
The Interplay of Tumor Vessels and Immune Cells Affects Immunotherapy of Glioblastoma


Abstract
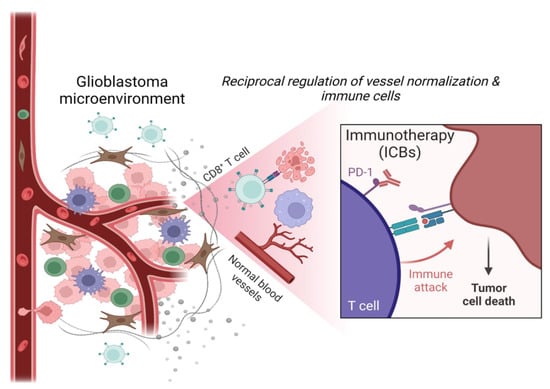
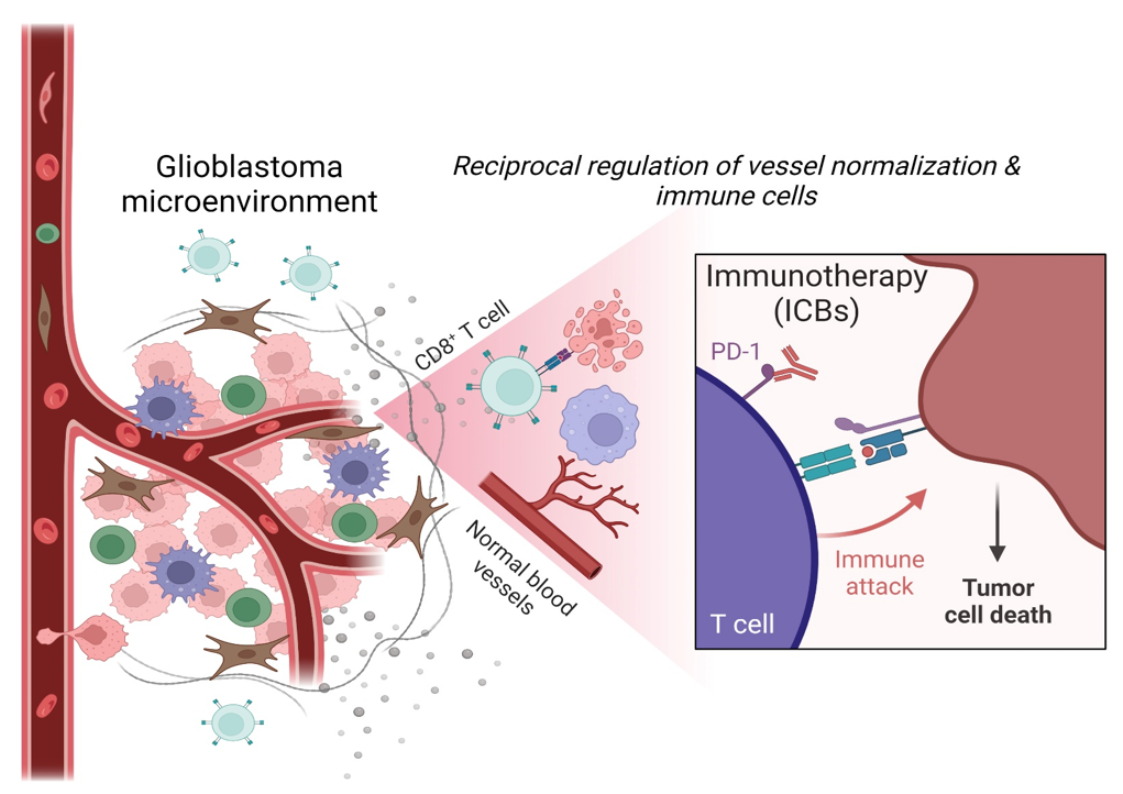
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
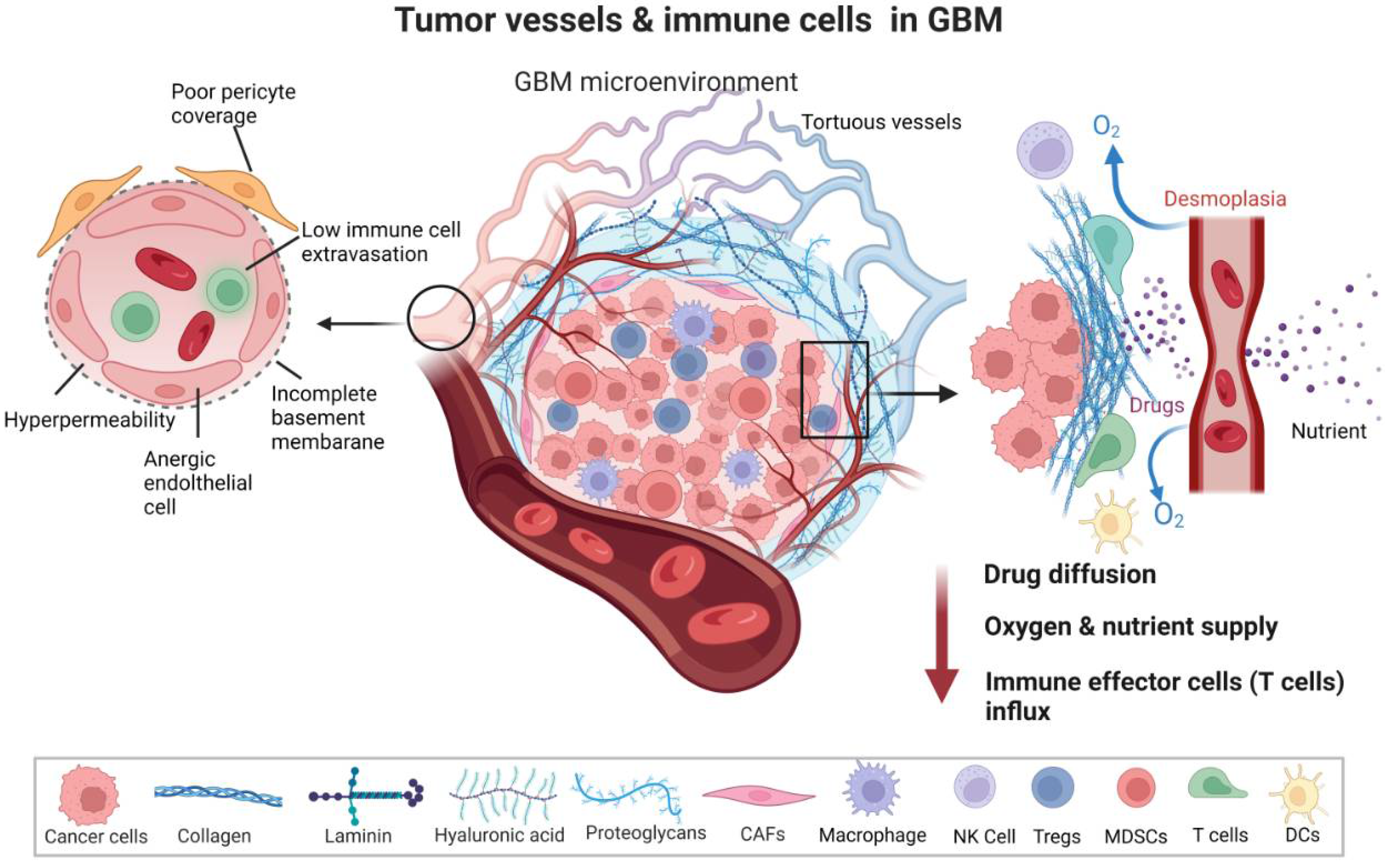
2. The Aberrant Vasculature in GBM
2.1. Structural and Functional Abnormalities of the GBM Vasculature
2.2. GBM Vasculature as Conduits of the Immune Escape and Therapy Resistance
2.3. Contribution of Tumor Vasculature to Local and Systemic Immunosuppression in the GBM Microenvironment
3. The Immune Landscape in GBM
3.1. Multifaceted Action of Glioma-Associated Myeloid Cells on TME and Antitumor Immunity
3.2. T Cells, NK Cells, and DCs in GBM
3.3. Failure of the Immune Checkpoint Blockade in GBM
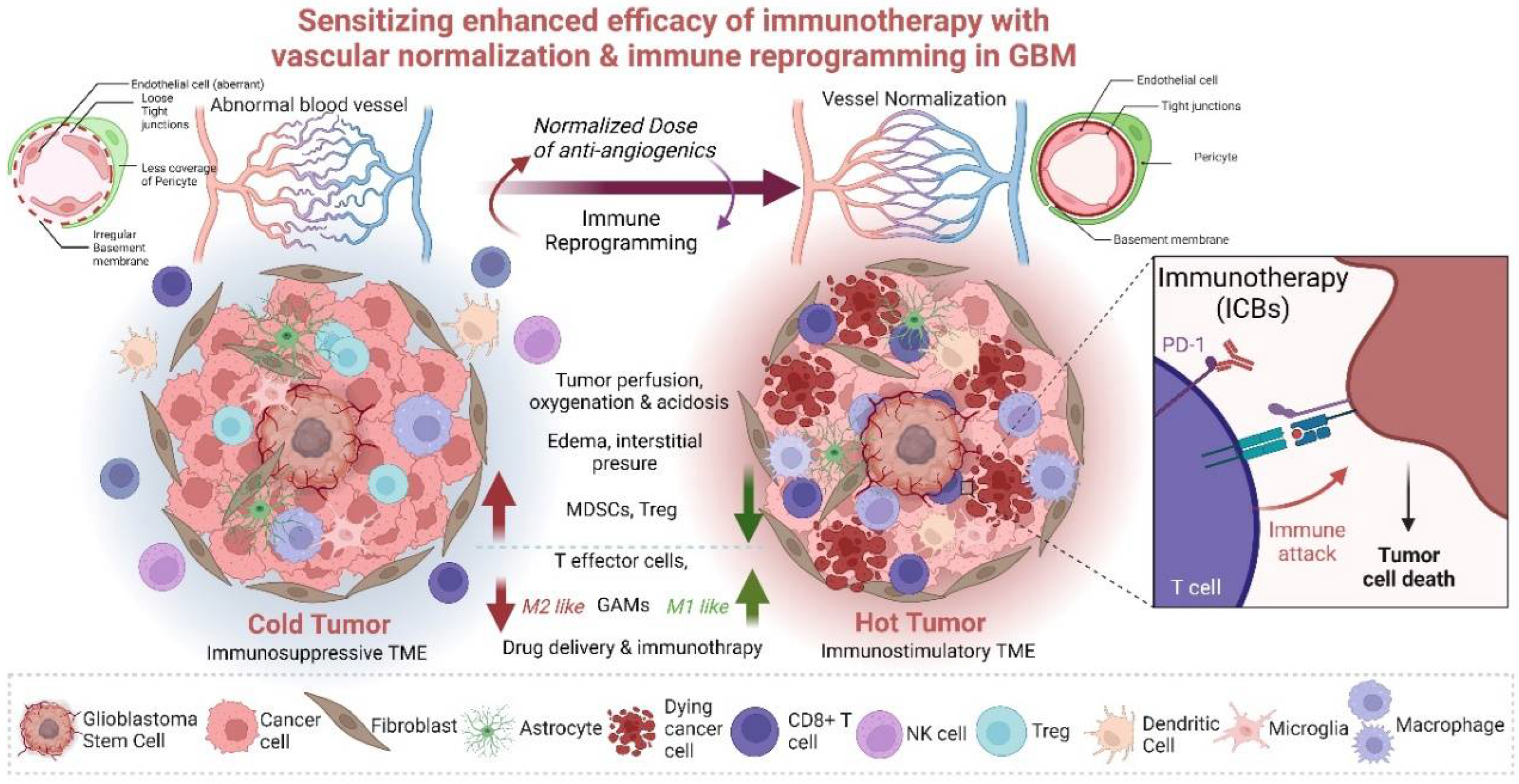
4. Reprograming of the GBM TME to Improve ICB Immunotherapy
5. Tumor Vessel Normalization to Improve ICB Immunotherapy in GBM
6. Perspectives and Further Directions
Author Contributions
Funding
Conflicts of Interest
References
- Weller, M.; van den Bent, M.; Hopkins, K.; Tonn, J.C.; Stupp, R.; Falini, A.; Cohen-Jonathan-Moyal, E.; Frappaz, D.; Henriksson, R.; Balana, C.; et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014, 15, e395–e403. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-Dense Temozolomide for Newly Diagnosed Glioblastoma: A Randomized Phase III Clinical Trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Capper, D. WHO 2016 Classification of Gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Rosińska, S.; Gavard, J. Tumor Vessels Fuel the Fire in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 6514. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Tabatabaei, P.; Visse, E.; Bergström, P.; Brännström, T.; Siesjö, P.; Bergenheim, A.T. Radiotherapy induces an immediate inflammatory reaction in malignant glioma: A clinical microdialysis study. J. Neurooncol. 2017, 131, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Giles, A.J.; Hutchinson, M.-K.; Sonnemann, H.M.; Jung, J.; Fecci, P.E.; Ratnam, N.M.; Zhang, W.; Song, H.; Bailey, R.; Davis, D.; et al. Dexamethasone-induced immunosuppression: Mechanisms and implications for immunotherapy. J. Immunother. Cancer 2018, 6, 51. [Google Scholar] [CrossRef]
- Karachi, A.; Dastmalchi, F.; Mitchell, D.A.; Rahman, M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro Oncol. 2018, 20, 1566–1572. [Google Scholar] [CrossRef]
- Ramirez, Y.P.; Weatherbee, J.L.; Wheelhouse, R.T.; Ross, A.H. Glioblastoma Multiforme Therapy and Mechanisms of Resistance. Pharmaceuticals 2013, 6, 1475–1506. [Google Scholar] [CrossRef]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Options Oncol. 2019, 20, 24. [Google Scholar] [CrossRef] [Green Version]
- Grossman, S.A.; Ye, X.; Lesser, G.; Sloan, A.; Carraway, H.; Desideri, S.; Piantadosi, S. Immunosuppression in Patients with High-Grade Gliomas Treated with Radiation and Temozolomide. Clin. Cancer Res. 2011, 17, 5473–5480. [Google Scholar] [CrossRef] [PubMed]
- Taylor, O.G.; Brzozowski, J.S.; Skelding, K.A. Glioblastoma Multiforme: An Overview of Emerging Therapeutic Targets. Front. Oncol. 2019, 9, 963. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.W.; Quail, D.F. Immunotherapy for Glioblastoma: Current Progress and Challenges. Front. Immunol. 2021, 12, 676301. [Google Scholar] [CrossRef]
