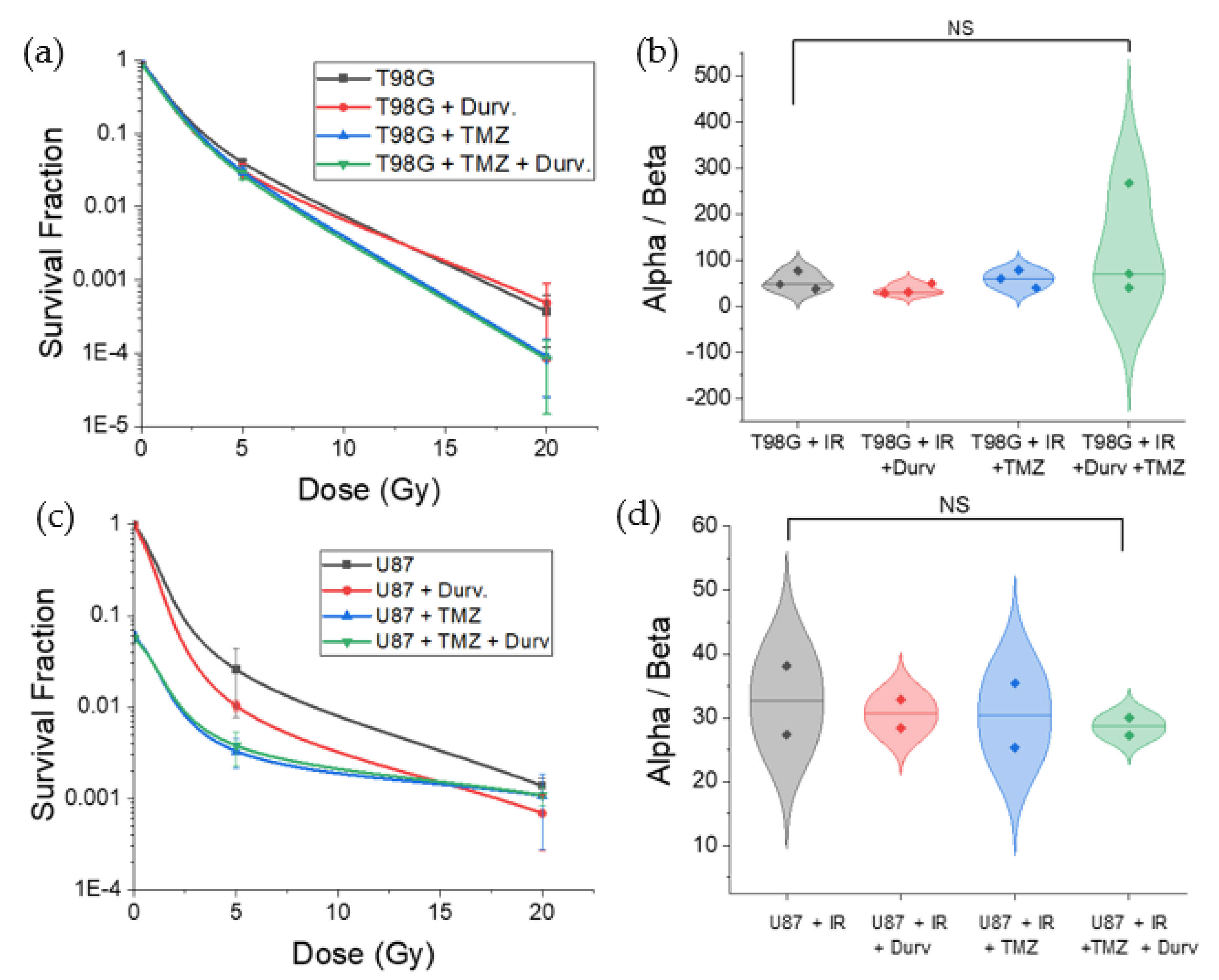
3.2. Radiation with Concurrent TMZ and Durvalumab Does Not Significantly Affect T98G Cell Survival Relative to Radiation Only
3.3. Radiation with Concurrent TMZ Decreases U87 MG Cell Survival
4. Discussion
4.1. Towards Patient-Adaptive Therapy
4.2. Effect of Radioimmunotherapy on Cell Migration
4.3. Effect of Radioimmunotherapy on Cell–Cell Adhesion
4.4. Effect of Radioimmunotherapy on Cell Survival
4.5. Limitations and Outlook
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A




References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Options Oncol. 2019, 20, 24. [Google Scholar] [CrossRef]
- Gzell, C.; Back, M.; Wheeler, H.; Bailey, D.; Foote, M. Radiotherapy in Glioblastoma: The Past, the Present and the Future. Clin. Oncol. 2017, 29, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Taphoorn, M.J.B.; Plaha, P. Advances in the management of glioblastoma. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Du, X.L.; Lu, G.; Zhu, J.-J. Survival benefit of glioblastoma patients after FDA approval of temozolomide concomitant with radiation and bevacizumab: A population-based study. Oncotarget 2017, 8, 44015–44031. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [PubMed]
- Karachi, A.; Dastmalchi, F.; Mitchell, D.; Rahman, M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro-Oncol. 2018, 20, 1566–1572. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Fidler, I.J. AACR Centennial Series: The Biology of Cancer Metastasis: Historical Perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef]
- Fidler, I.J.; Kripke, M.L. The challenge of targeting metastasis. Cancer Metastasis Rev. 2015, 34, 635–641. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Mu, N.; Jia, B.; Guo, Q.; Pan, L.; Zhu, M.; Zhang, W.; Zhang, K.; Li, W.; Li, M.; et al. Targeting radiation-tolerant persisted cells as a strategy for inhibiting radioresistance and recurrence in glioblastoma. Neuro Oncol. 2022, 24, 1056–1070. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Garcia, H.; Ramirez-Loera, C.; Malouff, T.D.; Seneviratne, D.S.; Palmer, J.D.; Trifiletti, D.M. Novel Strategies for Nanoparticle-Based Radiosensitization in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 9673. [Google Scholar] [CrossRef] [PubMed]
- Lehtipuro, S.; Nykter, M.; Granberg, K.J. Modes of immunosuppression in glioblastoma microenvironment. Oncotarget 2019, 10, 920–921. [Google Scholar] [CrossRef] [PubMed]
