
Hidradenitis Suppurativa: A Perspective on Genetic Factors Involved in the Disease

 ,
,  ,
,  ,
,  and
and 
Abstract
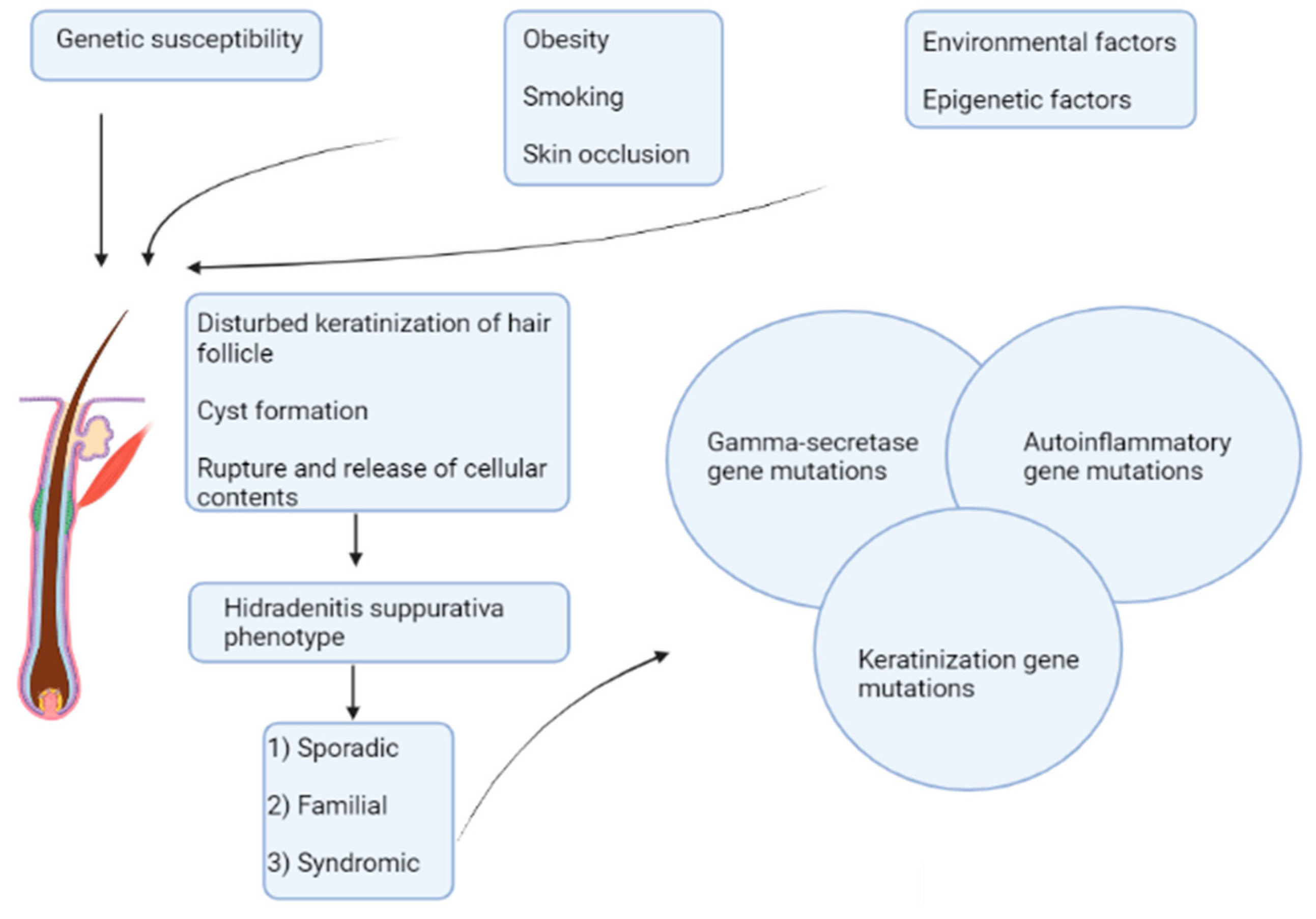
:1. Introduction
2. Clinical Features of Hidradenitis Suppurativa
3. Therapeutic Strategies for HS
4. Genetic Landscape of HS
4.1. Genetics of Familial Cases of HS
4.1.1. Gamma-Secretase Complex Gene Mutations
4.1.2. Mutations in Genes Other Than the Gamma-Secretase Ones
5. Genetics of Sporadic HS Cases
6. Genetics of Syndromic HS Cases
7. Genetics of HS in the Setting of Other Diseases
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kevin Phan, O.C.; Saxon, D. Smith Global prevalence of hidradenitis suppurativa and geographical variation—systematic review and meta-analysis. Biomed. Dermatol. 2020, 4, 2. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Benhadou, F.; Byrd, A.S.; Chandran, N.S.; Giamarellos-Bourboulis, E.J.; Fabbrocini, G.; Frew, J.W.; Fujita, H.; Gonzalez-Lopez, M.A.; Guillem, P.; et al. What causes hidradenitis suppurativa ?—15 years after. Exp. Dermatol. 2020, 29, 1154–1170. [Google Scholar] [CrossRef] [PubMed]
- Gasparic, J.; Theut Riis, P.; Jemec, G.B. Recognizing syndromic hidradenitis suppurativa: A review of the literature. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1809–1816. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Moltrasio, C.; Garcovich, S.; Marzano, A.V. PAPA spectrum disorders. G. Ital. Dermatol. Venereol. 2020, 155, 542–550. [Google Scholar] [CrossRef]
- Chen, W.; Ito, T.; Lin, S.H.; Song, Z.; Al-Khuzaei, S.; Jurik, A.G.; Plewig, G. Does SAPHO syndrome exist in dermatology? J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1501–1506. [Google Scholar] [CrossRef]
- Vural, S.; Baumgartner, M.; Lichtner, P.; Eckstein, G.; Hariry, H.; Chen, W.C.; Ruzicka, T.; Melnik, B.; Plewig, G.; Wagner, M.; et al. Investigation of gamma secretase gene complex mutations in German population with Hidradenitis suppurativa designate a complex polygenic heritage. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 1386–1392. [Google Scholar] [CrossRef]
- Marzano, A.V.; Genovese, G.; Moltrasio, C.; Tricarico, P.M.; Gratton, R.; Piaserico, S.; Garcovich, S.; Boniotto, M.; Brandao, L.; Moura, R.; et al. Whole-Exome Sequencing in 10 Unrelated Patients with Syndromic Hidradenitis Suppurativa: A Preliminary Step for a Genotype-Phenotype Correlation. Dermatology 2022, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, W.; Wen, W.; Sun, J.; Su, B.; Liu, B.; Ma, D.; Lv, D.; Wen, Y.; Qu, T.; et al. Gamma-secretase gene mutations in familial acne inversa. Science 2010, 330, 1065. [Google Scholar] [CrossRef]
- Wu, J.; Ge, H.; Fan, Y.; Zhen, Q.; Tang, L.; Sun, L. Novel Mutation of the NCSTN Gene Identified in a Chinese Acne Inversa Family. Ann. Dermatol. 2020, 32, 237–242. [Google Scholar] [CrossRef]
- Xiao, X.; He, Y.; Li, C.; Zhang, X.; Xu, H.; Wang, B. Nicastrin mutations in familial acne inversa impact keratinocyte proliferation and differentiation through the Notch and phosphoinositide 3-kinase/AKT signalling pathways. Br. J. Dermatol. 2016, 174, 522–532. [Google Scholar] [CrossRef]
