
Dexamethasone-Induced Adipose Tissue Redistribution and Metabolic Changes: Is Gene Expression the Main Factor? An Animal Model of Chronic Hypercortisolism
 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval and Animals
2.2. Chronic GC Treatment
2.3. Oral Glucose Tolerance Test (oGTT)
2.4. Euthanasia
2.5. Hormonal and Biochemical Analysis
2.6. Histological Analysis
2.7. RNA Isolation and Gene Expression Analysis
2.8. BAT Oxidative and Lipogenic Capacity
2.9. In Vivo Body Temperature Analysis
2.10. Statistical Analysis
3. Results
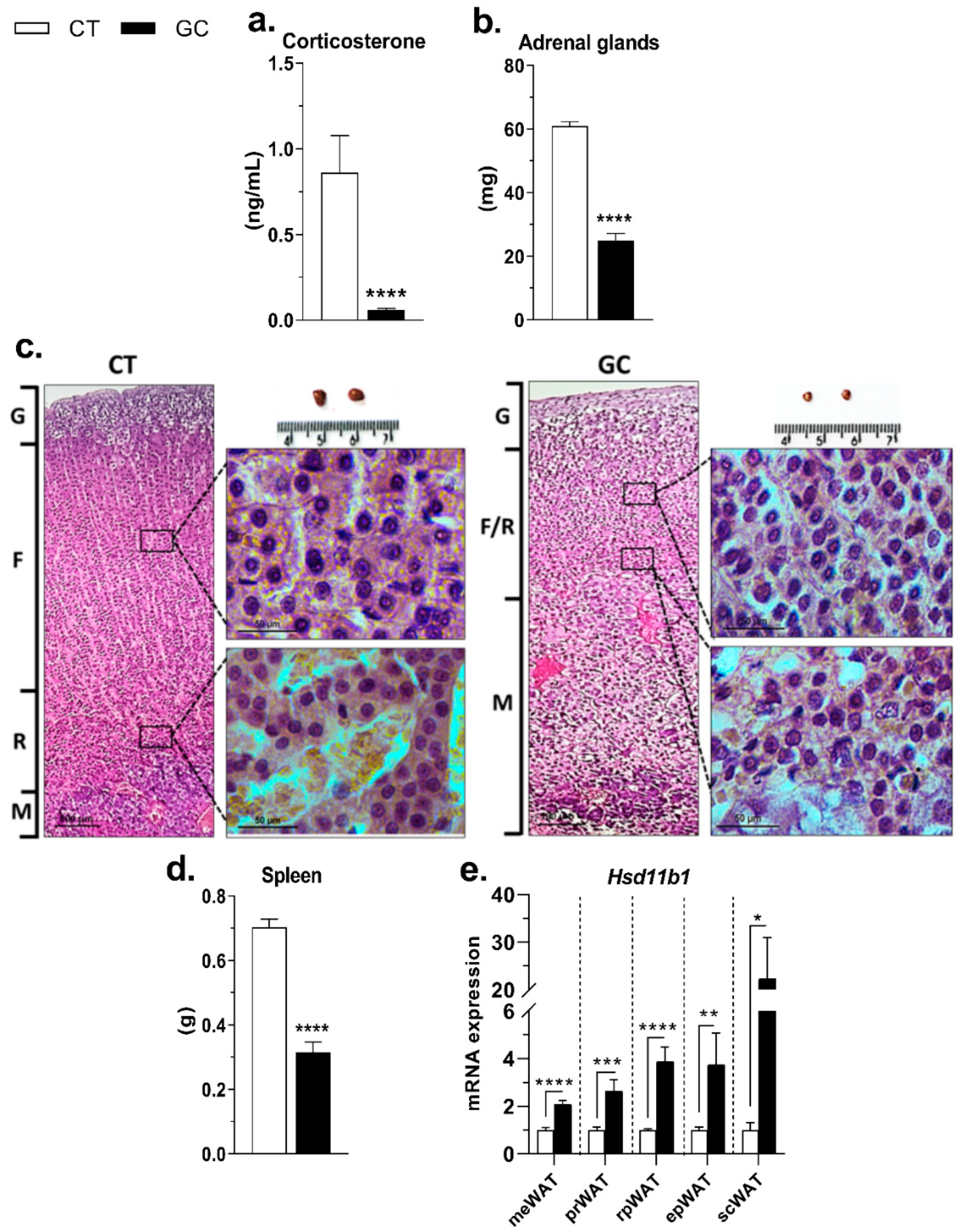
3.1. Inhibition of the HPA Axis
3.2. Hypercortisolism and Metabolic Changes
3.3. GC-Induced Adipose Tissue Redistribution
3.4. Direct and Permissive Actions by GCs in Gene Expression of WAT Lipolysis and Lipogenesis Pathways
3.5. BAT Whitening in Response to GC Excess
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peckett, A.J.; Wright, D.C.; Riddell, M.C. The Effects of Glucocorticoids on Adipose Tissue Lipid Metabolism. Metabolism 2011, 60, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids. Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Mitsugi, N.; Kimura, F. Simultaneous Determination of Blood Levels of Corticosterone and Growth Hormone in the Male Rat: Relation to Sleep-Wakefulness Cycle1. Neuroendocrinology 1985, 41, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, D.P.; Forbes, S.; Walker, B.R. Glucocorticoids and Fatty Acid Metabolism in Humans: Fuelling Fat Redistribution in the Metabolic Syndrome. J. Endocrinol. 2008, 197, 189–204. [Google Scholar] [CrossRef]
- Pivonello, R.; De Martino, M.C.; De Leo, M.; Simeoli, C.; Colao, A. Cushing’s Disease: The Burden of Illness. Endocrine 2017, 56, 10–18. [Google Scholar] [CrossRef]
- Nieman, L.K. Diagnosis of Cushing’s Syndrome in the Modern Era. Endocrinol. Metab. Clin. 2018, 47, 259–273. [Google Scholar] [CrossRef]
- Buttgereit, F.; Spies, C.M.; Bijlsma, J.W.J. Novel Glucocorticoids: Where Are We Now and Where Do We Want to Go? Clin. Exp. Rheumatol. 2015, 33, 29–33. [Google Scholar]
- Schoneveld, O.J.L.M.; Gaemers, I.C.; Lamers, W.H. Mechanisms of Glucocorticoid Signalling. Biochim. Biophys. Acta Gene Struct. Expr. 2004, 1680, 114–128. [Google Scholar] [CrossRef]
- Ahmed, M.H.; Hassan, A. Dexamethasone for the Treatment of Coronavirus Disease (COVID-19): A Review. SN Compr. Clin. Med. 2020, 2, 2637–2646. [Google Scholar] [CrossRef]
- Lester, M.; Sahin, A.; Pasyar, A. The Use of Dexamethasone in the Treatment of COVID-19. Ann. Med. Surg. 2020, 56, 218–219. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ghosh, R.; Vardhan, B.; Ojha, U.K.; Kalra, S. An Epidemic of Iatrogenic Cushing’s Syndrome in Anticipation in Post-COVID Era. J. Fam. Med. Prim. Care 2022, 11, 412. [Google Scholar] [CrossRef]