- Leonetti, A.; Wever, B.; Mazzaschi, G.; Assaraf, Y.G.; Rolfo, C.; Quaini, F.; Tiseo, M.; Giovannetti, E. Molecular basis and rationale for combining immune checkpoint inhibitors with chemotherapy in non-small cell lung cancer. Drug Resist. Updates 2019, 46, 100644. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.Y.; Choi, J.; Jackson, C.; Lim, M. Combination immunotherapy strategies for glioblastoma. J. Neuro Oncol. 2021, 151, 375–391. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; LaLonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.-J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Kaminska, B.; Ochocka, N.; Segit, P. Single-Cell Omics in Dissecting Immune Microenvironment of Malignant Gliomas-Challenges and Perspectives. Cells 2021, 10, 2264. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Chen, I.X.; Tong, R.; Ng, M.R.; Martin, J.D.; Naxerova, K.; Wu, M.W.; Huang, P.; Boucher, Y.; Kohane, D.S.; et al. Reprogramming the microenvironment with tumor-selective angiotensin blockers enhances cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2019, 116, 10674–10680. [Google Scholar] [CrossRef]
- Aslan, K.; Turco, V.; Blobner, J.; Sonner, J.K.; Liuzzi, A.R.; Núñez, N.G.; De Feo, D.; Kickingereder, P.V.; Fischer, M.; Green, E.; et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat. Commun. 2020, 11, 931. [Google Scholar] [CrossRef]
- Martin, J.D.; Seano, G.; Jain, R.K. Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu. Rev. Physiol. 2019, 81, 505–534. [Google Scholar] [CrossRef]
- Huang, Y.; Kim, B.Y.S.; Chan, C.K.; Hahn, S.M.; Weissman, I.L.; Jiang, W. Improving immune–vascular crosstalk for cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 195–203. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Jain, R.K. CD4 + T Cell Activation and Vascular Normalization: Two Sides of the Same Coin? Immunity 2017, 46, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Melo, V.; Bremer, E.; Martin, J.D. Towards Immunotherapy-Induced Normalization of the Tumor Microenvironment. Front. Cell Dev. Biol. 2022, 10, 908389. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, X.; Li, J. Manipulation of immune-vascular crosstalk: New strategies towards cancer treatment. Acta Pharm. Sin. B 2020, 10, 2018–2036. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Guyon, J.; Chapouly, C.; Andrique, L.; Bikfalvi, A.; Daubon, T. The Normal and Brain Tumor Vasculature: Morphological and Functional Characteristics and Therapeutic Targeting. Front. Physiol. 2021, 12, 622615. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Seano, G.; Jain, R.K. Vessel co-option in glioblastoma: Emerging insights and opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Tumor Vasculature: Improving Drug Delivery and Efficacy. Trends Cancer 2018, 4, 258–259. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Goldstein, A.; Wang, H.; Lo, H.C.; Kim, I.S.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Lamplugh, Z.; Fan, Y. Vascular Microenvironment, Tumor Immunity and Immunotherapy. Front. Immunol. 2021, 12, 811485. [Google Scholar] [CrossRef]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef]
