
Recurrent Glioblastoma Treatment: State of the Art and Future Perspectives in the Precision Medicine Era

 , ,
, ,  , , ,
, , ,
Abstract
:1. Introduction
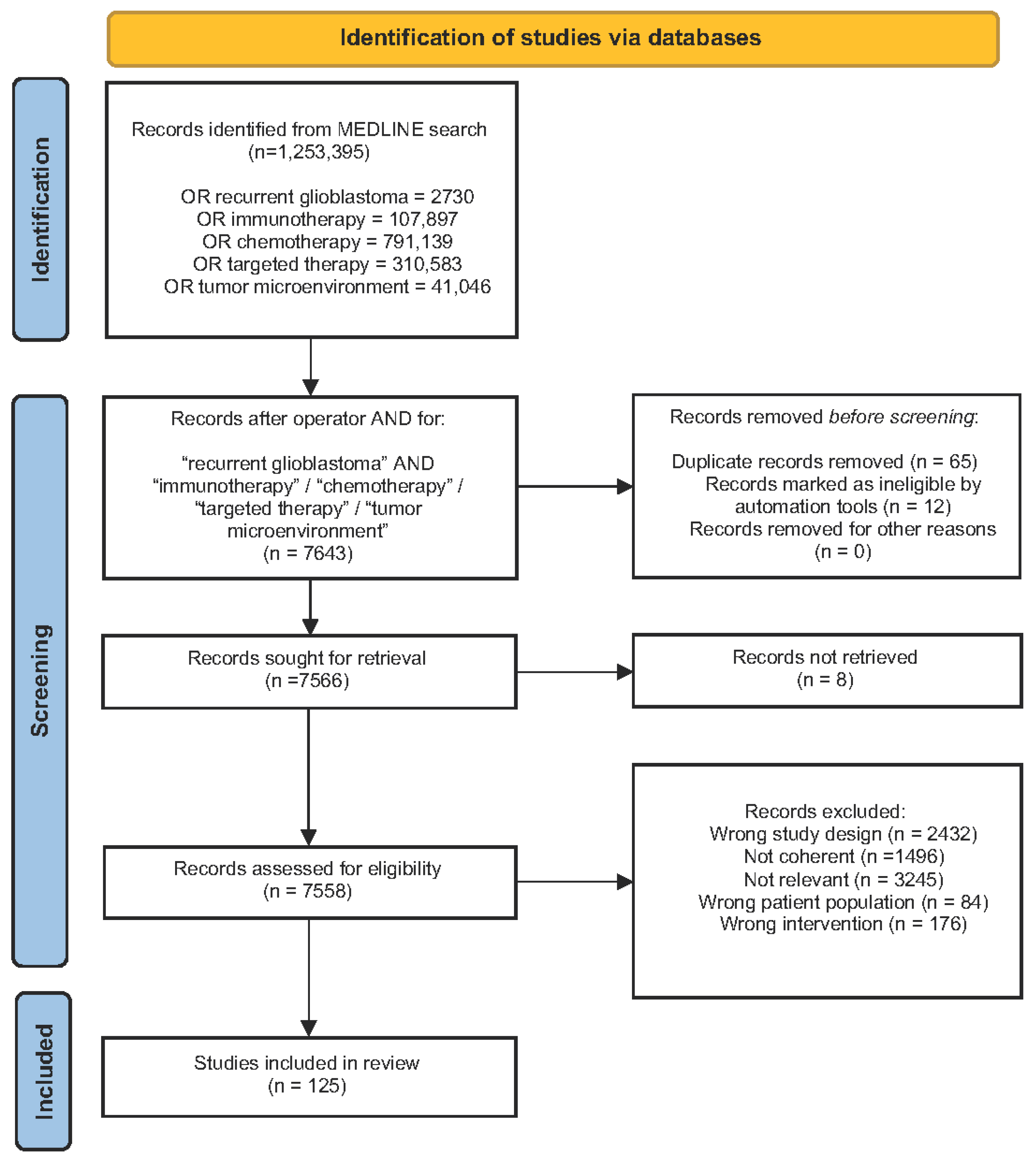
2. Materials and Methods
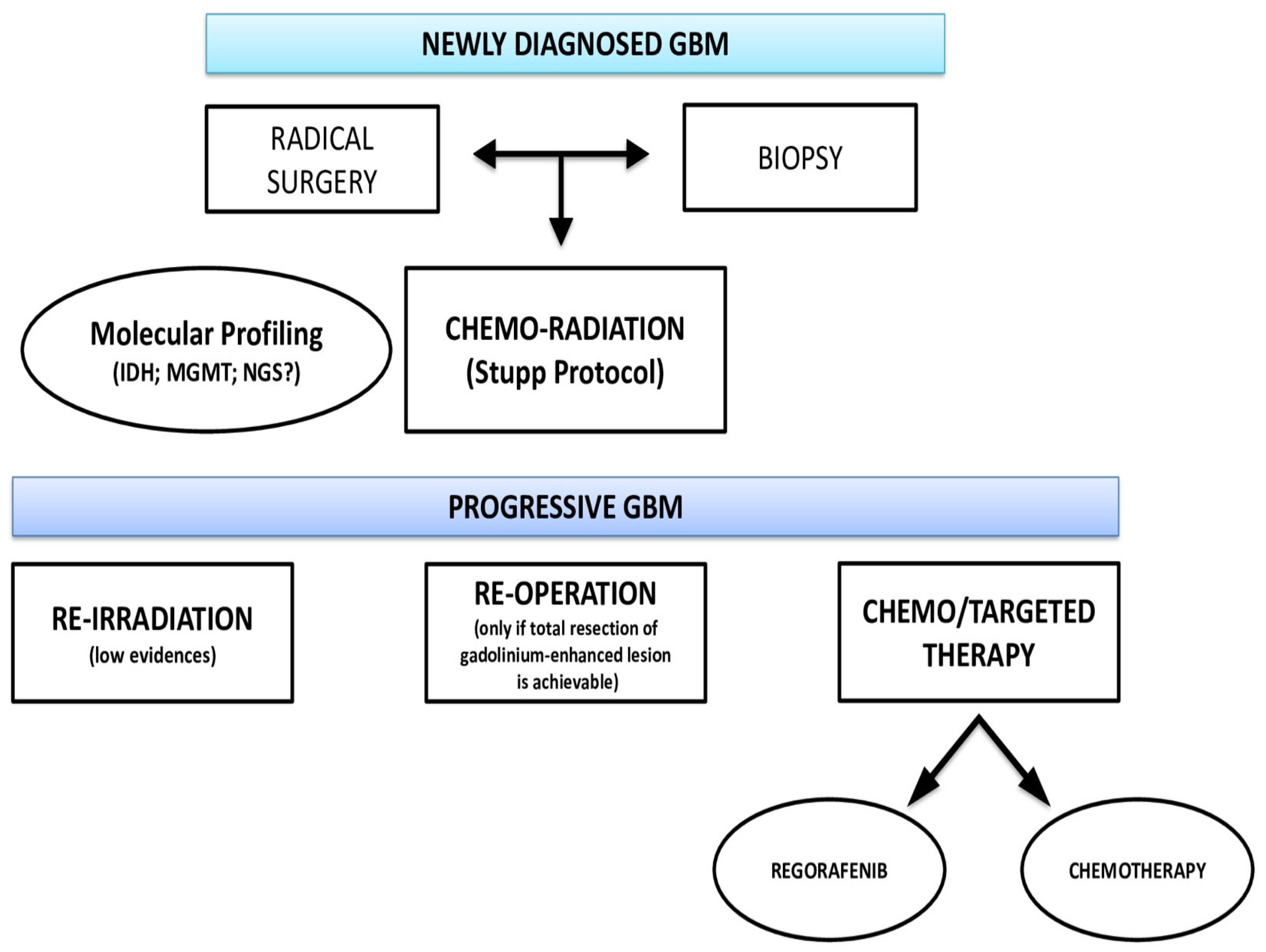
3. Locoregional Treatment of rGBM
4. Molecular Footprints of rGBM
4.1. O-6Methylguanine-DNA Methyltransferase
4.2. Vascular Endothelial Growth Factor
4.3. Epidermal Growth Factor Receptor
4.4. Telomerase Reverse Transcriptase
4.5. Platelet-Derived Growth Factor Receptor
4.6. Regorafenib, a Multi-Targeted Tyrosine Kinase Inhibitor
5. A Promising Future Direction: Immunotherapy
5.1. Adoptive T-Cell Therapy
5.2. Immune Checkpoint Inhibitors
5.3. Peptide Vaccines
5.4. Dendritic Cell Vaccines
5.5. Oncolytic Viruses
6. Current Guidelines for the Treatment of rGBM
6.1. CNS Guidelines on the Role of Radiation Therapy in rGBM
6.2. CNS Guidelines on the Role of Cytoreductive Surgery in rGBM
6.3. CNS Guidelines on the Role of Cytotoxic Therapies in rGBM (TMZ Monotherapy)
6.4. CNS Guidelines on the Role of Cytotoxic Therapies in rGBM (TMZ Combinations)
6.5. CNS Guidelines on the Role of Cytotoxic Therapies in rGBM (TTF)
6.6. EANO Guidelines for the Treatment of rGBM
7. Summary and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): Analysis of individual records for 37,513,025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Available online: https://www.nccn.org/ (accessed on 27 March 2022).