- The Nobel Assembly. Press Release: The Nobel Prize in Physiology or Medicine 2018. Available online: https://www.nobelprize.org/prizes/medicine/2018/summary/ (accessed on 5 May 2022).
- Dobosz, P.; Dzieciątkowski, T. The Intriguing History of Cancer Immunotherapy. Front. Immunol. 2019, 10, 2965. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunosuppression and autoimmune disease: Beyond immunosuppressive networks for tumour immunity. Immunology 2006, 119, 254–264. [Google Scholar] [CrossRef]
- Lee, Y.H.; Tai, D.; Yip, C.; Choo, S.P.; Chew, V. Combinational Immunotherapy for Hepatocellular Carcinoma: Radiotherapy, Immune Checkpoint Blockade and Beyond. Front. Immunol. 2020, 11, 568759. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, W.; Li, N.; Neri, S.; Sharma, A.; Jiang, W.; Lin, S.H. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front. Pharmacol. 2018, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Derer, A.; Frey, B.; Fietkau, R.; Gaipl, U.S. Immune-modulating properties of ionizing radiation: Rationale for the treatment of cancer by combination radiotherapy and immune checkpoint inhibitors. Cancer Immunol. Immunother. 2016, 65, 779–786. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef]
- Majc, B.; Novak, M.; Kopitar-Jerala, N.; Jewett, A.; Breznik, B. Immunotherapy of Glioblastoma: Current Strategies and Challenges in Tumor Model Development. Cells 2021, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Erkkinen, M.G.; Nestler, U.; Stupp, R.; Mehta, M.; Aldape, K.; Gilbert, M.R.; Black, P.M.; Loeffler, J.S. Temozolomide-Mediated Radiation Enhancement in Glioblastoma: A Report on Underlying Mechanisms. Clin. Cancer Res. 2006, 12, 4738–4746. [Google Scholar] [CrossRef] [PubMed]
- Pouessel, D.; Mervoyer, A.; Larrieu-Ciron, D.; Cabarrou, B.; Robert, M.; Frenel, J.; Olivier, P.; Mounier, M.; Moyal, E.C.-J. OS4.4 Hypofractionated stereotactic radiotherapy and anti-PDL1 Durvalumab combination in recurrent glioblastoma: Results of the phase I part of the phase I/II STERIMGLI trial. Neuro-Oncol. 2019, 21, iii10–iii11. [Google Scholar] [CrossRef]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.-Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncol. 2016, 18, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.J.; Homer, N.; O'Connor, B.D.; Chen, Z.; Eskin, A.; Lee, H.; Merriman, B.; Nelson, S.F. U87MG Decoded: The Genomic Sequence of a Cytogenetically Aberrant Human Cancer Cell Line. PLoS Genet. 2010, 6, e1000832. [Google Scholar] [CrossRef] [PubMed]