- Pacheco, C.; Martins, C.; Monteiro, J.; Baltazar, F.; Costa, B.M.; Sarmento, B. Glioblastoma Vasculature: From its Critical Role in Tumor Survival to Relevant in Vitro Modelling. Front. Drug Deliv. 2022, 2, 823412. [Google Scholar] [CrossRef]
- Kim, S.-J.; Lee, H.J.; Kim, M.S.; Choi, H.J.; He, J.; Wu, Q.; Aldape, K.; Weinberg, J.S.; Yung, W.A.; Conrad, C.A.; et al. Macitentan, a Dual Endothelin Receptor Antagonist, in Combination with Temozolomide Leads to Glioblastoma Regression and Long-term Survival in Mice. Clin. Cancer Res. 2015, 21, 4630–4641. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef]
- Orr, B.A.; Eberhart, C.G. Molecular Pathways: Not a Simple Tube—The Many Functions of Blood Vessels. Clin. Cancer Res. 2015, 21, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Bhaduri, A.; Di Lullo, E.; Jung, D.; Müller, S.; Crouch, E.E.; Espinosa, C.S.; Ozawa, T.; Alvarado, B.; Spatazza, J.; Cadwell, C.R.; et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell 2020, 26, 48–63.e6. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Guryanova, O.A.; Zhou, W.; Liu, C.; Huang, Z.; Fang, X.; Wang, X.; Chen, C.; Wu, Q.; He, Z.; et al. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci. Transl. Med. 2018, 10, eaah6816. [Google Scholar] [CrossRef] [PubMed]
- Mazor, G.; Levin, L.; Picard, D.; Ahmadov, U.; Carén, H.; Borkhardt, A.; Reifenberger, G.; Leprivier, G.; Remke, M.; Rotblat, B. The lncRNA TP73-AS1 is linked to aggressiveness in glioblastoma and promotes temozolomide resistance in glioblastoma cancer stem cells. Cell Death Dis. 2019, 10, 246. [Google Scholar] [CrossRef]
- Van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.; Wesseling, P.; Wurdinger, T.; de Vries, H. Overcoming the blood–brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.G.; Campanati, L.; Righy, C.; D’Andrea-Meira, I.; de Sampaio, E.; Spohr, T.C.L.; Porto-Carreiro, I.; Pereira, C.M.; Balça-Silva, J.; Kahn, S.A.; et al. Gliomas and the vascular fragility of the blood brain barrier. Front. Cell. Neurosci. 2014, 8, 418. [Google Scholar] [CrossRef]
- Boucher, Y.; Salehil, H.; Witwerl, B.; Harsh, G.R.; Jain, R.K. Interstitial fluid pressure in intracranial tumours in patients and in rodents. Br. J. Cancer 1997, 75, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Ratnam, N.M.; Gilbert, M.R.; Giles, A.J. Immunotherapy in CNS cancers: The role of immune cell trafficking. Neuro-Oncology 2019, 21, 37–46. [Google Scholar] [CrossRef]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.-C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Wang, J.; Ye, C.; Chen, C.; Xiong, H.; Xie, B.; Zhou, J.; Chen, Y.; Zheng, S.; Wang, L. Glucose transporter GLUT1 expression and clinical outcome in solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 16875–16886. [Google Scholar] [CrossRef] [Green Version]