- De Bonis, P.; Anile, C.; Pompucci, A.; Fiorentino, A.; Balducci, M.; Chiesa, S.; Lauriola, L.; Maira, G.; Mangiola, A. The influence of surgery on recurrence pattern of glioblastoma. Clin. Neurol. Neurosurg. 2013, 115, 37–43. [Google Scholar] [CrossRef]
- van Nifterik, K.A.; Elkhuizen, P.H.M.; van Andel, R.J.; Stalpers, L.J.A.; Leenstra, S.; Lafleur, M.V.M.; Vandertop, W.P.; Slotman, B.J.; Hulsebos, T.J.M.; Sminia, P. Genetic profiling of a distant second glioblastoma multiforme after radiotherapy: Recurrence or second primary tumor? J. Neurosurg. 2006, 105, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef]
- Giordano, G.; Parcesepe, P.; Bruno, G.; Piscazzi, A.; Lizzi, V.; Remo, A.; Pancione, M.; D’Andrea, M.R.; De Santis, E.; Coppola, L.; et al. Evidence-Based Second-Line Treatment in RAS Wild-Type/Mutated Metastatic Colorectal Cancer in the Precision Medicine Era. Int. J. Mol. Sci. 2021, 22, 7717. [Google Scholar] [CrossRef]
- Tan, A.C.; Tan, D.S.W. Targeted Therapies for Lung Cancer Patients with Oncogenic Driver Molecular Alterations. J. Clin. Oncol. 2022, 40, 611–625. [Google Scholar] [CrossRef]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Song, K.; Wu, S.; Hameed, N.U.F.; Kudulaiti, N.; Xu, H.; Qin, Z.-Y.; Wu, J.-S. The prognosis of glioblastoma: A large, multifactorial study. Br. J. Neurosurg. 2021, 35, 555–561. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Veritas Health Innovation. Covidence Systematic Review Software, Melbourne, Australia. Available online: www.covidence.org (accessed on 5 May 2022).
- Sanai, N.; Polley, M.Y.; McDermott, M.W.; Parsa, A.T.; Berger, M.S. An extent of resection threshold for newly diagnosed glioblastomas. J. Neurosurg. 2011, 115, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbagallo, G.M.; Jenkinson, M.D.; Brodbelt, A.R. ‘Recurrent’ glioblastoma multiforme, when should we reoperate? Br. J. Neurosurg. 2008, 22, 452–455. [Google Scholar] [CrossRef]
- Clarke, J.L.; Ennis, M.M.; Yung, W.K.A.; Chang, S.M.; Wen, P.Y.; Cloughesy, T.F.; DeAngelis, L.; Robins, H.I.; Lieberman, F.S.; Fine, H.A.; et al. Is surgery at progression a prognostic marker for improved 6-month progression-free survival or overall survival for patients with recurrent glioblastoma? Neuro Oncol. 2011, 13, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Woodroffe, R.W.; Zanaty, M.; Soni, N.; Mott, S.L.; Helland, L.C.; Pasha, A.; Maley, J.; Dhungana, N.; A Jones, K.; Monga, V.; et al. Survival after reoperation for recurrent glioblastoma. J. Clin. Neurosci. 2020, 73, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Robin, A.M.; Lee, I.; Kalkanis, S.N. Reoperation for Recurrent Glioblastoma Multiforme. Neurosurg. Clin. N. Am. 2017, 28, 407–428. [Google Scholar] [CrossRef]
- Yong, R.L.; Lonser, R.R. Surgery for glioblastoma multiforme: Striking a balance. World Neurosurg. 2011, 76, 528–530. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Fernandez, J.; Garcia-Pallero, M.; Blasco, G.; Penanes, J.R.; Gil-Simoes, R.; Pulido, P.; Sola, R.G. Usefulness of Reintervention in Recurrent Glioblastoma: An Indispensable Weapon for Increasing Survival. World Neurosurg. 2017, 108, 610–617. [Google Scholar] [CrossRef]
- Yaprak, G.; Isık, N.; Gemici, C.; Pekyurek, M.; Bıcakcı, B.C.; Demircioglu, F.; Tatarlı, N. Stereotactic Radiotherapy in Recurrent Glioblastoma: A Valid Salvage Treatment Option. Ster. Funct. Neurosurg. 2020, 98, 167–175. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, R.; Martin-Subero, J.I.; Rohde, V.; Kirsch, M.; Alaminos, M.; Fernández, A.F.; Ropero, S.; Schackert, G.; Esteller, M. A microarray-based DNA methylation study of glioblastoma multiforme. Epigenetics 2009, 4, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Joudi Mashhad, M.; Hassanian, S.M.; Anvari, K.; Avan, A. The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. J. Cell Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent Glioblastoma: From Molecular Landscape to New Treatment Perspectives. Cancers 2020, 13, 47. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Lee, E.; Yong, R.L.; Paddison, P.; Zhu, J. Comparison of glioblastoma (GBM) molecular classification methods. In Seminars in Cancer Biology; Academic Press: New York, NY, USA, 2018. [Google Scholar] [CrossRef]
- Draaisma, K.; Chatzipli, A.; Taphoorn, M.; Kerkhof, M.; Weyerbrock, A.; Sanson, M.; Hoeben, A.; Lukacova, S.; Lombardi, G.; Leenstra, S.; et al. Molecular Evolution of IDH Wild-Type Glioblastomas Treated with Standard of Care Affects Survival and Design of Precision Medicine Trials: A Report from the EORTC 1542 Study. J. Clin. Oncol. 2020, 38, 81–99. [Google Scholar] [CrossRef]
- Brandes, A.A.; Franceschi, E.; Paccapelo, A.; Tallini, G.; De Biase, D.; Ghimenton, C.; Danieli, D.; Zunarelli, E.; Lanza, G.; Silini, E.M.; et al. Role of MGMT Methylation Status at Time of Diagnosis and Recurrence for Patients with Glioblastoma: Clinical Implications. Oncology 2017, 22, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Cantero, D.; De Lope, R.; De La Presa, R.M.; Sepúlveda, J.M.; Borrás, J.M.; Castresana, J.S.; D’Haene, N.; García, J.F.; Salmon, I.; Mollejo, M.; et al. Molecular Study of Long-Term Survivors of Glioblastoma by Gene-Targeted Next-Generation Sequencing. J. Neuropathol. Exp. Neurol. 2018, 77, 710–716. [Google Scholar] [CrossRef]
