
Immune Response to Biofilm Growing Pulmonary Pseudomonas aeruginosa Infection


{kind=link}
{kind=link}
Abstract
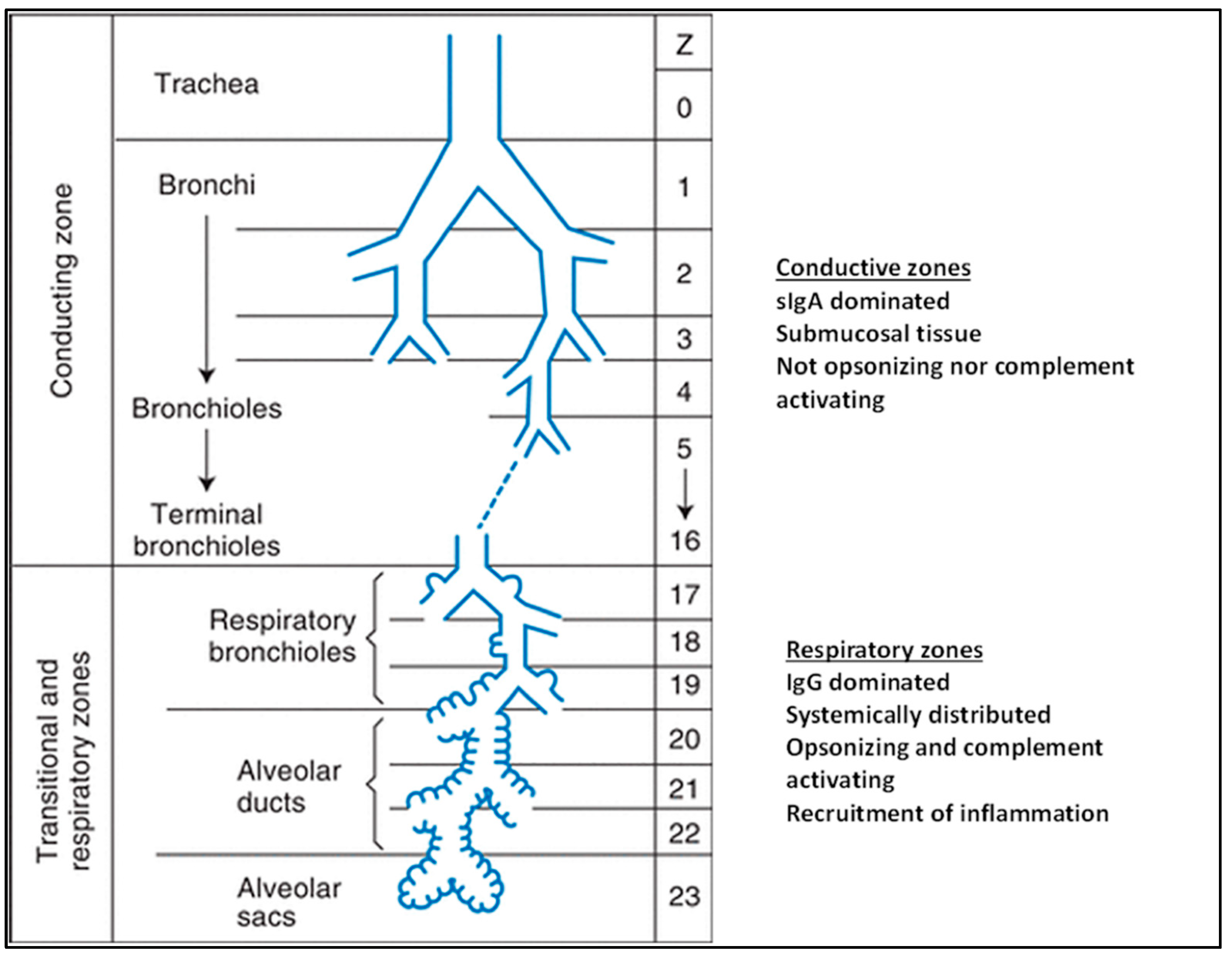
:1. Airways
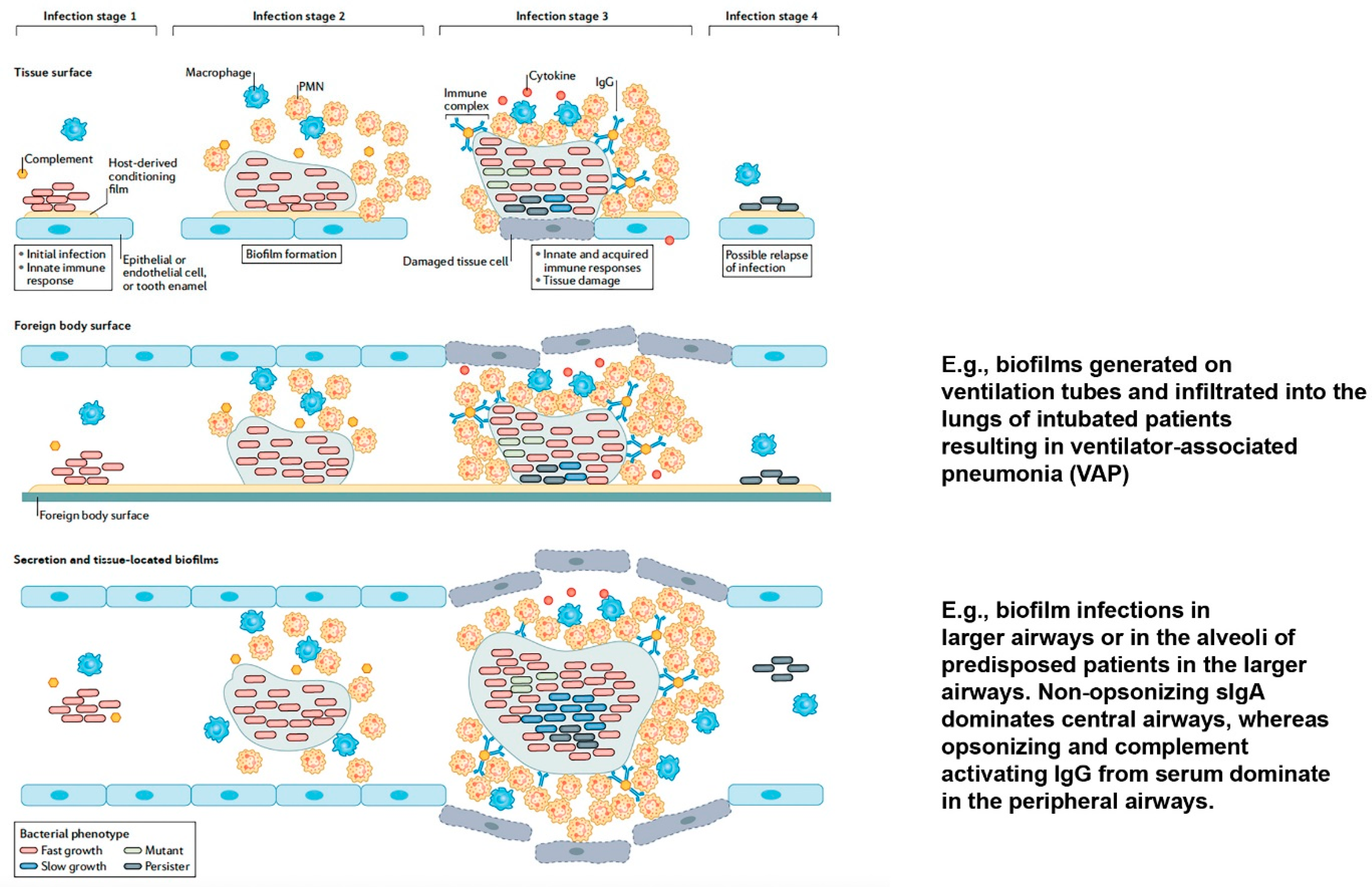
2. Biofilms
3. Biofilm and Initiating the Immune System
4. Complement System
5. T-Helper Cell Response
6. CFTR Modulators and Immune Response in CF
7. Prophylaxis of Pulmonary Pseudomonas aeruginosa Biofilm Infections
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Neupane, A.S.; Willson, M.; Chojnacki, A.K.; Vargas E Silva Castanheira, F.; Morehouse, C.; Carestia, A.; Keller, A.E.; Peiseler, M.; DiGiandomenico, A.; Kelly, M.M.; et al. Patrolling Alveolar Macrophages Conceal Bacteria from the Immune System to Maintain Homeostasis. Cell 2020, 183, 110–125.e11. [Google Scholar] [CrossRef] [PubMed]
- West, J.B. Pulmonary Physiology and Pathophysiology: An Integrated, Case-Based Approach; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Prussin, A.J., II; Garcia, E.B.; Marr, L.C. Total Virus and Bacteria Concentrations in Indoor and Outdoor Air. Environ. Sci. Technol. Lett. 2015, 2, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Boyaka, P.N.; Fujihashi, K. 20—Host Defenses at Mucosal Surfaces. In Clinical Immunology, 5th ed.; Rich, R.R., Fleisher, T.A., Shearer, W.T., Schroeder, H.W., Frew, A.J., Weyand, C.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 285–298.e1. ISBN 9780702068966. [Google Scholar]
- Kimura, S.; Mutoh, M.; Hisamoto, M.; Saito, H.; Takahashi, S.; Asakura, T.; Ishii, M.; Nakamura, Y.; Iida, J.; Hase, K.; et al. Airway M Cells Arise in the Lower Airway Due to RANKL Signaling and Reside in the Bronchiolar Epithelium Associated with iBALT in Murine Models of Respiratory Disease. Front. Immunol. 2019, 10, 1323. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Randall, T.D.; Silva-Sanchez, A. Inducible Bronchus-Associated Lymphoid Tissue: Taming Inflammation in the Lung. Front. Immunol. 2016, 7, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciofu, O.; Moser, C.; Jensen, P.Ø.; Høiby, N. Tolerance and resistance of microbial biofilms. Nat. Rev. Microbiol. 2022. [Google Scholar] [CrossRef]
- Qu, J.; Cai, Z.; Duan, X.; Zhang, H.; Cheng, H.; Han, S.; Yu, K.; Jiang, Z.; Zhang, Y.; Liu, Y.; et al. Pseudomonas aeruginosa modulates alginate biosynthesis and type VI secretion system in two critically ill COVID-19 patients. Cell Biosci. 2022, 12, 14. [Google Scholar] [CrossRef]
- Qu, J.; Cai, Z.; Liu, Y.; Duan, X.; Han, S.; Liu, J.; Zhu, Y.; Jiang, Z.; Zhang, Y.; Zhuo, C.; et al. Persistent Bacterial Coinfection of a COVID-19 Patient Caused by a Genetically Adapted Pseudomonas aeruginosa Chronic Colonizer. Front. Cell Infect. Microbiol. 2021, 11, 641920. [Google Scholar] [CrossRef]
- Faure, E.; Kwong, K.; Nguyen, D. Pseudomonas aeruginosa in Chronic Lung Infections: How to Adapt Within the Host? Front. Immunol. 2018, 9, 2416. [Google Scholar] [CrossRef] [Green Version]