- De Gooijer, M.C.; de Vries, N.A.; Buckle, T.; Buil, L.C.; Beijnen, J.H.; Boogerd, W.; van Tellingen, O. Improved Brain Penetration and Antitumor Efficacy of Temozolomide by Inhibition of ABCB1 and ABCG2. Neoplasia 2018, 20, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Dréan, A.; Rosenberg, S.; Lejeune, F.-X.; Goli, L.; Nadaradjane, A.A.; Guehennec, J.; Schmitt, C.; Verreault, M.; Bielle, F.; Mokhtari, K.; et al. ATP binding cassette (ABC) transporters: Expression and clinical value in glioblastoma. J. Neuro Oncol. 2018, 138, 479–486. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Askoxylakis, V.; Guo, Y.; Datta, M.; Kloepper, J.; Ferraro, G.B.; Bernabeu, M.O.; Fukumura, D.; McDannold, N.; Jain, R.K. Mechanisms of enhanced drug delivery in brain metastases with focused ultrasound-induced blood–tumor barrier disruption. Proc. Natl. Acad. Sci. USA 2018, 115, E8717–E8726. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Bak, M.; Melander, F.; Thomsen, M.S.; Burkhart, A.; Kempen, P.J.; Andresen, T.L.; Moos, T. Modulating the antibody density changes the uptake and transport at the blood-brain barrier of both transferrin receptor-targeted gold nanoparticles and liposomal cargo. J. Control. Release 2019, 295, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Guerit, S.; Liebner, S. Blood-Brain Barrier Breakdown Determines Differential Therapeutic Outcome in Genetically Diverse Forms of Medulloblastoma. Cancer Cell 2016, 29, 427–429. [Google Scholar] [CrossRef]
- Erel-Akbaba, G.; Carvalho, L.A.; Tian, T.; Zinter, M.; Akbaba, H.; Obeid, P.J.; Chiocca, E.A.; Weissleder, R.; Kantarci, A.G.; Tannous, B.A. Radiation-Induced Targeted Nanoparticle-Based Gene Delivery for Brain Tumor Therapy. ACS Nano 2019, 13, 4028–4040. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, M.B.; Garg, A.; Agostinis, P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Q.; Hong, Y. Tumor Vessel Normalization: A Window to Enhancing Cancer Immunotherapy. Technol. Cancer Res. Treat. 2020, 19, 1533033820980116. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Stylianopoulos, T.; Martin, J.D.; Popović, Z.; Chen, O.; Kamoun, W.S.; Bawendi, M.G.; Fukumura, D.; Jain, R.K. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat. Nanotechnol. 2012, 7, 383–388. [Google Scholar] [CrossRef]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.-J.; Kojima, N.; Lopez, P.A.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef]
- Wei, S.C.; Anang, N.-A.A.S.; Sharma, R.; Andrews, M.C.; Reuben, A.; Levine, J.H.; Cogdill, A.P.; Mancuso, J.J.; Wargo, J.A.; Pe’er, D.; et al. Combination anti–CTLA-4 plus anti–PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. USA 2019, 116, 22699–22709. [Google Scholar] [CrossRef] [PubMed]
- Barrett, D.M.; Grupp, S.A.; June, C.H. Chimeric Antigen Receptor– and TCR-Modified T Cells Enter Main Street and Wall Street. J. Immunol. 2015, 195, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Di Tacchio, M.; Macas, J.; Weissenberger, J.; Sommer, K.; Bähr, O.; Steinbach, J.P.; Senft, C.; Seifert, V.; Glas, M.; Herrlinger, U.; et al. Tumor Vessel Normalization, Immunostimulatory Reprogramming, and Improved Survival in Glioblastoma with Combined Inhibition of PD-1, Angiopoietin-2, and VEGF. Cancer Immunol. Res. 2019, 7, 1910–1927. [Google Scholar] [CrossRef]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e6, Erratum in Immunity 2018, 48, 599. [Google Scholar] [CrossRef]
- Kleber, S.; Sancho-Martinez, I.; Wiestler, B.; Beisel, A.; Gieffers, C.; Hill, O.; Thiemann, M.; Mueller, W.; Sykora, J.; Kuhn, A.; et al. Yes and PI3K Bind CD95 to Signal Invasion of Glioblastoma. Cancer Cell 2008, 13, 235–248. [Google Scholar] [CrossRef]