- Chai, E.A.R.; Li, G.; Liu, Y.; Zhang, K.; Zhao, Z.; Wu, F.; Chang, Y.; Pang, B.; Li, J.; Li, Y.; et al. Predictive value of MGMT promoter methylation on the survival of TMZ treated IDH-mutant glioblastoma. Cancer Biol. Med. 2021, 18, 272–282. [Google Scholar] [CrossRef]
- Friedman, H.S. Temozolomide in early stages of newly diagnosed malignant glioma and neoplastic meningitis. Semin. Oncol. 2000, 27, 35–40. [Google Scholar]
- Poon, M.T.C.; Bruce, M.; Simpson, J.E.; Hannan, C.J.; Brennan, P.M. Temozolomide sensitivity of malignant glioma cell lines—a systematic review assessing consistencies between in vitro studies. BMC Cancer. 2021, 21, 1240. [Google Scholar] [CrossRef]
- Taal, W.; Oosterkamp, H.M.; Walenkamp, A.M.E.; Dubbink, H.J.; Beerepoot, L.V.; Hanse, M.C.J.; Buter, J.; Honkoop, A.H.; Boerman, D.; De Vos, F.; et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): A randomised controlled phase 2 trial. Lancet Oncol. 2014, 15, 943–953. [Google Scholar] [CrossRef]
- Brandes, A.A.; Finocchiaro, G.; Zagonel, V.; Reni, M.; Caserta, C.; Fabi, A.; Clavarezza, M.; Maiello, E.; Eoli, M.; Lombardi, G.; et al. AVAREG: A phase II, randomized, noncomparative study of fotemustine or bevacizumab for patients with recurrent glioblastoma. Neuro Oncol. 2016, 18, 1304–1312. [Google Scholar] [CrossRef]
- Quinn, J.A.; Jiang, S.X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; Sampson, J.H.; et al. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J. Clin. Oncol. 2009, 27, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Tsuji, S.; Nakamura, S.; Egashira, Y.; Shimazawa, M.; Nakayama, N.; Yano, H.; Iwama, T.; Hara, H. Riluzole enhances the antitumor effects of temozolomide via suppression of MGMT expression in glioblastoma. J. Neurosurg. 2020, 134, 701–710. [Google Scholar] [CrossRef]
- Giordano, G.; Febbraro, A.; Venditti, M.; Campidoglio, S.; Olivieri, N.; Raieta, K.; Parcesepe, P.; Imbriani, G.C.; Remo, A.; Pancione, M. Targeting angiogenesis and tumor microenvironment in metastatic colorectal cancer: Role of aflibercept. Gastroenterol. Res. Pract. 2014, 2014, 526178. [Google Scholar] [CrossRef] [Green Version]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [Green Version]
- Hasselbalch, B.; Lassen, U.; Hansen, S.; Holmberg, M.; Mau-Sørensen, M.; Kosteljanetz, M.; Broholm, H.; Stockhausen, M.-T.; Poulsen, H.S. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: A phase II trial. Neuro Oncol. 2010, 12, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Sathornsumetee, S.; Desjardins, A.; Vredenburgh, J.J.; McLendon, R.E.; Marcello, J.; Herndon, J.E.; Mathe, A.; Hamilton, M.; Rich, J.N.; Norfleet, J.A.; et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro Oncol. 2010, 12, 1300–1310. [Google Scholar] [CrossRef] [Green Version]
- Galanis, E.; Anderson, S.K.; Lafky, J.M.; Uhm, J.H.; Giannini, C.; Kumar, S.K.; Kimlinger, T.K.; Northfelt, D.W.; Flynn, P.J.; Jaeckle, K.A.; et al. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): A north central cancer treatment group trial. Clin. Cancer Res. 2013, 19, 4816–4823. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandris, Q.G.; Martini, M.; Cenci, T.; DI Bonaventura, R.; Lauretti, L.; Stumpo, V.; Olivi, A.; Larocca, L.M.; Pallini, R.; Montano, N. Tailored therapy for recurrent glioblastoma. Report of a personalized molecular approach. J. Neurosurg. Sci. 2020. [Google Scholar] [CrossRef]
- Giordano, G.; Remo, A.; Porras, A.; Pancione, M. Immune Resistance and EGFR Antagonists in Colorectal Cancer. Cancers 2019, 11, 1089. [Google Scholar] [CrossRef] [Green Version]
- Sabbah, D.A.; Hajjo, R.; Sweidan, K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. [Google Scholar] [CrossRef]
- Talukdar, S.; Emdad, L.; Das, S.K.; Fisher, P.B. EGFR: An essential receptor tyrosine kinase-regulator of cancer stem cells. Adv. Cancer Res. 2020, 147, 161–188. [Google Scholar] [CrossRef]
- Liu, X.; Wang, P.; Zhang, C.; Ma, Z. Epidermal growth factor receptor (EGFR): A rising star in the era of precision medicine of lung cancer. Oncotarget 2017, 8, 50209–50220. [Google Scholar] [CrossRef] [Green Version]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Libermann, T.; Nusbaum, H.R.; Razon, N.; Kris, R.; Lax, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification and overexpression of the EGF receptor gene in primary human glioblastomas. J. Cell Sci. Suppl. 1985, 3, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Chi, A.S.; Cahill, D.P.; Reardon, D.A.; Wen, P.Y.; Mikkelsen, T.; Peereboom, D.M.; Wong, E.T.; Gerstner, E.R.; Dietrich, J.; Plotkin, S.R.; et al. Exploring Predictors of Response to Dacomitinib in EGFR-Amplified Recurrent Glioblastoma. JCO Precis. Oncol. 2020, 4, PO.19.00295. [Google Scholar] [CrossRef]
- Cruz Da Silva, E.; Mercier, M.C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef]
- Frederick, L.; Wang, X.Y.; Eley, G.; James, C.D. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000, 60, 1383–1387. [Google Scholar]
- Reardon, D.A.; Groves, M.D.; Wen, P.Y.; Nabors, L.; Mikkelsen, T.; Rosenfeld, S.; Raizer, J.; Barriuso, J.; McLendon, R.E.; Suttle, A.B.; et al. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin. Cancer Res. 2013, 19, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Bady, P.; Kamoshima, Y.; Kouwenhoven, M.C.M.; Delorenzi, M.; Lambiv, W.L.; Hamou, M.-F.; Matter, M.S.; Koch, A.; et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib—a phase II trial. Mol. Cancer Ther. 2011, 10, 1102–1112. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, A.; Wang, M.; Robins, H.I.; Lautenschlaeger, T.; Curran, W.J.; Brachman, D.G.; Schultz, C.J.; Choucair, A.; Dolled-Filhart, M.; Christiansen, J.; et al. RTOG 0211: A phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1206–1211. [Google Scholar] [CrossRef] [Green Version]