- Moss, J.; Ehrmantraut, M.E.; Banwart, B.D.; Frank, D.W.; Barbieri, J.T. Sera from adult patients with cystic fibrosis contain antibodies to Pseudomonas aeruginosa type III apparatus. Infect. Immun. 2001, 69, 1185–1188. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Bar-Meir, M.; McColley, S.; Cullina, J.; Potter, E.; Powers, C.; Prickett, M.; Seshadri, R.; Jovanovic, B.; Petrocheilou, A.; et al. Evolution of Pseudomonas aeruginosa type III secretion in cystic fibrosis: A paradigm of chronic infection. Transl. Res. 2008, 152, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Kimbrell, D.A.; Beutler, B. The evolution and genetics of innate immunity. Nat. Rev. Genet. 2001, 2, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.T.; Kharazmi, A.; Garred, P.; Kronborg, G.; Fomsgaard, A.; Mollnes, T.E.; Høiby, N. Complement activation by Pseudomonas aeruginosa biofilms. Microb. Pathog. 1993, 15, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Mutlu, G.M.; Budinger, G.R.; Perlman, H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 2013, 49, 503–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, R.F.; Myers, A.J.; Jarvela, J.; Flynn, J.; Rutledge, T.; Bonfield, T.; Lin, P.L. Diversity of Human and Macaque Airway Immune Cells at Baseline and during Tuberculosis Infection. Am. J. Respir. Cell Mol. Biol. 2016, 55, 899–908. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.R.; Hotten, D.F.; Malakhau, Y.; Volker, E.; Ghio, A.J.; Noble, P.W.; Kraft, M.; Hollingsworth, J.W.; Gunn, M.D.; Tighe, R.M. Flow Cytometric Analysis of Myeloid Cells in Human Blood, Bronchoalveolar Lavage, and Lung Tissues. Am. J. Respir. Cell Mol. Biol. 2016, 54, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Høiby, N.; Bjarnsholt, T.; Moser, C.; Bassi, G.L.; Coenye, T.; Donelli, G.; Hall-Stoodley, L.; Holá, V.; Imbert, C.; Kirketerp-Møller, K.; et al. ESCMID guideline for the diagnosis and treatment of biofilm infections 2014. Clin. Microbiol. Infect. 2015, 21 (Suppl. 1), S1–S25. [Google Scholar] [CrossRef] [Green Version]
- Alhede, M.; Lorenz, M.; Fritz, B.G.; Jensen, P.Ø.; Ring, H.C.; Bay, L.; Bjarnsholt, T. Bacterial aggregate size determines phagocytosis efficiency of polymorphonuclear leukocytes. Med. Microbiol. Immunol. 2020, 209, 669–680. [Google Scholar] [CrossRef]
- Secor, P.R.; Burgener, E.B.; Kinnersley, M.; Jennings, L.K.; Roman-Cruz, V.; Popescu, M.; Van Belleghem, J.D.; Haddock, N.; Copeland, C.; Michaels, L.A.; et al. Pf Bacteriophage and Their Impact on Pseudomonas Virulence, Mammalian Immunity, and Chronic Infections. Front. Immunol. 2020, 11, 244. [Google Scholar] [CrossRef] [Green Version]
- Rybtke, M.; Jensen, P.Ø.; Nielsen, C.H.; Tolker-Nielsen, T. The Extracellular Polysaccharide Matrix of Pseudomonas aeruginosa Biofilms Is a Determinant of Polymorphonuclear Leukocyte Responses. Infect. Immun. 2020, 89, e00631-20. [Google Scholar] [CrossRef]
- Moser, C.; Jensen, P.Ø.; Thomsen, K.; Kolpen, M.; Rybtke, M.; Lauland, A.S.; Trøstrup, H.; Tolker-Nielsen, T. Immune Responses to Pseudomonas aeruginosa Biofilm Infections. Front. Immunol. 2021, 12, 625597. [Google Scholar] [CrossRef]
- Singh, S.; Almuhanna, Y.; Alshahrani, M.Y.; Lowman, D.W.; Rice, P.J.; Gell, C.; Ma, Z.; Graves, B.; Jackson, D.; Lee, K.; et al. Carbohydrates from Pseudomonas aeruginosa biofilms interact with immune C-type lectins and interfere with their receptor function. NPJ Biofilms Microbiomes 2021, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Schiøtz, P.O.; Sørensen, H.; Høiby, M. Activated complement in the sputum from patients with cystic fibrosis. Acta Pathol. Microbiol. Scand. Sect. C Immunol. 1979, 87C, 1–5. [Google Scholar]