- Wisniewski, P.; Ellert-Miklaszewska, A.; Kwiatkowska, A.; Kaminska, B. Non-apoptotic Fas signaling regulates invasiveness of glioma cells and modulates MMP-2 activity via NFκB-TIMP-2 pathway. Cell Signal. 2010, 22, 212–220. [Google Scholar] [CrossRef]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Igney, F.H.; Behrens, C.K.; Krammer, P.H. Tumor counterattack-concept and reality. Eur. J. Immunol. 2000, 30, 725–731. [Google Scholar] [CrossRef]
- Didenko, V.v.; Ngo, H.N.; Minchew, C.; Baskin, D.S. Apoptosis of T lymphocytes invading glioblastomas multiforme: A possible tumor defense mechanism. J. Neurosurg. 2002, 96, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sagar; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef]
- Mohammad, M.G.; Tsai, V.W.; Ruitenberg, M.J.; Hassanpour, M.; Li, H.; Hart, P.H.; Breit, S.N.; Sawchenko, P.E.; Brown, D.A. Immune cell trafficking from the brain maintains CNS immune tolerance. J. Clin. Investig. 2014, 124, 1228–1241. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Bonnardel, J.; Guilliams, M. Developmental control of macrophage function. Curr. Opin. Immunol. 2018, 50, 64–74. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef]
- De Boeck, A.; Ahn, B.Y.; D’Mello, C.; Lun, X.; Menon, S.V.; Alshehri, M.M.; Szulzewsky, F.; Shen, Y.; Khan, L.; Dang, N.H.; et al. Glioma-derived IL-33 orchestrates an inflammatory brain tumor microenvironment that accelerates glioma progression. Nat. Commun. 2020, 11, 4997. [Google Scholar] [CrossRef]
- Zhang, B.; Gu, X.; Han, X.; Gao, Q.; Liu, J.; Guo, T.; Gao, D. Crosstalk between DNA methylation and histone acetylation triggers GDNF high transcription in glioblastoma cells. Clin. Epigenetics 2020, 12, 47. [Google Scholar] [CrossRef]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad—Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Wallmann, T.; Zhang, X.-M.; Wallerius, M.; Bolin, S.; Joly, A.-L.; Sobocki, C.; Leiss, L.; Jiang, Y.; Bergh, J.; Holland, E.C.; et al. Microglia Induce PDGFRB Expression in Glioma Cells to Enhance Their Migratory Capacity. iScience 2018, 9, 71–83. [Google Scholar] [CrossRef]
- Fanelli, G.; Grassini, D.; Ortenzi, V.; Pasqualetti, F.; Montemurro, N.; Perrini, P.; Naccarato, A.; Scatena, C. Decipher the Glioblastoma Microenvironment: The First Milestone for New Groundbreaking Therapeutic Strategies. Genes 2021, 12, 445. [Google Scholar] [CrossRef] [PubMed]
- Datsi, A.; Sorg, R.V. Dendritic Cell Vaccination of Glioblastoma: Road to Success or Dead End. Front. Immunol. 2021, 12, 4506. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef]
- Dubinski, D.; Wölfer, J.; Hasselblatt, M.; Schneider-Hohendorf, T.; Bogdahn, U.; Stummer, W.; Wiendl, H.; Grauer, O.M. CD4+T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol. 2015, 18, 807–818. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef]
- DiDomenico, J.; Lamano, J.B.; Oyon, D.; Li, Y.; Veliceasa, D.; Kaur, G.; Ampie, L.; Choy, W.; Lamano, J.B.; Bloch, O. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. OncoImmunology 2018, 7, e1448329. [Google Scholar] [CrossRef]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [Green Version]