- Halatsch, M.-E.; Gehrke, E.E.; Vougioukas, V.I.; Bötefür, I.C.; A-Borhani, F.; Efferth, T.; Gebhart, E.; Domhof, S.; Schmidt, U.; Buchfelder, M. Inverse correlation of epidermal growth factor receptor messenger RNA induction and suppression of anchorage-independent growth by OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in glioblastoma multiforme cell lines. J. Neurosurg. 2004, 100, 523–533. [Google Scholar] [CrossRef]
- Bent, M.V.D.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.-S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F.; et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro Oncol. 2020, 22, 684–693, Erratum in Neuro Oncol. 2021, 23, 1415. [Google Scholar] [CrossRef]
- Barthel, F.P.; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; Anderson, K.J.; Abiola, O.; Aldape, K.; Alfaro, K.D.; et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019, 576, 112–120. [Google Scholar] [CrossRef]
- Heidenreich, B.; Rachakonda, P.S.; Hosen, I.; Volz, F.; Hemminki, K.; Weyerbrock, A.; Kumar, R. TERT promoter mutations and telomere length in adult malignant gliomas and recurrences. Oncotarget 2015, 6, 10617–10633. [Google Scholar] [CrossRef] [Green Version]
- Killela, P.J.; Pirozzi, C.J.; Healy, P.; Reitman, Z.J.; Lipp, E.; Rasheed, B.A.; Yang, R.; Diplas, B.H.; Wang, Z.; Greer, P.K.; et al. Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget 2014, 5, 1515–1525. [Google Scholar] [CrossRef] [Green Version]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.W.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Miki, S.; Fujimoto, K.; Fukuoka, K.; Matsushita, Y.; Maida, Y.; Yasukawa, M.; Hayashi, M.; Shinkyo, R.; Kikuchi, K.; et al. Eribulin penetrates brain tumor tissue and prolongs survival of mice harboring intracerebral glioblastoma xenografts. Cancer Sci. 2019, 110, 2247–2257. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.S.M.; Gao, Y.; Chau, D.H.W.; Li, G.H.Y.; Lai, L.H.; Huang, P.T.; Huang, C.F.; Huang, J.J.; Chen, Y.; Kung, H.F.; et al. A novel glioblastoma cancer gene therapy using AAV-mediated long-term expression of human TERT C-terminal polypeptide. Cancer Gene Ther. 2007, 14, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.-C.; Lin, S.-Z.; Chen, Y.-L.; Chang, J.-S.; Ho, L.-I.; Liu, P.-Y.; Chang, L.-F.; Harn, Y.-C.; Chen, S.-P.; Sun, L.-Y.; et al. Butylidenephthalide suppresses human telomerase reverse transcriptase (TERT) in human glioblastomas. Ann. Surg. Oncol. 2011, 18, 3514–3527. [Google Scholar] [CrossRef]
- Ostman, A. PDGF receptors-mediators of autocrine tumor growth and regulators of tumor vasculature and stroma. Cytokine Growth Factor Rev. 2004, 15, 275–286. [Google Scholar] [CrossRef]
- Hermanson, M.; Funa, K.; Koopmann, J.; Maintz, D.; Waha, A.; Westermark, B.; Heldin, C.H.; Wiestler, O.D.; Louis, D.N.; von Deimling, A.; et al. Association of loss of heterozygosity on chromosome 17p with high platelet-derived growth factor alpha receptor expression in human malignant gliomas. Cancer Res. 1996, 56, 164–171. [Google Scholar]
- Wen, P.Y.; Yung, W.A.; Lamborn, K.R.; Dahia, P.L.; Wang, Y.; Peng, B.; Abrey, L.E.; Raizer, J.; Cloughesy, T.F.; Fink, K.; et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99-08. Clin. Cancer Res. 2006, 12, 4899–4907. [Google Scholar] [CrossRef] [Green Version]
- Raymond, E.; Brandes, A.; Dittrich, C.; Fumoleau, P.; Coudert, B.; Clement, P.M.; Frenay, M.; Rampling, R.; Stupp, R.; Kros, J.M.; et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: A European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J. Clin. Oncol. 2008, 26, 4659–4665. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Egorin, M.J.; Quinn, J.A.; Rich, J.N.; Gururangan, I.; Vredenburgh, J.J.; Desjardins, A.; Sathornsumetee, S.; Provenzale, J.M.; Herndon, J.E.; et al. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J. Clin. Oncol. 2005, 23, 9359–9368. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; Von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.-H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer. 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Hamed, H.A.; Tavallai, S.; Grant, S.; Poklepovic, A.; Dent, P. Sorafenib/regorafenib and lapatinib interact to kill CNS tumor cells. J. Cell Physiol. 2015, 230, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Khawaja, M.R.; Hong, D.S.; Amini, B.; Yungfang, J.; Liu, H.; Johnson, A.; Schrock, A.B.; Ali, S.M.; Sun, J.X.; et al. First-in-human trial of multikinase VEGF inhibitor regorafenib and anti-EGFR antibody cetuximab in advanced cancer patients. JCI Insight. 2017, 2, e90380. [Google Scholar] [CrossRef]
- Sidaway, P. Relapsed glioblastomas respond to regorafenib. Nat. Rev. Clin. Oncol. 2019, 16, 144. [Google Scholar] [CrossRef]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Associazione Italiana di Oncologia Medica. Neoplasie Cerebrali (Version 2019). Available online: https://www.aiom.it/wp-content/uploads/2019/10/2019_LG_AIOM_Cerebrali-1.pdf (accessed on 4 June 2022).
- National Comprehensive Cancer Network. Central Nervous System Cancers (Version 1.2022). Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1425 (accessed on 4 June 2022).