- Miskinyte, S.; Nassif, A.; Merabtene, F.; Ungeheuer, M.N.; Join-Lambert, O.; Jais, J.P.; Hovnanian, A. Nicastrin mutations in French families with hidradenitis suppurativa. J. Investig. Dermatol. 2012, 132, 1728–1730. [Google Scholar] [CrossRef] [PubMed]
- Pink, A.E.; Simpson, M.A.; Brice, G.W.; Smith, C.H.; Desai, N.; Mortimer, P.S.; Barker, J.N.; Trembath, R.C. PSENEN and NCSTN mutations in familial hidradenitis suppurativa (Acne Inversa). J. Investig. Dermatol. 2011, 131, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- Pink, A.E.; Simpson, M.A.; Desai, N.; Dafou, D.; Hills, A.; Mortimer, P.; Smith, C.H.; Trembath, R.C.; Barker, J.N.W. Mutations in the gamma-secretase genes NCSTN, PSENEN, and PSEN1 underlie rare forms of hidradenitis suppurativa (acne inversa). J. Investig. Dermatol. 2012, 132, 2459–2461. [Google Scholar] [CrossRef] [PubMed]
- Ralser, D.J.; Basmanav, F.B.; Tafazzoli, A.; Wititsuwannakul, J.; Delker, S.; Danda, S.; Thiele, H.; Wolf, S.; Busch, M.; Pulimood, S.A.; et al. Mutations in gamma-secretase subunit-encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J. Clin. Investig. 2017, 127, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Minami-Hori, M.; Nagahata, H.; Nozaki, H.; Iinuma, S.; Igawa, S.; Kanno, K.; Kishibe, M.; Kanazawa, N.; Ishida-Yamamoto, A. Novel PSTPIP1 gene mutation in pyoderma gangrenosum, acne and suppurative hidradenitis syndrome. J. Dermatol. 2018, 45, e213–e214. [Google Scholar] [CrossRef]
- Marzano, A.V.; Trevisan, V.; Gattorno, M.; Ceccherini, I.; De Simone, C.; Crosti, C. Pyogenic arthritis, pyoderma gangrenosum, acne, and hidradenitis suppurativa (PAPASH): A new autoinflammatory syndrome associated with a novel mutation of the PSTPIP1 gene. JAMA Dermatol. 2013, 149, 762–764. [Google Scholar] [CrossRef]
- Calderon-Castrat, X.; Bancalari-Diaz, D.; Roman-Curto, C.; Romo-Melgar, A.; Amoros-Cerdan, D.; Alcaraz-Mas, L.A.; Fernandez-Lopez, E.; Canueto, J. PSTPIP1 gene mutation in a pyoderma gangrenosum, acne and suppurative hidradenitis (PASH) syndrome. Br. J. Dermatol. 2016, 175, 194–198. [Google Scholar] [CrossRef]
- Jfri, A.; Litvinov, I.V.; Netchiporouk, E.; O’Brien, E. Novel variants of MEFV and NOD2 genes in familial hidradenitis suppurativa: A case report. SAGE Open Med. Case Rep. 2020, 8, 2050313X20953113. [Google Scholar] [CrossRef]
- Marzano, A.V.; Ceccherini, I.; Gattorno, M.; Fanoni, D.; Caroli, F.; Rusmini, M.; Grossi, A.; De Simone, C.; Borghi, O.M.; Meroni, P.L.; et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine 2014, 93, e187. [Google Scholar] [CrossRef]
- Ornelas, J.; Sivamani, R.; Awasthi, S. A Report of Two Patients with Darier Disease and Hidradenitis Suppurativa. Pediatr. Dermatol. 2016, 33, e265–e266. [Google Scholar] [CrossRef]
- Higgins, R.; Pink, A.; Hunger, R.; Yawalkar, N.; Navarini, A.A. Generalized Comedones, Acne, and Hidradenitis Suppurativa in a Patient with an FGFR2 Missense Mutation. Front. Med. 2017, 4, 16. [Google Scholar] [CrossRef]
- Maintz, L.; Betz, R.C.; Allam, J.P.; Wenzel, J.; Jaksche, A.; Friedrichs, N.; Bieber, T.; Novak, N. Keratitis-ichthyosis-deafness syndrome in association with follicular occlusion triad. Eur. J. Dermatol. 2005, 15, 347–352. [Google Scholar] [PubMed]