- Turnis, M.E.; Sawant, D.V.; Szymczak-Workman, A.L.; Andrews, L.P.; Delgoffe, G.M.; Yano, H.; Beres, A.J.; Vogel, P.; Workman, C.J.; Vignali, D.A. Interleukin-35 Limits Anti-Tumor Immunity. Immunity 2016, 44, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Ross, W.G.; Agbai, O.N.; Frazier, R.; Figler, R.A.; Rieger, J.; Linden, J.; Ernst, P.B. The A2BAdenosine Receptor Impairs the Maturation and Immunogenicity of Dendritic Cells. J. Immunol. 2009, 182, 4616–4623. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tian, Z.; Wei, H. Developmental and Functional Control of Natural Killer Cells by Cytokines. Front. Immunol. 2017, 8, 930. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yang, W.; Zhang, H.; Ren, Y.; Fang, Z.; Yuan, C.; Yao, Z. The Prognostic Landscape of Tumor-Infiltrating Immune Cells and Immune Checkpoints in Glioblastoma. Technol. Cancer Res. Treat. 2019, 18, 1533033819869949. [Google Scholar] [CrossRef]
- Golán, I.; Rodríguez de la Fuente, L.; Costoya, J.A. NK Cell-Based Glioblastoma Immunotherapy. Cancers 2018, 10, 522. [Google Scholar] [CrossRef]
- De Leo, A.; Ugolini, A.; Veglia, F. Myeloid Cells in Glioblastoma Microenvironment. Cells 2020, 10, 18. [Google Scholar] [CrossRef]
- Mittal, D.; Vijayan, D.; Putz, E.M.; Aguilera, A.R.; Markey, K.A.; Straube, J.; Kazakoff, S.; Nutt, S.L.; Takeda, K.; Hill, G.R.; et al. Interleukin-12 from CD103+ Batf3-Dependent Dendritic Cells Required for NK-Cell Suppression of Metastasis. Cancer Immunol. Res. 2017, 5, 1098–1108. [Google Scholar] [CrossRef]
- Böttcher, J.P.; e Sousa, C.R. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef]
- Wang, J.; Liu, P.; Xin, S.; Wang, Z.; Li, J. Nrf2 suppresses the function of dendritic cells to facilitate the immune escape of glioma cells. Exp. Cell Res. 2017, 360, 66–73. [Google Scholar] [CrossRef]
- Ugolini, A.; Tyurin, V.A.; Tyurina, Y.Y.; Tcyganov, E.N.; Donthireddy, L.; Kagan, V.E.; Gabrilovich, D.I.; Veglia, F. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight 2020, 5, e138581. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, O.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 2017, 8, 2122. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhao, Q.; Gabrusiewicz, K.; Kong, L.-Y.; Xia, X.; Wang, J.; Ott, M.; Xu, J.; Davis, R.E.; Huo, L.; et al. FGL2 promotes tumor progression in the CNS by suppressing CD103+ dendritic cell differentiation. Nat. Commun. 2019, 10, 448. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef]
- Jain, S.; Chalif, E.J.; Aghi, M.K. Interactions Between Anti-Angiogenic Therapy and Immunotherapy in Glioblastoma. Front. Oncol. 2022, 11, 5702. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Cuzzubbo, S.; McArthur, S.; Durrant, L.G.; Adhikaree, J.; Tinsley, C.J.; Pockley, A.G.; McArdle, S.E.B. Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front. Immunol. 2020, 11, 582106. [Google Scholar] [CrossRef]
- Bausart, M.; Préat, V.; Malfanti, A. Immunotherapy for glioblastoma: The promise of combination strategies. J. Exp. Clin. Cancer Res. 2022, 41, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Prithviraj, P.; Shrestha, R.; Sharma, R.; Anaka, M.; Bridle, K.; Kannourakis, G.; Crawford, D.; Jayachandran, A. Prognostic Role of Immune Checkpoint Regulators in Cholangiocarcinoma: A Pilot Study. J. Clin. Med. 2021, 10, 2191. [Google Scholar] [CrossRef]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Saung, M.T.; Muth, S.; Ding, D.; Thomas, D.L.; Blair, A.B.; Tsujikawa, T.; Coussens, L.; Jaffee, E.M.; Zheng, L. Targeting myeloid-inflamed tumor with anti-CSF-1R antibody expands CD137+ effector T-cells in the murine model of pancreatic cancer. J. Immunother. Cancer 2018, 6, 118. [Google Scholar] [CrossRef]