- Pier, G.B.; Coleman, F.; Grout, M.; Franklin, M.; Ohman, D.E. Role of alginate O acetylation in resistance of mucoid Pseudomonas aeruginosa to opsonic phagocytosis. Infect. Immun. 2001, 69, 1895–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, N.; Lee, B.; Hentzer, M.; Rasmussen, T.B.; Song, Z.; Johansen, H.K.; Givskov, M.; Høiby, N. Azithromycin blocks quorum sensing and alginate polymer formation and increases the sensitivity to serum and stationary-growth-phase killing of Pseudomonas aeruginosa and attenuates chronic P. aeruginosa lung infection in Cftr−/− mice. Antimicrob. Agents Chemother. 2007, 51, 3677–3687. [Google Scholar] [CrossRef] [Green Version]
- Schulz-Kuhnt, A.; Greif, V.; Hildner, K.; Knipfer, L.; Döbrönti, M.; Zirlik, S.; Fuchs, F.; Atreya, R.; Zundler, S.; López-Posadas, R.; et al. ILC2 Lung-Homing in Cystic Fibrosis Patients: Functional Involvement of CCR6 and Impact on Respiratory Failure. Front. Immunol. 2020, 11, 691. [Google Scholar] [CrossRef]
- Hagner, M.; Albrecht, M.; Guerra, M.; Braubach, P.; Halle, O.; Zhou-Suckow, Z.; Butz, S.; Jonigk, D.; Hansen, G.; Schultz, C.; et al. IL-17A from innate and adaptive lymphocytes contributes to inflammation and damage in cystic fibrosis lung disease. Eur. Respir. J. 2021, 57, 1900716. [Google Scholar] [CrossRef]
- Brady, R.A.; Leid, J.G.; Calhoun, J.H.; Costerton, J.W.; Shirtliff, M.E. Osteomyelitis and the role of biofilms in chronic infection. FEMS Immunol. Med. Microbiol. 2008, 52, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Schaudinn, C.; Gorur, A.; Keller, D.; Sedghizadeh, P.P.; Costerton, J.W. Periodontitis: An archetypical biofilm disease. J. Am. Dent. Assoc. 2009, 140, 978–986. [Google Scholar] [CrossRef]
- Moser, C.; Pedersen, H.T.; Lerche, C.J.; Kolpen, M.; Line, L.; Thomsen, K.; Høiby, N.; Jensen, P.Ø. Biofilms and host response—Helpful or harmful. APMIS 2017, 125, 320–338. [Google Scholar] [CrossRef] [Green Version]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Lai, L.; Alaverdi, N.; Maltais, L.; Morse, H.C., III. Mouse cell surface antigens: Nomenclature and immunophenotyping. J. Immunol. 1998, 160, 3861–3868. [Google Scholar] [PubMed]
- Kaliński, P.; Hilkens, C.M.; Wierenga, E.A.; Kapsenberg, M.L. T-cell priming by type-1 and type-2 polarized dendritic cells: The concept of a third signal. Immunol. Today 1999, 20, 561–567. [Google Scholar] [CrossRef]
- Moser, C.; Kjaergaard, S.; Pressler, T.; Kharazmi, A.; Koch, C.; Høiby, N. The immune response to chronic Pseudomonas aeruginosa lung infection in cystic fibrosis patients is predominantly of the Th2 type. APMIS 2000, 108, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.B.; Hsu, Y.P.; Olds, L. Cytokine dysregulation in activated cystic fibrosis (CF) peripheral lymphocytes. Clin. Exp. Immunol. 2000, 120, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Damlund, D.S.; Christophersen, L.; Jensen, P.Ø.; Alhede, M.; Høiby, N.; Moser, C. Activation of pulmonary and lymph node dendritic cells during chronic Pseudomonas aeruginosa lung infection in mice. APMIS 2016, 124, 500–507. [Google Scholar] [CrossRef]
- Skindersoe, M.E.; Zeuthen, L.H.; Brix, S.; Fink, L.N.; Lazenby, J.; Whittall, C.; Williams, P.; Diggle, S.P.; Froekiaer, H.; Cooley, M.; et al. Pseudomonas aeruginosa quorum-sensing signal molecules interfere with dendritic cell-induced T-cell proliferation. FEMS Immunol. Med. Microbiol. 2009, 55, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.Ø.; Givskov, M.; Bjarnsholt, T.; Moser, C. The immune system vs. Pseudomonas aeruginosa biofilms. FEMS Immunol. Med. Microbiol. 2010, 59, 292–305. [Google Scholar] [CrossRef] [Green Version]