- Pedraz, J.; Penas, P.F.; Garcia-Diez, A. Pachyonychia congenita and hidradenitis suppurativa: No response to infliximab therapy. J. Eur. Acad. Dermatol. Venereol. 2008, 22, 1500–1501. [Google Scholar] [CrossRef] [PubMed]
- Marzuillo, P.; Piccolo, V.; Mascolo, M.; Apicella, A.; Argenziano, G.; Della Vecchia, N.; Guarino, S.; Miraglia Del Giudice, E.; La Manna, A. Patients affected by dent disease 2 could be predisposed to hidradenitis suppurativa. J. Eur. Acad. Dermatol. Venereol. 2018, 32, e309–e311. [Google Scholar] [CrossRef] [PubMed]
- Basmanav, F.B.; Oprisoreanu, A.M.; Pasternack, S.M.; Thiele, H.; Fritz, G.; Wenzel, J.; Grosser, L.; Wehner, M.; Wolf, S.; Fagerberg, C.; et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am. J. Hum. Genet. 2014, 94, 135–143. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Desai, N.; Emtestam, L.; Hunger, R.E.; Ioannides, D.; Juhasz, I.; Lapins, J.; Matusiak, L.; Prens, E.P.; Revuz, J.; et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 619–644. [Google Scholar] [CrossRef]
- Onderdijk, A.J.; van der Zee, H.H.; Esmann, S.; Lophaven, S.; Dufour, D.N.; Jemec, G.B.; Boer, J. Depression in patients with hidradenitis suppurativa. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 473–478. [Google Scholar] [CrossRef]
- van der Zee, H.H.; Jemec, G.B. New insights into the diagnosis of hidradenitis suppurativa: Clinical presentations and phenotypes. J. Am. Acad. Dermatol. 2015, 73, S23–S26. [Google Scholar] [CrossRef]
- Kimball, A.B.; Kerdel, F.; Adams, D.; Mrowietz, U.; Gelfand, J.M.; Gniadecki, R.; Prens, E.P.; Schlessinger, J.; Zouboulis, C.C.; van der Zee, H.H.; et al. Adalimumab for the treatment of moderate to severe Hidradenitis suppurativa: A parallel randomized trial. Ann. Intern. Med. 2012, 157, 846–855. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Tzellos, T.; Kyrgidis, A.; Jemec, G.B.E.; Bechara, F.G.; Giamarellos-Bourboulis, E.J.; Ingram, J.R.; Kanni, T.; Karagiannidis, I.; Martorell, A.; et al. Development and validation of the International Hidradenitis Suppurativa Severity Score System (IHS4), A novel dynamic scoring system to assess HS severity. Br. J. Dermatol. 2017, 177, 1401–1409. [Google Scholar] [CrossRef]
- Dudink, K.; Aarts, P.; Ardon, C.B.; Vossen, A.; Koster, S.B.L.; Van den Bosch, J.F.; Prens, E.P.; van der Zee, H.H. Prevalence and Clinical Characteristics of Hidradenitis Suppurativa Phenotypes in a Large Dutch Cohort. Dermatology 2021, 238, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Frew, J.W.; Hawkes, J.E.; Sullivan-Whalen, M.; Gilleaudeau, P.; Krueger, J.G. Inter-rater reliability of phenotypes and exploratory genotype-phenotype analysis in inherited hidradenitis suppurativa. Br. J. Dermatol. 2019, 181, 566–571. [Google Scholar] [CrossRef]
- Markota Cagalj, A.; Marinovic, B.; Bukvic Mokos, Z. New and Emerging Targeted Therapies for Hidradenitis Suppurativa. Int. J. Mol. Sci. 2022, 23, 3753. [Google Scholar] [CrossRef] [PubMed]
- Marzano, A.V.; Genovese, G.; Casazza, G.; Moltrasio, C.; Dapavo, P.; Micali, G.; Sirna, R.; Gisondi, P.; Patrizi, A.; Dini, V.; et al. Evidence for a ‘window of opportunity’ in hidradenitis suppurativa treated with adalimumab: A retrospective, real-life multicentre cohort study. Br. J. Dermatol. 2021, 184, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Pearson, G. Axillary hidradenitis reconstruction using a dermal regeneration template. Wounds 2022, 34, 43–46. [Google Scholar] [CrossRef]