- Annabi, B.; Laflamme, C.; Sina, A.; Lachambre, M.-P.; Béliveau, R. A MT1-MMP/NF-κB signaling axis as a checkpoint controller of COX-2 expression in CD133(+) U87 glioblastoma cells. J. Neuroinflammation 2009, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Kiseleva, L.N.; Kartashev, A.V.; Vartanyan, N.L.; Pinevich, A.A.; Samoilovich, M.P. A172 and T98G cell lines characteristics. Cell Tissue Biol. 2016, 10, 341–348. [Google Scholar] [CrossRef]
- Murad, H.; Alghamian, Y.; Aljapawe, A.; Madania, A. Effects of ionizing radiation on the viability and proliferative behavior of the human glioblastoma T98G cell line. BMC Res. Notes 2018, 11, 330. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Pandey, A.; Tripathi, S.C.; Mai, J.; Hanash, S.M.; Shen, H.; Mitra, S.; Rostomily, R.C. Combinatorial Effect of PLK1 Inhibition with Temozolomide and Radiation in Glioblastoma. Cancers 2021, 13, 5114. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Li, X.; Chen, Y.; Liu, R.; Ren, C.; Du, S. The efficacy of hypofractionated radiotherapy (HFRT) with concurrent and adjuvant temozolomide in newly diagnosed glioblastoma: A meta-analysis. Cancer Radiother. 2021, 25, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Campian, J.L.; Piotrowski, A.F.; Ye, X.; Hakim, F.T.; Rose, J.; Yan, X.-Y.; Lu, Y.; Gress, R.; Grossman, S.A. Serial changes in lymphocyte subsets in patients with newly diagnosed high grade astrocytomas treated with standard radiation and temozolomide. J. Neuro-Oncol. 2017, 135, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Shaverdian, N.; Thor, M.; Shepherd, A.F.; Offin, M.D.; Jackson, A.; Wu, A.J.; Gelblum, D.Y.; Yorke, E.D.; Ii, C.B.S.; Chaft, J.E.; et al. Radiation pneumonitis in lung cancer patients treated with chemoradiation plus durvalumab. Cancer Med. 2020, 9, 4622–4631. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.E.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; Cho, B.C.; et al. Three-Year Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC—Update from PACIFIC. J. Thorac. Oncol. 2020, 15, 288–293. [Google Scholar] [CrossRef]
- Reardon, D.A.; Kaley, T.J.; Dietrich, J.; Lim, M.; Dunn, G.P.; Gan, H.K.; Cloughesy, T.F.; Clarke, J.L.; Park, A.J.; Macri, M.J.; et al. Phase 2 study to evaluate the clinical efficacy and safety of MEDI4736 (durvalumab) in patients with glioblastoma (GBM). J. Clin. Oncol. 2016, 34, TPS2080. [Google Scholar] [CrossRef]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef]
- Medscape. Temozolomide (Rx). 2021. Available online: https://reference.medscape.com/drug/temodar-temozolomide-342229#91 (accessed on 13 January 2021).