- Hoiby, N.; Flensborg, E.W.; Beck, B.; Friis, B.; Jacobsen, S.V.; Jacobsen, L. Pseudomonas aeruginosa infection in cystic fibrosis. Diagnostic and prognostic significance of Pseudomonas aeruginosa precipitins determined by means of crossed immunoelectrophoresis. Scand. J. Respir. Dis. 1977, 58, 65–79. [Google Scholar]
- Johansen, H.K.; Nørregaard, L.; Gøtzsche, P.C.; Pressler, T.; Koch, C.; Høiby, N. Antibody response to Pseudomonas aeruginosa in cystic fibrosis patients: A marker of therapeutic success?—A 30-year cohort study of survival in Danish CF patients after onset of chronic P. aeruginosa lung infection. Pediatr. Pulmonol. 2004, 37, 427–432. [Google Scholar] [CrossRef]
- Hartl, D.; Griese, M.; Kappler, M.; Zissel, G.; Reinhardt, D.; Rebhan, C.; Schendel, D.J.; Krauss-Etschmann, S. Pulmonary T(H)2 response in Pseudomonas aeruginosa–infected patients with cystic fibrosis. J. Allergy Clin. Immunol. 2006, 117, 204–211. [Google Scholar] [CrossRef]
- Moss, R.B.; Mayer-Hamblett, N.; Wagener, J.; Daines, C.; Hale, K.; Ahrens, R.; Gibson, R.L.; Anderson, P.; Retsch-Bogart, G.; Nasr, S.Z.; et al. Randomized, double-blind, placebo-controlled, dose-escalating study of aerosolized interferon gamma-1b in patients with mild to moderate cystic fibrosis lung disease. Pediatr. Pulmonol. 2005, 39, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Brazova, J.; Sediva, A.; Pospisilova, D.; Vavrova, V.; Pohunek, P.; Macek, M., Jr.; Bartunkova, J.; Lauschmann, H. Differential cytokine profile in children with cystic fibrosis. Clin. Immunol. 2005, 115, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Johansen, H.K.; Song, Z.; Hougen, H.P.; Rygaard, J.; Høiby, N. Chronic Pseudomonas aeruginosa lung infection is more severe in Th2 responding BALB/c mice compared to Th1 responding C3H/HeN mice. APMIS 1997, 105, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Hougen, H.P.; Song, Z.; Rygaard, J.; Kharazmi, A.; Høiby, N. Early immune response in susceptible and resistant mice strains with chronic Pseudomonas aeruginosa lung infection determines the type of T-helper cell response. APMIS 1999, 107, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Jensen, P.O.; Kobayashi, O.; Hougen, H.P.; Song, Z.; Rygaard, J.; Kharazmi, A.; Hoby, N. Improved outcome of chronic Pseudomonas aeruginosa lung infection is associated with induction of a Th1-dominated cytokine response. Clin. Exp. Immunol. 2002, 127, 206–213. [Google Scholar] [CrossRef]
- Mauch, R.M.; Jensen, P.Ø.; Moser, C.; Levy, C.E.; Høiby, N. Mechanisms of humoral immune response against Pseudomonas aeruginosa biofilm infection in cystic fibrosis. J. Cyst. Fibros. 2018, 17, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Eagar, T.N.; Miller, S.D. 16—Helper T-cell subsets and control of the inflammatory response. In Clinical Immunology, 5th ed.; Rich, R.R., Fleisher, T.A., Shearer, W.T., Schroeder, H.W., Frew, A.J., Weyand, C.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 235–245. ISBN 9780723436911. [Google Scholar]