- Ujiie, H.; Rosmarin, D.; Schon, M.P.; Stander, S.; Boch, K.; Metz, M.; Maurer, M.; Thaci, D.; Schmidt, E.; Cole, C.; et al. Unmet Medical Needs in Chronic, Non-communicable Inflammatory Skin Diseases. Front. Med. 2022, 9, 875492. [Google Scholar] [CrossRef] [PubMed]
- Del Duca, E.; Morelli, P.; Bennardo, L.; Di Raimondo, C.; Nistico, S.P. Cytokine Pathways and Investigational Target Therapies in Hidradenitis Suppurativa. Int. J. Mol. Sci. 2020, 21, 8436. [Google Scholar] [CrossRef] [PubMed]
- Brandao, L.; Moura, R.; Tricarico, P.M.; Gratton, R.; Genovese, G.; Moltrasio, C.; Garcovich, S.; Boniotto, M.; Crovella, S.; Marzano, A.V. Altered keratinization and vitamin D metabolism may be key pathogenetic pathways in syndromic hidradenitis suppurativa: A novel whole exome sequencing approach. J. Dermatol. Sci. 2020, 99, 17–22. [Google Scholar] [CrossRef]
- Jemec, G.B. Clinical practice. Hidradenitis suppurativa. N. Engl. J. Med. 2012, 366, 158–164. [Google Scholar] [CrossRef]
- Kjaersgaard Andersen, R.; Clemmensen, S.B.; Larsen, L.A.; Hjelmborg, J.V.B.; Odum, N.; Jemec, G.B.E.; Christensen, K. Evidence of gene-gene interaction in hidradenitis suppurativa: A nationwide registry study of Danish twins. Br. J. Dermatol. 2022, 186, 78–85. [Google Scholar] [CrossRef]
- van Straalen, K.R.; Prens, E.P.; Willemsen, G.; Boomsma, D.I.; van der Zee, H.H. Contribution of Genetics to the Susceptibility to Hidradenitis Suppurativa in a Large, Cross-sectional Dutch Twin Cohort. JAMA Dermatol. 2020, 156, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, J.S.; Fitzsimmons, E.M.; Gilbert, G. Familial hidradenitis suppurativa: Evidence in favour of single gene transmission. J. Med. Genet. 1984, 21, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, J.S.; Guilbert, P.R.; Fitzsimmons, E.M. Evidence of genetic factors in hidradenitis suppurativa. Br. J. Dermatol. 1985, 113, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Von Der Werth, J.M.; Williams, H.C.; Raeburn, J.A. The clinical genetics of hidradenitis suppurativa revisited. Br. J. Dermatol. 2000, 142, 947–953. [Google Scholar] [CrossRef]
- Gao, M.; Wang, P.G.; Cui, Y.; Yang, S.; Zhang, Y.H.; Lin, D.; Zhang, K.Y.; Liang, Y.H.; Sun, L.D.; Yan, K.L.; et al. Inversa acne (hidradenitis suppurativa): A case report and identification of the locus at chromosome 1p21.1–1q25.3. J. Investig. Dermatol. 2006, 126, 1302–1306. [Google Scholar] [CrossRef]
- Jurisch-Yaksi, N.; Sannerud, R.; Annaert, W. A fast growing spectrum of biological functions of gamma-secretase in development and disease. Biochim. Biophys. Acta 2013, 1828, 2815–2827. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhou, H.; Walian, P.J.; Jap, B.K. The discovery and role of CD147 as a subunit of gamma-secretase complex. Drug News Perspect. 2006, 19, 133–138. [Google Scholar] [CrossRef]
- Shirotani, K.; Edbauer, D.; Prokop, S.; Haass, C.; Steiner, H. Identification of distinct gamma-secretase complexes with different APH-1 variants. J. Biol. Chem. 2004, 279, 41340–41345. [Google Scholar] [CrossRef]