- Amoozgar, Z.; Kloepper, J.; Ren, J.; Tay, R.E.; Kazer, S.W.; Kiner, E.; Krishnan, S.; Posada, J.M.; Ghosh, M.; Mamessier, E.; et al. Targeting Treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nat. Commun. 2021, 12, 2582. [Google Scholar] [CrossRef]
- Shitara, K.; Nishikawa, H. Regulatory T cells: A potential target in cancer immunotherapy. Ann. N. Y. Acad. Sci. 2016, 1417, 104–115. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Park, J.-S.; Kim, I.-K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.-C.; et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell 2016, 30, 953–967. [Google Scholar] [CrossRef] [Green Version]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Khan, K.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Jain, R.K.; Duda, D.G. The Current Landscape of Immune Checkpoint Blockade in Hepatocellular Carcinoma. JAMA Oncol. 2021, 7, 113–123. [Google Scholar] [CrossRef]
- Mazzone, M.; Bergers, G. Regulation of Blood and Lymphatic Vessels by Immune Cells in Tumors and Metastasis. Annu. Rev. Physiol. 2019, 81, 535–560. [Google Scholar] [CrossRef]
- Datta, M.; Coussens, L.M.; Nishikawa, H.; Hodi, F.S.; Jain, R.K. Reprogramming the Tumor Microenvironment to Improve Immunotherapy: Emerging Strategies and Combination Therapies. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Mpekris, F.; Voutouri, C.; Baish, J.W.; Duda, D.G.; Munn, L.L.; Stylianopoulos, T.; Jain, R.K. Combining microenvironment normalization strategies to improve cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2020, 117, 3728–3737. [Google Scholar] [CrossRef]
- Zheng, X.; Fang, Z.; Liu, X.; Deng, S.; Zhou, P.; Wang, X.; Zhang, C.; Yin, R.; Hu, H.; Chen, X.; et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J. Clin. Investig. 2018, 128, 2104–2115. [Google Scholar] [CrossRef]
- Chen, I.X.; Chauhan, V.P.; Posada, J.; Ng, M.R.; Wu, M.W.; Adstamongkonkul, P.; Huang, P.; Lindeman, N.; Langer, R.; Jain, R.K. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 4558–4566. [Google Scholar] [CrossRef]
- Zhao, Y.; Cao, J.; Melamed, A.; Worley, M.; Gockley, A.; Jones, D.; Nia, H.T.; Zhang, Y.; Stylianopoulos, T.; Kumar, A.S.; et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2019, 116, 2210–2219. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Hamzah, J.; Jugold, M.; Kiessling, F.; Rigby, P.J.; Manzur, M.; Marti, H.H.; Rabie, T.; Kaden, S.; Gröne, H.-J.; Hämmerling, G.J.; et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 2008, 453, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassará, A.; Wyser Rmili, C.; Kiialainen, A.; Kienast, Y.; Mueller, H.-J.; Ooi, C.-H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9, eaak9670. [Google Scholar] [CrossRef]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti–PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef]
- Wang, N.; Jain, R.K.; Batchelor, T.T. New Directions in Anti-Angiogenic Therapy for Glioblastoma. Neurotherapeutics 2017, 14, 321–332. [Google Scholar] [CrossRef]