- Lin, A.J.; Campian, J.L.; Hui, C.; Rudra, S.; Rao, Y.J.; Thotala, D.; Hallahan, D.; Huang, J. Impact of concurrent versus adjuvant chemotherapy on the severity and duration of lymphopenia in glioma patients treated with radiation therapy. J. Neuro-Oncol. 2018, 136, 403–411. [Google Scholar] [CrossRef]
- De Sousa Linhares, A.; Battin, C.; Jutz, S.; Leitner, J.; Hafner, C.; Tobias, J.; Wiedermann, U.; Kundi, M.; Zlabinger, G.J.; Grabmeier-Pfistershammer, K.; et al. Therapeutic PD-L1 antibodies are more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling. Sci. Rep. 2019, 9, 11472. [Google Scholar] [CrossRef]
- Ratliff, M.; Kim, H.; Qi, H.; Kim, M.; Ku, B.; Azorin, D.D.; Hausmann, D.; Khajuria, R.K.; Patel, A.; Maier, E.; et al. Patient-Derived Tumor Organoids for Guidance of Personalized Drug Therapies in Recurrent Glioblastoma. Int. J. Mol. Sci. 2022, 23, 6572. [Google Scholar] [CrossRef]
- Giaever, I.; Keese, C.R. Micromotion of mammalian cells measured electrically. Proc. Natl. Acad. Sci. USA 1991, 88, 7896–7900. [Google Scholar] [CrossRef] [PubMed]
- Robilliard, L.D.; Yu, J.; Jun, S.-M.; Anchan, A.; Finlay, G.; Angel, C.E.; Graham, E.S. Can ECIS Biosensor Technology Be Used to Measure the Cellular Responses of Glioblastoma Stem Cells? Biosensors 2021, 11, 498. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, T.; Marschner, J.; Zhao, Z.B.; Świderska, M.K.; Anders, H.-J. Electric cell-substrate impedance sensing in kidney research. Nephrol. Dial. Transpl. 2019, 36, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Wegener, J.; Keese, C.R.; Giaever, I. Electric Cell–Substrate Impedance Sensing (ECIS) as a Noninvasive Means to Monitor the Kinetics of Cell Spreading to Artificial Surfaces. Exp. Cell Res. 2000, 259, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Palliyage, G.H.; Hussein, N.; Mimlitz, M.; Weeder, C.; Alnasser, M.H.A.; Singh, S.; Ekpenyong, A.; Tiwari, A.K.; Chauhan, H. Novel Curcumin-Resveratrol Solid Nanoparticles Synergistically Inhibit Proliferation of Melanoma Cells. Pharm. Res. 2021, 38, 851–871. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.G.; Martin, T.A. Electric Cell-Substrate Impedance Sensing and Cancer Metastasis; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar] [CrossRef]
- Hong, J.; Kandasamy, K.; Marimuthu, M.; Choi, C.S.; Kim, S. Electrical cell-substrate impedance sensing as a non-invasive tool for cancer cell study. Analyst 2011, 136, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Renken, C.; Keese, C.; Giaever, I. Automated assays for quantifying cell migration. BioTechniques 2010, 49, 844. [Google Scholar] [CrossRef]
- Kahlert, U.; Koch, K.; Suwala, A.K.; Hartmann, R.; Cheng, M.; Maciaczyk, D.; Willbold, D.; Eberhart, C.G.; Glunde, K.; Maciaczyk, J. The effect of neurosphere culture conditions on the cellular metabolism of glioma cells. Folia Neuropathol. 2015, 3, 219–225. [Google Scholar] [CrossRef]
- Bar, E.E.; Lin, A.; Mahairaki, V.; Matsui, W.; Eberhart, C.G. Hypoxia Increases the Expression of Stem-Cell Markers and Promotes Clonogenicity in Glioblastoma Neurospheres. Am. J. Pathol. 2010, 177, 1491–1502. [Google Scholar] [CrossRef]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; Van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 7th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012. [Google Scholar]