- Stockinger, B.; Veldhoen, M. Differentiation and function of Th17 T cells. Curr. Opin. Immunol. 2007, 19, 281–286. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Zhou, L.; Littman, D.R. Transcriptional regulation of Th17 cell differentiation. Semin. Immunol. 2007, 19, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Decraene, A.; Willems-Widyastuti, A.; Kasran, A.; De Boeck, K.; Bullens, D.M.; Dupont, L.J. Elevated expression of both mRNA and protein levels of IL-17A in sputum of stable Cystic Fibrosis patients. Respir. Res. 2010, 11, 177. [Google Scholar] [CrossRef] [Green Version]
- Tiringer, K.; Treis, A.; Fucik, P.; Gona, M.; Gruber, S.; Renner, S.; Dehlink, E.; Nachbaur, E.; Horak, F.; Jaksch, P.; et al. A Th17- and Th2-skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 2013, 187, 621–629. [Google Scholar] [CrossRef]
- Bayes, H.K.; Bicknell, S.; MacGregor, G.; Evans, T.J. T helper cell subsets specific for Pseudomonas aeruginosa in healthy individuals and patients with cystic fibrosis. PLoS ONE 2014, 9, e90263. [Google Scholar] [CrossRef] [PubMed]
- Bruscia, E.M.; Bonfield, T.L. Innate and Adaptive Immunity in Cystic Fibrosis. Clin. Chest Med. 2016, 37, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Rieber, N.; Brand, A.; Hector, A.; Graepler-Mainka, U.; Ost, M.; Schäfer, I.; Wecker, I.; Neri, D.; Wirth, A.; Mays, L.; et al. Flagellin induces myeloid-derived suppressor cells: Implications for Pseudomonas aeruginosa infection in cystic fibrosis lung disease. J. Immunol. 2013, 190, 1276–1284. [Google Scholar] [CrossRef] [Green Version]
- Öz, H.H.; Zhou, B.; Voss, P.; Carevic, M.; Schroth, C.; Frey, N.; Rieber, N.; Hector, A.; Hartl, D. Pseudomonas aeruginosa Airway Infection Recruits and Modulates Neutrophilic Myeloid-Derived Suppressor Cells. Front. Cell. Infect. Microbiol. 2016, 6, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aanaes, K.; Johansen, H.K.; Poulsen, S.S.; Pressler, T.; Buchwald, C.; Høiby, N. Secretory IgA as a diagnostic tool for Pseudomonas aeruginosa respiratory colonization. J. Cyst. Fibros. 2013, 12, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, A.M.; Lecocq, M.; Noel, S.; Detry, B.; Carlier, F.M.; Aboubakar Nana, F.; Bouzin, C.; Leal, T.; Vermeersch, M.; De Rose, V.; et al. Lung immunoglobulin A immunity dysregulation in cystic fibrosis. EBioMedicine 2020, 60, 102974. [Google Scholar] [CrossRef]
- Pilette, C.; Godding, V.; Kiss, R.; Delos, M.; Verbeken, E.; Decaestecker, C.; De Paepe, K.; Vaerman, J.P.; Decramer, M.; Sibille, Y. Reduced epithelial expression of secretory component in small airways correlates with airflow obstruction in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 163, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Millares, L.; Martí, S.; Ardanuy, C.; Liñares, J.; Santos, S.; Dorca, J.; García-Nuñez, M.; Quero, S.; Monsó, E. Specific IgA against Pseudomonas aeruginosa in severe COPD. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 2807–2811. [Google Scholar] [CrossRef] [Green Version]
- Ladjemi, M.Z.; Martin, C.; Lecocq, M.; Detry, B.; Nana, F.A.; Moulin, C.; Weynand, B.; Fregimilicka, C.; Bouzin, C.; Thurion, P.; et al. Increased IgA Expression in Lung Lymphoid Follicles in Severe Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2019, 199, 592–602. [Google Scholar] [CrossRef]
- Ladjemi, M.Z.; Lecocq, M.; Weynand, B.; Bowen, H.; Gould, H.J.; Van Snick, J.; Detry, B.; Pilette, C. Increased IgA production by B-cells in COPD via lung epithelial interleukin-6 and TACI pathways. Eur. Respir. J. 2015, 45, 980–993. [Google Scholar] [CrossRef] [Green Version]