- Hur, J.-Y.; Gertsik, N.; Johnson, D.; Li, Y.-M. Chapter 4—γ-Secretase Inhibitors: From Chemical Probes to Drug Development. In Developing Therapeutics for Alzheimer’s Disease; Elsevier: Amsterdam, The Netherlands, 2016; pp. 63–76. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Luo, W.J.; Wang, H.; Lin, P.; Vetrivel, K.S.; Liao, F.; Li, F.; Wong, P.C.; Farquhar, M.G.; Thinakaran, G.; et al. Nicastrin is critical for stability and trafficking but not association of other presenilin/gamma-secretase components. J. Biol. Chem. 2005, 280, 17020–17026. [Google Scholar] [CrossRef]
- Shah, S.; Lee, S.F.; Tabuchi, K.; Hao, Y.H.; Yu, C.; LaPlant, Q.; Ball, H.; Dann, C.E., 3rd; Sudhof, T.; Yu, G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell 2005, 122, 435–447. [Google Scholar] [CrossRef]
- Francis, R.; McGrath, G.; Zhang, J.; Ruddy, D.A.; Sym, M.; Apfeld, J.; Nicoll, M.; Maxwell, M.; Hai, B.; Ellis, M.C.; et al. aph-1 and pen-2 are Required for Notch Pathway Signaling, γ-Secretase Cleavage of βAPP, and Presenilin Protein Accumulation. Dev. Cell 2002, 3, 85–97. [Google Scholar] [CrossRef]
- Hasegawa, H.; Sanjo, N.; Chen, F.; Gu, Y.J.; Shier, C.; Petit, A.; Kawarai, T.; Katayama, T.; Schmidt, S.D.; Mathews, P.M.; et al. Both the sequence and length of the C terminus of PEN-2 are critical for intermolecular interactions and function of presenilin complexes. J. Biol. Chem. 2004, 279, 46455–46463. [Google Scholar] [CrossRef] [PubMed]
- Laudon, H.; Hansson, E.M.; Melen, K.; Bergman, A.; Farmery, M.R.; Winblad, B.; Lendahl, U.; von Heijne, G.; Naslund, J. A nine-transmembrane domain topology for presenilin 1. J. Biol. Chem. 2005, 280, 35352–35360. [Google Scholar] [CrossRef] [PubMed]
- Thinakaran, G.; Borchelt, D.R.; Lee, M.K.; Slunt, H.H.; Spitzer, L.; Kim, G.; Ratovitsky, T.; Davenport, F.; Nordstedt, C.; Seeger, M.; et al. Endoproteolysis of Presenilin 1 and Accumulation of Processed Derivatives In Vivo. Neuron 1996, 17, 181–190. [Google Scholar] [CrossRef]
- Ratovitski, T.; Slunt, H.H.; Thinakaran, G.; Price, D.L.; Sisodia, S.S.; Borchelt, D.R. Endoproteolytic processing and stabilization of wild-type and mutant presenilin. J. Biol. Chem. 1997, 272, 24536–24541. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.F.; Shah, S.; Yu, C.; Wigley, W.C.; Li, H.; Lim, M.; Pedersen, K.; Han, W.; Thomas, P.; Lundkvist, J.; et al. A conserved GXXXG motif in APH-1 is critical for assembly and activity of the gamma-secretase complex. J. Biol. Chem. 2004, 279, 4144–4152. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Nishimura, M.; Arawaka, S.; Levitan, D.; Zhang, L.; Tandon, A.; Song, Y.Q.; Rogaeva, E.; Chen, F.; Kawarai, T.; et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 2000, 407, 48–54. [Google Scholar] [CrossRef]
- Okuyama, R.; Tagami, H.; Aiba, S. Notch signaling: Its role in epidermal homeostasis and in the pathogenesis of skin diseases. J. Dermatol. Sci. 2008, 49, 187–194. [Google Scholar] [CrossRef]
- Moltrasio, C.; Romagnuolo, M.; Marzano, A.V. Epigenetic Mechanisms of Epidermal Differentiation. Int. J. Mol. Sci. 2022, 23, 4874. [Google Scholar] [CrossRef]