- Keese, C.R.; Bhawe, K.; Wegener, J.; Giaever, I. Real-Time Impedance Assay to Follow the Invasive Activities of Metastatic Cells in Culture. BioTechniques 2002, 33, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, C.M.; Oei, A.L.; Crezee, J.; Bel, A.; Franken, N.A.P.; Stalpers, L.J.A.; Kok, H.P. The alfa and beta of tumours: A review of parameters of the linear-quadratic model, derived from clinical radiotherapy studies. Radiat. Oncol. 2018, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Ramakrishna, R.; Magge, R.; Wernicke, A.G. Advances in Radiotherapy for Glioblastoma. Front. Neurol. 2018, 8, 748. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Marcu, L.G.; Hull, A.; Bezak, E. Radioimmunotherapy of glioblastoma multiforme—Current status and future prospects. Crit. Rev. Oncol. 2021, 163, 103395. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.; Taube, J.M. PD-1/PD-L1 inhibitors. Curr. Opin. Pharmacol. 2015, 23, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Prathivadhi-Bhayankaram, S.V.; Ning, J.; Mimlitz, M.; Taylor, C.; Gross, E.; Nichols, M.; Guck, J.; Ekpenyong, A.E. Chemotherapy impedes in vitro microcirculation and promotes migration of leukemic cells with impact on metastasis. Biochem. Biophys. Res. Commun. 2016, 479, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Merrick, M.; Mimlitz, M.J.; Weeder, C.; Akhter, H.; Bray, A.; Walther, A.; Nwakama, C.; Bamesberger, J.; Djam, H.; Abid, K.; et al. In vitro radiotherapy and chemotherapy alter migration of brain cancer cells before cell death. Biochem. Biophys. Rep. 2021, 27, 101071. [Google Scholar] [CrossRef]
- Shankar, A.; Kumar, S.; Iskander, A.; Varma, N.R.; Janic, B.; Decarvalho, A.; Mikkelsen, T.; Frank, J.; Ali, M.M.; Knight, R.; et al. Subcurative radiation significantly increases cell proliferation, invasion, and migration of primary glioblastoma multiforme in vivo. Chin. J. Cancer 2014, 33, 148–158. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Yu, J.; Han, L.; Yang, F.; Zhao, M.; Zhou, H.; Hu, L. SOCS5 contributes to temozolomide resistance in glioblastoma by regulating Bcl-2-mediated autophagy. Bioengineered 2022, 13, 14125–14137. [Google Scholar] [CrossRef]
- Stupp, R.; Dietrich, P.-Y.; Kraljevic, S.O.; Pica, A.; Maillard, I.; Maeder, P.; Meuli, R.; Janzer, R.; Pizzolato, G.; Miralbell, R.; et al. Promising Survival for Patients With Newly Diagnosed Glioblastoma Multiforme Treated With Concomitant Radiation Plus Temozolomide Followed by Adjuvant Temozolomide. J. Clin. Oncol. 2002, 20, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Gene Funct. Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walter, Y.; Hubbard, A.; Benoit, A.; Jank, E.; Salas, O.; Jordan, D.; Ekpenyong, A. Development of In Vitro Assays for Advancing Radioimmunotherapy against Brain Tumors. Biomedicines 2022, 10, 1796. https://doi.org/10.3390/biomedicines10081796
Walter Y, Hubbard A, Benoit A, Jank E, Salas O, Jordan D, Ekpenyong A. Development of In Vitro Assays for Advancing Radioimmunotherapy against Brain Tumors. Biomedicines. 2022; 10(8):1796. https://doi.org/10.3390/biomedicines10081796
Chicago/Turabian StyleWalter, Yohan, Anne Hubbard, Allie Benoit, Erika Jank, Olivia Salas, Destiny Jordan, and Andrew Ekpenyong. 2022. "Development of In Vitro Assays for Advancing Radioimmunotherapy against Brain Tumors" Biomedicines 10, no. 8: 1796. https://doi.org/10.3390/biomedicines10081796
APA StyleWalter, Y., Hubbard, A., Benoit, A., Jank, E., Salas, O., Jordan, D., & Ekpenyong, A. (2022). Development of In Vitro Assays for Advancing Radioimmunotherapy against Brain Tumors. Biomedicines, 10(8), 1796. https://doi.org/10.3390/biomedicines10081796