- Lombardi, G.; Caccese, M.; Padovan, M.; Cerretti, G.; Pintacuda, G.; Manara, R.; Di Sarra, F.; Zagonel, V. Regorafenib in Recurrent Glioblastoma Patients: A Large and Monocentric Real-Life Study. Cancers 2021, 13, 4731. [Google Scholar] [CrossRef]
- Delgado-López, P.D.; Corrales-García, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef]
- Daubon, T.; Hemadou, A.; Romero Garmendia, I.; Saleh, M. Glioblastoma Immune Landscape and the Potential of New Immunotherapies. Front. Immunol. 2020, 11, 585616. [Google Scholar] [CrossRef]
- Chuang, D.F.; Lin, X. Targeted Therapies for the Treatment of Glioblastoma in Adults. Curr. Oncol. Rep. 2019, 21, 61. [Google Scholar] [CrossRef]
- Chen, L.; Qin, D.; Guo, X.; Wang, Q.; Li, J. Putting Proteomics into Immunotherapy for Glioblastoma. Front. Immunol. 2021, 12, 593255. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Options Oncol. 2019, 20, 24. [Google Scholar] [CrossRef] [Green Version]
- Zanders, E.D.; Svensson, F.; Bailey, D.S. Therapy for glioblastoma: Is it working? Drug Discov. Today 2019, 24, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Li, X.; Li, Y.; Zhang, J.; Zong, Z.; Zhang, H. Current Immunotherapies for Glioblastoma Multiforme. Front. Immunol. 2021, 11, 603911. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Curry, W.T.; Carter, B.S.; Maus, M.V. Chimeric antigen receptor T-cell immunotherapy for glioblastoma: Practical insights for neurosurgeons. Neurosurg. Focus 2018, 44, E13. [Google Scholar] [CrossRef] [Green Version]
- Commissioner Oot. Press Announcements—FDA Approves CAR-T Cell Therapy to Treat Adults with Certain Types of Large B-Cell Lymphoma. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-car-t-cell-therapy-treat-adults-certain-types-large-b-cell-lymphoma (accessed on 4 June 2022).
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2015, 108, djv375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Choi, B.D.; Maus, M.V.; June, C.H.; Sampson, J.H. Immunotherapy for Glioblastoma: Adoptive T-cell Strategies. Clin. Cancer Res. 2019, 25, 2042–2048. [Google Scholar] [CrossRef]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.-F.; Orange, J.S.; Sumazin, P.; Man, T.-K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- Preusser, M.; Lim, M.; Hafler, D.A.; Reardon, D.A.; Sampson, J.H. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 2015, 11, 504–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef] [Green Version]
- Desai, K.; Hubben, A.; Ahluwalia, M. The Role of Checkpoint Inhibitors in Glioblastoma. Target Oncol. 2019, 14, 375–394. [Google Scholar] [CrossRef]
- Reardon, D.A.; Neuberg, D.S.; Keskin, D.B.; Tirosh, I.; Anandappa, A.; Mathewson, N.D.; Sun, J.; Shukla, S.A.; Gjini, E.; Li, S.; et al. Effect of dexamethasone in glioblastoma (GBM) patients on systemic and intratumoral T-cell responses induced by personalized neoantigen-targeting vaccine. In Proceedings of the 2018 ASCO Annual Meeting, Chicago, IL, USA, 31 May–4 June 2018. [Google Scholar]
- Arbour, K.C.; Mezquita, L.; Long, N.; Rizvi, H.; Auclin, E.; Ni, A.; Martínez-Bernal, G.; Ferrara, R.; Lai, W.V.; Hendriks, L.E.; et al. Impact of Baseline Steroids on Efficacy of Programmed Cell Death-1 and Programmed Death-Ligand 1 Blockade in Patients with Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2872–2878. [Google Scholar] [CrossRef]
- Ishikawa, E.; Yamamoto, T.; Matsumura, A. Prospect of Immunotherapy for Glioblastoma: Tumor Vaccine, Immune Checkpoint Inhibitors and Combination Therapy. Neurol. Med. Chir. 2017, 57, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; De Andrea, C.; De Cerio, A.L.-D.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; De Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro Oncol. 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Heimberger, A.B.; E Crotty, L.; E Archer, G.; Hess, K.R.; Wikstrand, C.J.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin. Cancer Res. 2003, 9, 4247–4254. [Google Scholar] [PubMed]
- Reardon, D.A.; Schuster, J.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; Desjardins, A.; Ashby, L.S.; et al. ReACT: Overall survival from a randomized phase II study of rindopepimut (CDX- 110) plus bevacizumab in relapsed glioblastoma. J. Clin. Oncol. 2015, 33, 2009. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Boydell, E.; Marinari, E.; Migliorini, D.; Dietrich, P.Y.; Patrikidou, A.; Dutoit, V. Exploratory Study of the Effect of IMA950/Poly-ICLC Vaccination on Response to Bevacizumab in Relapsing High-Grade Glioma Patients. Cancers 2019, 11, 464. [Google Scholar] [CrossRef] [Green Version]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [Green Version]
- Cho, D.-Y.; Lin, S.-Z.; Yang, W.-K.; Hsu, D.-M.; Lee, H.-C.; Lee, W.-Y.; Liu, S.-P. Recent advances of dendritic cells (DCs)-based immunotherapy for malignant gliomas. Cell Transplant. 2009, 18, 977–983. [Google Scholar] [CrossRef]
- Okada, H.; Kalinski, P.; Ueda, R.; Hoji, A.; Kohanbash, G.; Donegan, T.E.; Mintz, A.H.; Engh, J.A.; Bartlett, D.L.; Brown, C.K.; et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol. 2011, 29, 330–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, M.; Liau, L.M. Introduction: Brain tumor immunotherapy. J. Neurooncol. 2015, 123, 321–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Black, K.L.; Yu, J.S. Sensitization of malignant glioma to chemotherapy through dendritic cell vaccination. Expert Rev. Vaccines. 2006, 5, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Nassiri, F.; Wang, J.; Peruzzi, P.; Zadeh, G. Viral and other therapies for recurrent glioblastoma: Is a 24-month durable response unusual? Neuro Oncol. 2019, 21, 14–25. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Ziu, M.; Goyal, S.; Olson, J.J. Congress of Neurological Surgeons systematic review and evidence-based guidelines update on the role of radiation therapy in the management of progressive and recurrent glioblastoma in adults. J. Neurooncol. 2021, 158, 255–264. [Google Scholar] [CrossRef]
- Germano, I.M.; Ziu, M.; Wen, P.; Ormond, D.R.; Olson, J.J. Congress of Neurological Surgeons systematic review and evidence-based guidelines update on the role of cytotoxic chemotherapy and other cytotoxic therapies in the management of progressive glioblastoma in adults. J. Neurooncol. 2022, 158, 225–253. [Google Scholar] [CrossRef]