- Nguyen, B.C.; Lefort, K.; Mandinova, A.; Antonini, D.; Devgan, V.; Della Gatta, G.; Koster, M.I.; Zhang, Z.; Wang, J.; Tommasi di Vignano, A.; et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006, 20, 1028–1042. [Google Scholar] [CrossRef]
- Pan, Y.; Lin, M.H.; Tian, X.; Cheng, H.T.; Gridley, T.; Shen, J.; Kopan, R. gamma-secretase functions through Notch signaling to maintain skin appendages but is not required for their patterning or initial morphogenesis. Dev. Cell. 2004, 7, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; Plewig, G. Impaired Notch-MKP-1 signalling in hidradenitis suppurativa: An approach to pathogenesis by evidence from translational biology. Exp. Dermatol. 2013, 22, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Hessam, S.; Gambichler, T.; Skrygan, M.; Scholl, L.; Sand, M.; Meyer, T.; Stockfleth, E.; Bechara, F.G. Increased expression profile of NCSTN, Notch and PI3K/AKT3 in hidradenitis suppurativa. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 203–210. [Google Scholar] [CrossRef]
- Garcovich, S.; Tricarico, P.M.; Nait-Meddour, C.; Giovanardi, G.; Peris, K.; Crovella, S.; Boniotto, M. Novel nicastrin mutation in hidradenitis suppurativa-Dowling-Degos disease clinical phenotype: More than just clinical overlap? Br. J. Dermatol. 2020, 183, 758–759. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; Plewig, G. Impaired Notch signalling: The unifying mechanism explaining the pathogenesis of hidradenitis suppurativa (acne inversa). Br. J. Dermatol. 2013, 168, 876–878. [Google Scholar] [CrossRef]
- Shai Padeh, Y.B.; Ozen, S. Familial Mediterranean Fever. In Textbook of Autoinflammation; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar] [CrossRef]
- Vural, S.; Gundogdu, M.; Kundakci, N.; Ruzicka, T. Familial Mediterranean fever patients with hidradenitis suppurativa. Int. J. Dermatol. 2017, 56, 660–663. [Google Scholar] [CrossRef]
- Goncharov, T.; Hedayati, S.; Mulvihill, M.M.; Izrael-Tomasevic, A.; Zobel, K.; Jeet, S.; Fedorova, A.V.; Eidenschenk, C.; deVoss, J.; Yu, K.; et al. Disruption of XIAP-RIP2 Association Blocks NOD2-Mediated Inflammatory Signaling. Mol. Cell. 2018, 69, 551–565. [Google Scholar] [CrossRef]
- Uniken Venema, W.T.; Voskuil, M.D.; Dijkstra, G.; Weersma, R.K.; Festen, E.A. The genetic background of inflammatory bowel disease: From correlation to causality. J. Pathol. 2017, 241, 146–158. [Google Scholar] [CrossRef]
- Yao, Q.; Li, E.; Shen, B. Autoinflammatory disease with focus on NOD2-associated disease in the era of genomic medicine. Autoimmunity 2019, 52, 48–56. [Google Scholar] [CrossRef]
- Savva, A.; Kanni, T.; Damoraki, G.; Kotsaki, A.; Giatrakou, S.; Grech, I.; Katoulis, A.; Papadavid, E.; Giamarellos-Bourboulis, E.J. Impact of Toll-like receptor-4 and tumour necrosis factor gene polymorphisms in patients with hidradenitis suppurativa. Br. J. Dermatol. 2013, 168, 311–317. [Google Scholar] [CrossRef]