- Corthésy, B. Multi-faceted functions of secretory IgA at mucosal surfaces. Front. Immunol. 2013, 4, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauch, R.M.; Rossi, C.L.; Nolasco da Silva, M.T.; Bianchi Aiello, T.; Ribeiro, J.D.; Ribeiro, A.F.; Høiby, N.; Levy, C.E. Secretory IgA-mediated immune response in saliva and early detection of Pseudomonas aeruginosa in the lower airways of pediatric cystic fibrosis patients. Med. Microbiol. Immunol. 2019, 208, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Fomsgaard, A.; Conrad, R.S.; Galanos, C.; Shand, G.H.; Høiby, N. Comparative immunochemistry of lipopolysaccharides from typable and polyagglutinable Pseudomonas aeruginosa strains isolated from patients with cystic fibrosis. J. Clin. Microbiol. 1988, 26, 821–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, M.Y.; McGroarty, E.J.; Kropinski, A.M.; MacDonald, L.A.; Pedersen, S.S.; Høiby, N.; Lam, J.S. Occurrence of a common lipopolysaccharide antigen in standard and clinical strains of Pseudomonas aeruginosa. J. Clin. Microbiol. 1989, 27, 962–967. [Google Scholar] [CrossRef] [Green Version]
- Likavcanova, E.; Lagacé, J. Quantitative analysis of immunoglobulin G subclass responses to Pseudomonas aeruginosa antigens in cystic fibrosis. J. Med. Microbiol. 1992, 36, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Rehm, B.H.; Grabert, E.; Hein, J.; Winkler, U.K. Antibody response of rabbits and cystic fibrosis patients to an alginate-specific outer membrane protein of a mucoid strain of Pseudomonas aeruginosa. Microb. Pathog. 1994, 16, 43–51. [Google Scholar] [CrossRef]
- Hoiby, N. Pseudomonas aeruginosa infection in cystic fibrosis. Diagnostic and prognostic significance of Pseudomonas aeruginosa precipitins determined by means of crossed immunoelectrophoresis. A survey. Acta Pathol. Microbiol. Scand. Suppl. 1977, 1–96. [Google Scholar]
- Pressler, T.; Karpati, F.; Granström, M.; Knudsen, P.K.; Lindblad, A.; Hjelte, L.; Olesen, H.V.; Meyer, P.; Høiby, N.; Scandinavian CF Study Consortium. Diagnostic significance of measurements of specific IgG antibodies to Pseudomonas aeruginosa by three different serological methods. J. Cyst. Fibros. 2009, 8, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Pressler, T.; Frederiksen, B.; Skov, M.; Garred, P.; Koch, C.; Høiby, N. Early rise of anti-pseudomonas antibodies and a mucoid phenotype of pseudomonas aeruginosa are risk factors for development of chronic lung infection—A case control study. J. Cyst. Fibros. 2006, 5, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Ciofu, O.; Bagge, N.; Høiby, N. Antibodies against beta-lactamase can improve ceftazidime treatment of lung infection with beta-lactam-resistant Pseudomonas aeruginosa in a rat model of chronic lung infection. APMIS 2002, 110, 881–891. [Google Scholar] [CrossRef]
- Schwensen, H.F.; Moser, C.; Perch, M.; Pressler, T.; Høiby, N. Pseudomonas aeruginosa antibody response in cystic fibrosis decreases rapidly following lung transplantation. J. Cyst. Fibros. 