- Patrick, H.H.; Sherman, J.H.; Elder, J.B.; Olson, J.J. Congress of neurological surgeons systematic review and evidence-based guidelines update on the role of cytoreductive surgery in the management of progressive glioblastoma in adults. J. Neurooncol. 2022, 158, 167–177. [Google Scholar] [CrossRef]
- Ryken, T.C.; Kalkanis, S.N.; Buatti, J.M.; Olson, J.J.; AANS/CNS Joint Guidelines Committee. The role of cytoreductive surgery in the management of progressive glioblastoma: A systematic review and evidence-based clinical practice guideline. J. Neurooncol. 2014, 118, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.J. Congress of Neurological Surgeons systematic review and evidence-based guidelines for the treatment of adults with progressive glioblastoma update: Introduction and methods. J. Neurooncol. 2021, 158, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Straube, C.; Antoni, S.; Gempt, J.; Zimmer, C.; Meyer, B.; Schlegel, J.; Schmidt-Graf, F.; Combs, S.E. Re-irradiation in elderly patients with glioblastoma: A single institution experience. J. Neurooncol. 2019, 142, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Bräutigam, E.; Lampl, C.; Track, C.; Nieder, C.; Pichler, J.; Hammer, J.; Geinitz, H. Re-irradiation of recurrent glioblastoma as part of a sequential multimodality treatment concept. Clin. Transl. Oncol. 2019, 21, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Straube, C.; Elpula, G.; Gempt, J.; Gerhardt, J.; Bette, S.; Zimmer, C.; Schmidt-Graf, F.; Meyer, B.; Combs, S.E. Re-irradiation after gross total resection of recurrent glioblastoma: Spatial pattern of recurrence and a review of the literature as a basis for target volume definition. Rebestrahlung nach kompletter Resektion eines rezidivierten Glioblastoms: Räumliches Rezidivmuster und Review der Literatur als Grundlage für das Zielvolumen. Strahlenther. Onkol. 2017, 193, 897–909. [Google Scholar] [CrossRef]
- Zwirner, K.; Paulsen, F.; Schittenhelm, J.; Borchers, C.; Skardelly, M.; Zips, D.; Eckert, F. Prognostic parameters and outcome after re-irradiation for progressive glioblastoma. Acta. Neurol. Scand. 2017, 136, 239–245. [Google Scholar] [CrossRef]
- Hasan, S.; Chen, E.; Lanciano, R.M.; Yang, J.; Hanlon, A.; Lamond, J.; Arrigo, S.; Ding, W.; Mikhail, M.; Ghaneie, A.; et al. Salvage Fractionated Stereotactic Radiotherapy with or without Chemotherapy and Immunotherapy for Recurrent Glioblastoma Multiforme: A Single Institution Experience. Front. Oncol. 2015, 5, 106. [Google Scholar] [CrossRef] [Green Version]
- Pinzi, V.; Orsi, C.; Marchetti, M.; Milanesi, I.M.; Bianchi, L.C.; DiMeco, F.; Cuccarini, V.; Farinotti, M.; Ferroli, P.; Finocchiaro, G.; et al. Radiosurgery reirradiation for high-grade glioma recurrence: A retrospective analysis. Neurol. Sci. 2015, 36, 1431–1440. [Google Scholar] [CrossRef]
- Greenspoon, J.; Sharieff, W.; Hirte, H.; Overholt, A.; DeVillers, R.; Gunnarsson, T.; Whitton, A. Fractionated stereotactic radiosurgery with concurrent temozolomide chemotherapy for locally recurrent glioblastoma multiforme: A prospective cohort study. Onco Targets Ther. 2014, 7, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Yazici, G.; Cengiz, M.; Özyigit, G.; Eren, G.; Yildiz, F.; Akyol, F.; Gurkaynak, M.; Zorlu, F. Hypofractionated stereotactic reirradiation for recurrent glioblastoma. J. Neurooncol. 2014, 120, 117–123. [Google Scholar] [CrossRef]
- Ciammella, P.; Podgornii, A.; Galeandro, M.; D’Abbiero, N.; Pisanello, A.; Botti, A.; Cagni, E.; Iori, M.; Iotti, C. Hypofractionated stereotactic radiation therapy for recurrent glioblastoma: Single institutional experience. Radiat. Oncol. 2013, 8, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchorska, B.; Weller, M.; Tabatabai, G.; Senft, C.; Hau, P.; Sabel, M.C.; Herrlinger, U.; Ketter, R.; Schlegel, U.; Marosi, C.; et al. Complete resection of contrast-enhancing tumor volume is associated with improved survival in recurrent glioblastoma-results from the DIRECTOR trial. Neuro Oncol. 2016, 18, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Yong, R.L.; Wu, T.; Mihatov, N.; Shen, M.J.; Brown, M.; Zaghloul, K.A.; Park, G.E.; Park, J.K. Residual tumor volume and patient survival following reoperation for recurrent glioblastoma. J. Neurosurg. 2014, 121, 802–809. [Google Scholar] [CrossRef]
- Hager, J.; Herrmann, E.; Kammerer, S.; Dinc, N.; Won, S.-Y.; Senft, C.; Seifert, V.; Marquardt, G.; Quick-Weller, J. Impact of resection on overall survival of recurrent Glioblastoma in elderly patients. Clin. Neurol. Neurosurg. 2018, 174, 21–25. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.S.; Cloney, M.B.; Sonabend, A.M.; Zacharia, B.; Nazarian, M.N.; Iwamoto, F.M.; Sisti, M.B.; Bruce, J.N.; McKhann, G.M. The Safety of Surgery in Elderly Patients with Primary and Recurrent Glioblastoma. World Neurosurg. 2015, 84, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.T.; Timmons, J.; Callahan, A.; O’Loughlin, L.; Giarusso, B.; Alsop, D.C. Phase I study of low-dose metronomic temozolomide for recurrent malignant gliomas. BMC Cancer 2016, 16, 914. [Google Scholar] [CrossRef] [Green Version]
- van Vugt, V.A.; Piccioni, D.E.; Brown, B.D.; Brown, T.; Saria, M.G.; Juarez, T.; Kesari, S. Retrospective analysis of safety and feasibility of a 3 days on/11 days off temozolomide dosing regimen in recurrent adult malignant gliomas. CNS Oncol. 2014, 3, 257–265. [Google Scholar] [CrossRef]
- Han, S.J.; Rolston, J.D.; Molinaro, A.M.; Clarke, J.L.; Prados, M.D.; Chang, S.M.; Berger, M.S.; DeSilva, A.; Butowski, N.A. Phase II trial of 7 days on/7 days off temozolmide for recurrent high-grade glioma. Neuro Oncol. 2014, 16, 1255–1262. [Google Scholar] [CrossRef] [Green Version]
- Omuro, A.; Chan, T.A.; Abrey, L.E.; Khasraw, M.; Reiner, A.; Kaley, T.; DeAngelis, L.; Lassman, A.B.; Nolan, C.; Gavrilovic, I.; et al. Phase II trial of continuous low-dose temozolomide for patients with recurrent malignant glioma. Neuro Oncol. 2013, 15, 242–250. [Google Scholar] [CrossRef]
- Archavlis, E.; Tselis, N.; Birn, G.; Ulrich, P.; Baltas, D.; Zamboglou, N. Survival analysis of HDR brachytherapy versus reoperation versus temozolomide alone: A retrospective cohort analysis of recurrent glioblastoma multiforme. BMJ Open 2013, 3, e002262. [Google Scholar] [CrossRef] [Green Version]