- Giatrakos, S.; Huse, K.; Kanni, T.; Tzanetakou, V.; Kramer, M.; Grech, I.; Papadavid, E.; Katoulis, A.; Stavrianeas, N.; Nothnagel, M.; et al. Haplotypes of IL-12Rbeta1 impact on the clinical phenotype of hidradenitis suppurativa. Cytokine 2013, 62, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Giamarellos-Bourboulis, E.J.; Platzer, M.; Karagiannidis, I.; Kanni, T.; Nikolakis, G.; Ulrich, J.; Bellutti, M.; Gollnick, H.; Bauer, M.; Zouboulis, C.C.; et al. High Copy Numbers of beta-Defensin Cluster on 8p23.1, Confer Genetic Susceptibility, and Modulate the Physical Course of Hidradenitis Suppurativa/Acne Inversa. J. Investig. Dermatol. 2016, 136, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Abstracts of the 7th European Hidradenitis Suppurativa Foundation (EHSF) Congress, 7–9 February 2018, Rotterdam, The Netherlands. Exp. Dermatol. 2018, 27, 5–32. [CrossRef]
- Braun-Falco, M.; Kovnerystyy, O.; Lohse, P.; Ruzicka, T. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)--a new autoinflammatory syndrome distinct from PAPA syndrome. J. Am. Acad. Dermatol. 2012, 66, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Andre, M.F.; Aumaitre, O.; Grateau, G.; Chamaillard, M.; Costedoat-Chalumeau, N.; Cardoso, M.C.; Henry-Berger, J.; Ramakrishna, B.S.; Delpech, M.; Piette, J.C.; et al. Longest form of CCTG microsatellite repeat in the promoter of the CD2BP1/PSTPIP1 gene is associated with aseptic abscesses and with Crohn disease in French patients. Dig. Dis. Sci. 2010, 55, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Detection of immune danger signals by NALP3. J. Leukoc. Biol. 2008, 83, 507–511. [Google Scholar] [CrossRef]
- Booshehri, L.M.; Hoffman, H.M. CAPS and NLRP3. J. Clin. Immunol. 2019, 39, 277–286. [Google Scholar] [CrossRef]
- Vural, S.; Gundogdu, M.; Gokpinar Ili, E.; Durmaz, C.D.; Vural, A.; Steinmuller-Magin, L.; Kleinhempel, A.; Holdt, L.M.; Ruzicka, T.; Giehl, K.A.; et al. Association of pyrin mutations and autoinflammation with complex phenotype hidradenitis suppurativa: A case-control study. Br. J. Dermatol. 2019, 180, 1459–1467. [Google Scholar] [CrossRef]
- Kimball, A.B.; Okun, M.M.; Williams, D.A.; Gottlieb, A.B.; Papp, K.A.; Zouboulis, C.C.; Armstrong, A.W.; Kerdel, F.; Gold, M.H.; Forman, S.B.; et al. Two Phase 3 Trials of Adalimumab for Hidradenitis Suppurativa. N. Engl. J. Med. 2016, 375, 422–434. [Google Scholar] [CrossRef]
- Garcia-Vega, L.; O’Shaughnessy, E.M.; Jan, A.; Bartholomew, C.; Martin, P.E. Connexin 26 and 43 play a role in regulating proinflammatory events in the epidermis. J. Cell Physiol. 2019, 234, 15594–15606. [Google Scholar] [CrossRef] [PubMed]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Moran, B.; Sweeney, C.M.; Hughes, R.; Malara, A.; Kirthi, S.; Tobin, A.M.; Kirby, B.; Fletcher, J.M. Hidradenitis Suppurativa is Characterized by Dysregulation of the Th17:Treg Cell Axis, which is Corrected by Anti-TNF Therapy. J. Investig. Dermatol. 2017, 137, 2389–2395. [Google Scholar] [CrossRef] [PubMed]
- Duchatelet, S.; Miskinyte, S.; Join-Lambert, O.; Ungeheuer, M.N.; Frances, C.; Nassif, A.; Hovnanian, A. First nicastrin mutation in PASH (pyoderma gangrenosum, acne and suppurative hidradenitis) syndrome. Br. J. Dermatol. 2015, 173, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, H.; Wang, B. Is SAPHO Syndrome Linked to PASH Syndrome and Hidradenitis Suppurativa by Nicastrin Mutation? A Case Report. J. Rheumatol. 2018, 45, 1605–1607. [Google Scholar] [CrossRef]
- Betz, R.C.; Planko, L.; Eigelshoven, S.; Hanneken, S.; Pasternack, S.M.; Bussow, H.; Van Den Bogaert, K.; Wenzel, J.; Braun-Falco, M.; Rutten, A.; et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am. J. Hum. Genet. 2006, 78, 510–519. [Google Scholar] [CrossRef]