2020, 19, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Skov, M.; Hansen, C.R.; Pressler, T. Cystic fibrosis—An example of personalized and precision medicine. APMIS 2019, 127, 352–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, D.S.; Grimwood, K.; Carzino, R.; Carlin, J.B.; Olinsky, A.; Phelan, P.D. Lower respiratory infection and inflammation in infants with newly diagnosed cystic fibrosis. BMJ 1995, 310, 1571–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, D.S.; Grimwood, K.; Carlin, J.B.; Carzino, R.; Gutièrrez, J.P.; Hull, J.; Olinsky, A.; Phelan, E.M.; Robertson, C.F.; Phelan, P.D. Lower airway inflammation in infants and young children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 1997, 156 Pt 1, 1197–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, D.S.; Hook, S.M.; Jamsen, K.M.; Nixon, G.M.; Carzino, R.; Carlin, J.B.; Robertson, C.F.; Grimwood, K. Lower airway inflammation in infants with cystic fibrosis detected by newborn screening. Pediatr. Pulmonol. 2005, 40, 500–510. [Google Scholar] [CrossRef]
- Hubeau, C.; Puchelle, E.; Gaillard, D. Distinct pattern of immune cell population in the lung of human fetuses with cystic fibrosis. J. Allergy Clin. Immunol. 2001, 108, 524–529. [Google Scholar] [CrossRef]
- Stoltz, D.A.; Meyerholz, D.K.; Pezzulo, A.A.; Ramachandran, S.; Rogan, M.P.; Davis, G.J.; Hanfland, R.A.; Wohlford-Lenane, C.; Dohrn, C.L.; Bartlett, J.A.; et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci. Transl. Med. 2010, 2, 29ra31. [Google Scholar] [CrossRef] [Green Version]
- Keown, K.; Brown, R.; Doherty, D.F.; Houston, C.; McKelvey, M.C.; Creane, S.; Linden, D.; McAuley, D.F.; Kidney, J.C.; Weldon, S.; et al. Airway Inflammation and Host Responses in the Era of CFTR Modulators. Int. J. Mol. Sci. 2020, 21, 6379. [Google Scholar] [CrossRef]
- Painter, R.G.; Valentine, V.G.; Lanson, N.A., Jr.; Leidal, K.; Zhang, Q.; Lombard, G.; Thompson, C.; Viswanathan, A.; Nauseef, W.M.; Wang, G.; et al. CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry 2006, 45, 10260–10269. [Google Scholar] [CrossRef] [Green Version]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef]
- Alanin, M.C.; Aanaes, K.; Høiby, N.; Pressler, T.; Skov, M.; Nielsen, K.G.; Taylor-Robinson, D.; Waldmann, E.; Krogh Johansen, H.; von Buchwald, C. Sinus surgery postpones chronic Gram-negative lung infection: Cohort study of 106 patients with cystic fibrosis. Rhinology 2016, 54, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Warr, G.W.; Magor, K.E.; Higgins, D.A. IgY: Clues to the origins of modern antibodies. Immunol. Today 1995, 16, 392–398. [Google Scholar] [CrossRef]
- Bhanushali, J.K.; Gilbert, J.M.; McDougald, L.R. Simple method to purify chicken immunoglobulin G. Poult. Sci. 1994, 73, 1158–1161. [Google Scholar] [CrossRef]
- Nilsson, E.; Kollberg, H.; Johannesson, M.; Wejåker, P.E.; Carlander, D.; Larsson, A. More than 10 years’ continuous oral treatment with specific immunoglobulin Y for the prevention of Pseudomonas aeruginosa infections: A case report. J. Med. Food 2007, 10, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.; Larsson, A.; Olesen, H.V.; Wejåker, P.E.; Kollberg, H. Good effect of IgY against Pseudomonas aeruginosa infections in cystic fibrosis patients. Pediatr. Pulmonol. 2008, 43, 892–899. [Google Scholar] [CrossRef]