- Poulsen, H.S.; Urup, T.; Michaelsen, S.; Staberg, M.; Lassen, U.; Villingshoej, M. The impact of bevacizumab treatment on survival and quality of life in newly diagnosed glioblastoma patients. Cancer Manag. Res. 2014, 6, 373–387. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, aaa0984. [Google Scholar] [CrossRef]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Ii, J.E.H.; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic Escape After Prolonged Progression-Free Survival With Epidermal Growth Factor Receptor Variant III Peptide Vaccination in Patients With Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef]
- Fisher, J.; Adamson, D. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neuro Oncol. 2020, 151, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Fritzell, S.; Sandén, E.; Eberstål, S.; Visse, E.; Darabi, A.; Siesjö, P. Intratumoral temozolomide synergizes with immunotherapy in a T cell-dependent fashion. Cancer Immunol. Immunother. 2013, 62, 1463–1474. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Desai, R.; Abel, M.L.; Riccione, K.A.; Batich, K.A.; Shen, S.H.; Chongsathidkiet, P.; Gedeon, P.; Elsamadicy, A.A.; Snyder, D.J.; et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. OncoImmunology 2018, 7, e1434464. [Google Scholar] [CrossRef]
- Karachi, A.; Yang, C.; Dastmalchi, F.; Sayour, E.J.; Huang, J.; Azari, H.; Long, Y.; Flores, C.; Mitchell, D.A.; Rahman, M. Modulation of temozolomide dose differentially affects T-cell response to immune checkpoint inhibition. Neuro-Oncology 2019, 21, 730–741. [Google Scholar] [CrossRef]
- Pérez, J.E.; Kopecky, J.; Visse, E.; Darabi, A.; Siesjö, P. Convection-enhanced delivery of temozolomide and whole cell tumor immunizations in GL261 and KR158 experimental mouse gliomas. BMC Cancer 2020, 20, 7. [Google Scholar] [CrossRef]
- Von Roemeling, C.A.; Wang, Y.; Qie, Y.; Yuan, H.; Zhao, H.; Liu, X.; Yang, Z.; Yang, M.; Deng, W.; Bruno, K.A.; et al. Therapeutic modulation of phagocytosis in glioblastoma can activate both innate and adaptive antitumour immunity. Nat. Commun. 2020, 11, 1508. [Google Scholar] [CrossRef]
- Dai, B.; Qi, N.; Li, J.; Zhang, G. Temozolomide combined with PD-1 Antibody therapy for mouse orthotopic glioma model. Biochem. Biophys. Res. Commun. 2018, 501, 871–876. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, M.; Lenkiewicz, A.M.; Kaminska, B. The Interplay of Tumor Vessels and Immune Cells Affects Immunotherapy of Glioblastoma. Biomedicines 2022, 10, 2292. https://doi.org/10.3390/biomedicines10092292
Ghosh M, Lenkiewicz AM, Kaminska B. The Interplay of Tumor Vessels and Immune Cells Affects Immunotherapy of Glioblastoma. Biomedicines. 2022; 10(9):2292. https://doi.org/10.3390/biomedicines10092292
Chicago/Turabian StyleGhosh, Mitrajit, Anna M. Lenkiewicz, and Bozena Kaminska. 2022. "The Interplay of Tumor Vessels and Immune Cells Affects Immunotherapy of Glioblastoma" Biomedicines 10, no. 9: 2292. https://doi.org/10.3390/biomedicines10092292
APA StyleGhosh, M., Lenkiewicz, A. M., & Kaminska, B. (2022). The Interplay of Tumor Vessels and Immune Cells Affects Immunotherapy of Glioblastoma. Biomedicines, 10(9), 2292. https://doi.org/10.3390/biomedicines10092292