- Norden, A.D.; Lesser, G.J.; Drappatz, J.; Ligon, K.L.; Hammond, S.N.; Lee, E.Q.; Reardon, D.R.; Fadul, C.E.; Plotkin, S.R.; Batchelor, T.T.; et al. Phase 2 study of dose-intense temozolomide in recurrent glioblastoma. Neuro Oncol. 2013, 15, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, T.; Arakawa, Y.; Ueba, T.; Oda, M.; Nishida, N.; Akiyama, Y.; Tsukahara, T.; Iwasaki, K.; Mikuni, N.; Miyamoto, S. Phase I/II Study of Temozolomide Plus Nimustine Chemotherapy for Recurrent Malignant Gliomas: Kyoto Neuro-oncology Group. Neurol. Med. Chir. 2017, 57, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liang, L.; Yang, T.; Qiao, Y.; Xia, Y.; Liu, L.; Li, C.; Lu, P.; Jiang, X. A pilot clinical study of apatinib plus irinotecan in patients with recurrent high-grade glioma: Clinical Trial/Experimental Study. Medicine 2017, 96, e9053. [Google Scholar] [CrossRef]
- Reynés, G.; Martínez-Sales, V.; Vila, V.; Balaña, C.; Pérez-Segura, P.; Vaz, M.A.; Benavides, M.; Gallego, O.; Palomero, I.; Gil-Gil, M.; et al. Phase II trial of irinotecan and metronomic temozolomide in patients with recurrent glioblastoma. Anticancer Drugs 2016, 27, 133–137. [Google Scholar] [CrossRef] [Green Version]
- Di Cristofori, A.; Zarino, B.; Fanizzi, C.; Fornara, G.A.; Bertani, G.; Rampini, P.; Carrabba, G.; Caroli, M. Analysis of factors influencing the access to concomitant chemo-radiotherapy in elderly patients with high grade gliomas: Role of MMSE, age and tumor volume. J. Neurooncol. 2017, 134, 377–385. [Google Scholar] [CrossRef]
- Franceschi, E.; Stupp, R.; Bent, M.J.V.D.; van Herpen, C.; Donadey, F.L.; Gorlia, T.; Hegi, M.; Lhermitte, B.; Strauss, L.C.; Allgeier, A.; et al. EORTC 26083 phase I/II trial of dasatinib in combination with CCNU in patients with recurrent glioblastoma. Neuro Oncol. 2012, 14, 1503–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, J.; Juratli, T.A.; Radev, Y.; Daubner, D.; Soucek, S.; Schackert, G.; Krex, D. Safety and effectiveness of bis-chloroethylnitrosourea wafer chemotherapy in elderly patients with recurrent glioblastoma. Oncology 2017, 93, 43–50. [Google Scholar] [CrossRef]
- Marinelli, A.; Lamberti, G.; Cerbone, L.; Cordua, N.; Buonerba, C.; Peluso, G.; Di Lorenzo, G.; De Placido, S. High-dose fotemustine in temozolomide-pretreated glioblastoma multiforme patients: A phase I/II trial. Medicine 2018, 97, e11254. [Google Scholar] [CrossRef]
- Pérez-Segura, P.; Manneh, R.; Ceballos, I.; García, A.; Benavides, M.; Fuster, J.; Vaz, M.A.; Cano, J.M.; Berros, J.P.; Covela, M.; et al. GEINOFOTE: Efficacy and safety of fotemustine in patients with high-grade recurrent gliomas and poor performance status. Clin. Transl. Oncol. 2016, 18, 805–812. [Google Scholar] [CrossRef]
- Lombardi, G.; Bellu, L.; Pambuku, A.; Della Puppa, A.; Fiduccia, P.; Farina, M.; D’Avella, D.; Zagonel, V. Clinical outcome of an alternative fotemustine schedule in elderly patients with recurrent glioblastoma: A mono-institutional retrospective study. J. Neurooncol. 2016, 128, 481–486. [Google Scholar] [CrossRef]
- Santoni, M.; Scoccianti, S.; Lolli, I.; Fabrini, M.G.; Silvano, G.; Detti, B.; Perrone, F.; Savio, G.; Iacovelli, R.; Burattini, L.; et al. Efficacy and safety of second-line fotemustine in elderly patients with recurrent glioblastoma. J. Neurooncol. 2013, 113, 397–401. [Google Scholar] [CrossRef] [Green Version]
- De Felice, F.; Bulzonetti, N.; Musio, D.; D’Elia, A.; Salvati, M.; Tombolini, V. Low-dose fotemustine as second-line chemotherapy for recurrent glioblastoma multiforme. Anticancer Drugs Res. 2013, 33, 4013–4016. [Google Scholar]
- Carrieri, F.A.; Smack, C.; Siddiqui, I.; Kleinberg, L.R.; Tran, P.T. Tumor Treating Fields: At the Crossroads Between Physics and Biology for Cancer Treatment. Front. Oncol. 2020, 10, 575992. [Google Scholar] [CrossRef]
- Lu, G.; Rao, M.; Zhu, P.; Liang, B.; El-Nazer, R.T.; Fonkem, E.; Bhattacharjee, M.B.; Zhu, J.-J. Triple-drug Therapy with Bevacizumab, Irinotecan, and Temozolomide Plus Tumor Treating Fields for Recurrent Glioblastoma: A Retrospective Study. Front. Neurol. 2019, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Kesari, S.; Ram, Z.; EF-14 Trial Investigators. Tumor-treating fields plus chemotherapy versus chemotherapy alone for glioblastoma at first recurrence: A post hoc analysis of the EF-14 trial. CNS Oncol. 2017, 6, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Ansstas, G.; Tran, D.D. Treatment with Tumor-Treating Fields Therapy and Pulse Dose Bevacizumab in Patients with Bevacizumab-Refractory Recurrent Glioblastoma: A Case Series. Case Rep. Neurol. 2016, 8, 1–9. [Google Scholar] [CrossRef]
- Rulseh, A.M.; Keller, J.; Klener, J.; Šroubek, J.; Dbalý, V.; Syrůček, M.; Tovaryš, F.; Vymazal, J. Long-term survival of patients suffering from glioblastoma multiforme treated with tumor-treating fields. World J. Surg. Oncol. 2012, 10, 220. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Wong, E.T.; Kanner, A.A.; Steinberg, D.; Engelhard, H.; Heidecke, V.; Kirson, E.D.; Taillibert, S.; Liebermann, F.; Dbalý, V.; et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: A randomised phase III trial of a novel treatment modality. Eur. J. Cancer 2012, 48, 2192–2202. [Google Scholar] [CrossRef] [Green Version]
- Harrabi, S.B.; Bougatf, N.; Mohr, A.; Haberer, T.; Herfarth, K.; Combs, S.E.; Debus, J.; Adeberg, S. Dosimetric advantages of proton therapy over conventional radiotherapy with photons in young patients and adults with low-grade glioma. Dosimetrische Vorteile der Protonentherapie gegenüber der konventionellen Strahlentherapie mit Photonen bei jungen Patienten und Erwachsenen mit niedriggradigem Gliom. Strahlenther. Onkol. 2016, 192, 759–769. [Google Scholar] [CrossRef] [Green Version]
- Malouff, T.D.; Peterson, J.L.; Mahajan, A.; Trifiletti, D.M. Carbon ion radiotherapy in the treatment of gliomas: A review. J. Neurooncol. 2019, 145, 191–199. [Google Scholar] [CrossRef]
- Yung, W.A.; Prados, M.D.; Yaya-Tur, R.; Rosenfeld, S.S.; Brada, M.; Friedman, H.S.; Albright, R.; Olson, J.; Chang, S.M.; O’Neill, A.M.; et al. Multicenter phase II trial of temozolomide in patients with anaplastic astrocytoma or anaplastic oligoastrocytoma at first relapse. J. Clin. Oncol. 1999, 17, 2762–2771. [Google Scholar] [CrossRef]