- Thomsen, K.; Christophersen, L.; Bjarnsholt, T.; Jensen, P.Ø.; Moser, C.; Høiby, N. Anti-Pseudomonas aeruginosa IgY Antibodies Induce Specific Bacterial Aggregation and Internalization in Human Polymorphonuclear Neutrophils. Infect. Immun. 2015, 83, 2686–2693. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, K.; Christophersen, L.; Jensen, P.Ø.; Bjarnsholt, T.; Moser, C.; Høiby, N. Anti-Pseudomonas aeruginosa IgY antibodies promote bacterial opsonization and augment the phagocytic activity of polymorphonuclear neutrophils. Hum. Vaccines Immunother. 2016, 12, 1690–1699. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, K.; Christophersen, L.; Bjarnsholt, T.; Jensen, P.Ø.; Moser, C.; Høiby, N. Anti-Pseudomonas aeruginosa IgY antibodies augment bacterial clearance in a murine pneumonia model. J. Cyst. Fibros. 2016, 15, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, K.; Christophersen, L.; Lerche, C.J.; Holmgaard, D.B.; Calum, H.; Høiby, N.; Moser, C. Azithromycin potentiates avian IgY effect against Pseudomonas aeruginosa in a murine pulmonary infection model. Int. J. Antimicrob. Agents 2021, 57, 106213. [Google Scholar] [CrossRef]
- Schuster, A.; Bend, J.; Høiby, N.; Verde, P.E.; Rottmann, A.; Larsson, A. Clinical study to evaluate an anti-Pseudomonas aeruginosa igY gargling solution. JCF 2019, 18 (Suppl. 1), WS12-5. [Google Scholar]
- Abe, C.; Homma, J.Y. IgE antibody production to elastase toxoid of Pseudomonas aeruginosa in mice. Jpn. J. Exp. Med. 1981, 51, 71–73. [Google Scholar] [PubMed]
- Salomonsen, C.M.; Chriél, M.; Jensen, T.H.; Rangstrup-Christensen, L.; Høiby, N.; Hammer, A.S. Effect of infectious dose and season on development of hemorrhagic pneumonia in mink caused by Pseudomonas aeruginosa. Can. J. Vet. Res. 2013, 77, 221–225. [Google Scholar] [PubMed]
- Bruderer, U.; Cryz, S.J., Jr.; Schaad, U.B.; Deusinger, M.; Que, J.U.; Lang, A.B. Affinity constants of naturally acquired and vaccine-induced anti-Pseudomonas aeruginosa antibodies in healthy adults and cystic fibrosis patients. J. Infect. Dis. 1992, 166, 344–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döring, G.; Pier, G.B. Vaccines and immunotherapy against Pseudomonas aeruginosa. Vaccine 2008, 26, 1011–1024. [Google Scholar] [CrossRef]
- Döring, G.; Meisner, C.; Stern, M.; Flagella Vaccine Trial Study Group. A double-blind randomized placebo-controlled phase III study of a Pseudomonas aeruginosa flagella vaccine in cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2007, 104, 11020–11025. [Google Scholar] [CrossRef] [Green Version]
- Proesmans, M.; Balinska-Miskiewicz, W.; Dupont, L.; Bossuyt, X.; Verhaegen, J.; Høiby, N.; de Boeck, K. Evaluating the “Leeds criteria” for Pseudomonas aeruginosa infection in a cystic fibrosis centre. Eur. Respir. J. 2006, 27, 937–943. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomsen, K.; Høiby, N.; Jensen, P.Ø.; Ciofu, O.; Moser, C. Immune Response to Biofilm Growing Pulmonary Pseudomonas aeruginosa Infection. Biomedicines 2022, 10, 2064. https://doi.org/10.3390/biomedicines10092064
Thomsen K, Høiby N, Jensen PØ, Ciofu O, Moser C. Immune Response to Biofilm Growing Pulmonary Pseudomonas aeruginosa Infection. Biomedicines. 2022; 10(9):2064. https://doi.org/10.3390/biomedicines10092064
Chicago/Turabian StyleThomsen, Kim, Niels Høiby, Peter Østrup Jensen, Oana Ciofu, and Claus Moser. 2022. "Immune Response to Biofilm Growing Pulmonary Pseudomonas aeruginosa Infection" Biomedicines 10, no. 9: 2064. https://doi.org/10.3390/biomedicines10092064
APA StyleThomsen, K., Høiby, N., Jensen, P. Ø., Ciofu, O., & Moser, C. (2022). Immune Response to Biofilm Growing Pulmonary Pseudomonas aeruginosa Infection. Biomedicines, 10(9), 2064. https://doi.org/10.3390/biomedicines10092064