- Pavithra, S.; Pai, H.; Mallya, H.; Pai, G.S. Nevus comedonicus syndrome. Indian J. Dermatol. 2011, 56, 771–772. [Google Scholar] [CrossRef]
- Melnik, B.C. Role of FGFR2-signaling in the pathogenesis of acne. Dermato Endocrinol. 2009, 1, 141–156. [Google Scholar] [CrossRef]
- Bezdicka, M.; Langer, J.; Hacek, J.; Zieg, J. Dent Disease Type 2 as a Cause of Focal Segmental Glomerulosclerosis in a 6-Year-Old Boy: A Case Report. Front. Pediatr. 2020, 8, 583230. [Google Scholar] [CrossRef]


{kind=link}
| Gene | Variant Segregation | Main Study Groups |
|---|---|---|
| NCSTN | Sporadic | Vural et al. 2021 [6] |
| Syndromic HS | Marzano et al. 2022 [7] | |
| Familial | Wang et al. 2010 [8]; Wu et al. 2020 [9]; Xiao et al. 2016 [10]; Miskinyte et al.2012 [11]; Pink et al. 2011 [12] | |
| PSENEN | Familial | Pink et al. 2012 [13] |
| Familial syndromic HS | Pink et al. 2012 [13] | |
| HS + DDD | Ralser et al. 2017 [14] | |
| PSTPIP1 | Familial syndromic HS | Saito et al. 2018 [15] |
| Sporadic syndromic HS | Marzano et al. 2013 [16]; Calderon-Castrat et al. 2016 [17] | |
| MEFV | Familial | Jfri et al. 2020 [18] |
| Sporadic syndromic HS + FMF | Marzano et al. 2014 [19] | |
| NLRP3 | Sporadic syndromic HS | Marzano et al. 2014 [19] |
| NOD2 | Familial | Jfri et al. 2020 [18] |
| Sporadic syndromic HS | Marzano et al. 2014 [19] | |
| Sporadic syndromic HS | Marzano et al. 2022 [7] | |
| MPO | Sporadic syndromic HS | Marzano et al. 2022 [7] |
| NLRC4 | Sporadic syndromic HS | Marzano et al. 2022 [7] |
| OTULIN | Sporadic syndromic HS | Marzano et al. 2022 [7] |
| ATP2A2 | Darier’s disease + HS | Ornelas et al. 2016 [20] |
| FGFR2 | Nevus Comedonicus + HS | Higgins et al. 2017 [21] |
| GJB2 | Sporadic syndromic HS | Marzano et al. 2022 [7] |
| Sporadic KID + HS | Maintz et al. 2005 [22] | |
| IL1RN | Sporadic syndromic HS + FMF | Marzano et al. 2014 [19] |
| KRT6 | Pachyonychia Congenita + HS | Pedraz et al. 2008 [23] |
| OCRL1 | Dent disease + HS | Marzuillo et al. 2018 [24] |
| POFUT1 | HS + DDD | Basmanav et al. 2014 [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moltrasio, C.; Tricarico, P.M.; Romagnuolo, M.; Marzano, A.V.; Crovella, S. Hidradenitis Suppurativa: A Perspective on Genetic Factors Involved in the Disease. Biomedicines 2022, 10, 2039. https://doi.org/10.3390/biomedicines10082039
Moltrasio C, Tricarico PM, Romagnuolo M, Marzano AV, Crovella S. Hidradenitis Suppurativa: A Perspective on Genetic Factors Involved in the Disease. Biomedicines. 2022; 10(8):2039. https://doi.org/10.3390/biomedicines10082039
Chicago/Turabian StyleMoltrasio, Chiara, Paola Maura Tricarico, Maurizio Romagnuolo, Angelo Valerio Marzano, and Sergio Crovella. 2022. "Hidradenitis Suppurativa: A Perspective on Genetic Factors Involved in the Disease" Biomedicines 10, no. 8: 2039. https://doi.org/10.3390/biomedicines10082039
APA StyleMoltrasio, C., Tricarico, P. M., Romagnuolo, M., Marzano, A. V., & Crovella, S. (2022). Hidradenitis Suppurativa: A Perspective on Genetic Factors Involved in the Disease. Biomedicines, 10(8), 2039. https://doi.org/10.3390/biomedicines10082039