- Brada, M.; Stenning, S.; Gabe, R.; Thompson, L.C.; Levy, D.; Rampling, R.; Erridge, S.; Saran, F.; Gattamaneni, R.; Hopkins, K.; et al. Temozolomide versus procarbazine, lomustine, and vincristine in recurrent high-grade glioma. J. Clin. Oncol. 2010, 28, 4601–4608. [Google Scholar] [CrossRef] [Green Version]
- van den Bent, M.J.; Klein, M.; Smits, M.; Reijneveld, J.C.; French, P.J.; Clement, P.; de Vos, F.Y.; Wick, A.; Mulholland, P.J.; Taphoorn, M.J.; et al. Bevacizumab and temozolomide in patients with first recurrence of WHO grade II and III glioma, without 1p/19q co-deletion (TAVAREC): A randomised controlled phase 2 EORTC trial. Lancet Oncol. 2018, 19, 1170–1179. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| No. of clinical trials | 103 |
| Phase | Phase 1: 52 (50.49%) |
| Phase 2: 46 (44.66%) | |
| Phase 3: 5 (4.85%) | |
| No. of arms | 1: 56 (54.37%) |
| 2: 32 (31.07%) | |
| >2: 15 (14.56%) | |
| No. of enrolled patients | <50: 66 (64.08%) |
| 50–100: 24 (23.3%) | |
| 100–200: 10 (9.71%) | |
| 200–500: 2 (1.94%) | |
| >500: 1 (0.97%) | |
| No. of systemic therapies as a therapeutical investigation | 1: 50 (48.54%) |
| 2: 41 (39.81%) | |
| 3: 9 (8.74%) | |
| >3: 3 (2.91%) | |
| Combinations | Trials including radiotherapy: 21 (20.39%) |
| Trials including surgery: 32 (31.08%) | |
| Trials including radiotherapy and surgery: 7 (6.8%) | |
| Treatment allocation | Randomized: 22 (21.36%) |
| Non-randomized: 24 (23.3%) | |
| n/a: 57 (55.34%) | |
| Masking | None: 95 (92.23%) |
| Single: 1 (0.97%) | |
| Double: 4 (3.88%) | |
| Other: 3 (2.91%) | |
| Interventional model | Parallel assignment: 26 (25.24%) |
| Sequential assignment: 20 (19.42%) | |
| Single-group assignment: 56 (54.37%) | |
| Crossover assignment: 1 (0.97%) | |
| Country | USA: 61 (59.22%) |
| International: 14 (13.59) | |
| China: 12 (11.65%) | |
| Norway: 3 (2.91%) | |
| Germany: 2 (1.94%) | |
| Others: 11 (10.68%) | |
| Estimated date of completion | 2022–2025: 90 (87.38%) |
| 2026–2030: 12 (11.65%) | |
| Beyond 2030: 1 (0.97%) | |
| Most represented primary endpoints | Treatment-related adverse effects: 34 (33.01%) |
| PFS: 27 (26,21%) | |
| Dose-limiting toxicity: 26 (25.24%) OS: 22 (21.36%) |
| Drug Group | Agent | Trial Phase | Effect |
|---|---|---|---|
| Alkylating agents | TMZ (Temozolomide) | I: 7, II: 8, III: 2 | DNA-alkylating agent, whose effect mostly occurs at the N7 or O6 positions of guanine residues. DNA modification may induce the death of tumoral cells. The drug efficacy might be hindered by the enzyme MGMT. |
| Lomustine | I: 2, II: 3, III: 1 | Bifunctional alkylating agent (effect both on DNA and RNA). In DNA, it creates interstrand cross-links. Owing to its ability to carmaboylate on aminoacidic residues of proteins, its effect might be further increased by inhibiting several key enzymatic processes. | |
| VAL-083 | I: 0, II: 0, III: 1 | Bi-functional alkylating agent—its effects are expressed through cross-linking with an epoxide group along all phases of the cell cycle. | |
| Anti-angiogenic | Bevacizumab | I: 1, II: 7, III: 2 | Inhibitor of VEGF-A, causing the inhibition of angiogenesis |
| Immune checkpoint inhibitors | Nivolumab | I: 4 II: 3 III: 0 | Preventing PD-L1-induced T-cell inactivation by binding PD-1 to its extracellular domain |
| Ipilimumab | I: 4, II: 1, III: 0 | Avoiding T-cell inactivation by binding CTLA-4 receptors | |
| Pembrolizumab | I: 1, II: 3, III: 0 | Preventing PD-L1-induced T-cell inactivation by binding PD-1 on its extracellular domain | |
| PARP inhibitor | Niraparib | I: 1, II: 2, III: 0 | Preventing tumor cells’ DNA reparation, and consequently inducing tumor cell death by inhibiting PARP1/2 |
| Adoptive T-cell therapy | CAR-T B7-H3 | I: 3, II: 1, III: 0 | Allows the T-cells to recognize B7-H3 in order to increase the immunological response |
| Topoisomerase inhibitor | Irinotecan | I: 2, II: 1, III: 0 | Traps a subset of topoisomerase-1-DNA, avoiding tumor cells’ DNA replication |
| Autologous dendritic cell | ADCTA | I: 0, II: 0, III: 1 | Elicitation of antigen-specific, CD4/CD8 cytotoxic T-cells’ responses and induction of IFN-γ secretion |
| FASN inhibitor | ASC40 | I: 0, II: 0, III: 1 | Induction of the depletion of long-chain fatty acids, consequently leading to cell death by inhibiting FASN, which is preferentially expressed in malignant tissues |
| PI3K/mTOR inhibitor | Paxalisib | I: 0, II: 0, III: 1 | Inhibition of cell growth/survival by specifically inhibiting PI3K in the PI3K/AKT kinase signaling pathway |
| VEGFR2-TIE2 tyrosine kinase inhibitor | Regorafenib | I: 0, II: 0, III: 1 | Anti-angiogenic activity by inhibiting VEGFR2-TIE2 tyrosine kinase |
| JAK1/3 inhibitor | Tofacitinib | I: 0, II: 0, III: 1 | Influence on DNA transcription by inhibiting JAK1/JAK3 and interfering with the JAK-STAT pathway |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leone, A.; Colamaria, A.; Fochi, N.P.; Sacco, M.; Landriscina, M.; Parbonetti, G.; de Notaris, M.; Coppola, G.; De Santis, E.; Giordano, G.; et al. Recurrent Glioblastoma Treatment: State of the Art and Future Perspectives in the Precision Medicine Era. Biomedicines 2022, 10, 1927. https://doi.org/10.3390/biomedicines10081927
Leone A, Colamaria A, Fochi NP, Sacco M, Landriscina M, Parbonetti G, de Notaris M, Coppola G, De Santis E, Giordano G, et al. Recurrent Glioblastoma Treatment: State of the Art and Future Perspectives in the Precision Medicine Era. Biomedicines. 2022; 10(8):1927. https://doi.org/10.3390/biomedicines10081927
Chicago/Turabian StyleLeone, Augusto, Antonio Colamaria, Nicola Pio Fochi, Matteo Sacco, Matteo Landriscina, Giovanni Parbonetti, Matteo de Notaris, Giulia Coppola, Elena De Santis, Guido Giordano, and et al. 2022. "Recurrent Glioblastoma Treatment: State of the Art and Future Perspectives in the Precision Medicine Era" Biomedicines 10, no. 8: 1927. https://doi.org/10.3390/biomedicines10081927
APA StyleLeone, A., Colamaria, A., Fochi, N. P., Sacco, M., Landriscina, M., Parbonetti, G., de Notaris, M., Coppola, G., De Santis, E., Giordano, G., & Carbone, F. (2022). Recurrent Glioblastoma Treatment: State of the Art and Future Perspectives in the Precision Medicine Era. Biomedicines, 10(8), 1927. https://doi.org/10.3390/biomedicines10081927