- Rebuffat, A.G.; Tam, S.; Nawrocki, A.R.; Baker, M.E.; Frey, B.M.; Frey, F.J.; Odermatt, A. The 11-Ketosteroid 11-Ketodexamethasone Is a Glucocorticoid Receptor Agonist. Mol. Cell. Endocrinol. 2004, 214, 27–37. [Google Scholar] [CrossRef]
- Caprio, M.; Fève, B.; Claës, A.; Viengchareun, S.; Lombès, M.; Zennaro, M.-C. Pivotal Role of the Mineralocorticoid Receptor in Corticosteroid-Induced Adipogenesis. FASEB J. 2007, 21, 2185–2194. [Google Scholar] [CrossRef]
- Beaupere, C.; Liboz, A.; Fève, B.; Blondeau, B.; Guillemain, G. Molecular Mechanisms of Glucocorticoid-Induced Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 623. [Google Scholar] [CrossRef]
- He, Y.; Yi, W.; Suino-Powell, K.; Zhou, X.E.; Tolbert, W.D.; Tang, X.; Yang, J.; Yang, H.; Shi, J.; Hou, L.; et al. Structures and Mechanism for the Design of Highly Potent Glucocorticoids. Cell Res. 2014, 24, 713. [Google Scholar] [CrossRef]
- Heitzer, M.D.; Wolf, I.M.; Sanchez, E.R.; Witchel, S.F.; DeFranco, D.B. Glucocorticoid Receptor Physiology. Rev. Endocr. Metab. Disord. 2007, 8, 321–330. [Google Scholar] [CrossRef]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid Receptor Signaling in Health and Disease. Trends Pharmacol. Sci. 2013, 34, 518. [Google Scholar] [CrossRef]
- Lecoq, L.; Vincent, P.; Lavoie-Lamoureux, A.; Lavoie, J.P. Genomic and Non-Genomic Effects of Dexamethasone on Equine Peripheral Blood Neutrophils. Vet. Immunol. Immunopathol. 2009, 128, 126–131. [Google Scholar] [CrossRef]
- Clark, A.R. Anti-Inflammatory Functions of Glucocorticoid-Induced Genes. Mol. Cell. Endocrinol. 2007, 275, 79–97. [Google Scholar] [CrossRef]
- Feelders, R.A.; Pulgar, S.J.; Kempel, A.; Pereira, A.M. The Burden of Cushing’s Disease: Clinical and Health-Related Quality of Life Aspects. Eur. J. Endocrinol. 2012, 167, 311–326. [Google Scholar] [CrossRef]
- Nieman, L.K. Cushing’s Syndrome: Update on Signs, Symptoms and Biochemical Screening. Eur. J. Endocrinol. 2015, 173, M33–M38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.E.; Peckett, A.J.; D’Souza, A.M.; Hawke, T.J.; Riddell, M.C. Adipogenic and Lipolytic Effects of Chronic Glucocorticoid Exposure. Am. J. Physiol. Cell Physiol. 2011, 300, C198–C209. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, Y.D.C.; Bond, A.; Sen, A.; Franklin, D.M.; Lea-Currie Renee, Y.; Sujkowski, D.; Ellis, P.N.; Wilkison, W.O.; Gimble, J.M. Thiazolidinediones and Glucocorticoids Synergistically Induce Differentiation of Human Adipose Tissue Stromal Cells: Biochemical, Cellular, and Molecular Analysis. Metab. -Clin. Exp. 2001, 50, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J. Hormonal Regulation of Adipogenesis. Compr. Physiol. 2017, 7, 1151–1195. [Google Scholar] [CrossRef]
- Chimin, P.; Farias, T.D.S.M.; Torres-Leal, F.L.; Bolsoni-Lopes, A.; Campaña, A.B.; Andreotti, S.; Lima, F.B. Chronic Glucocorticoid Treatment Enhances Lipogenic Activity in Visceral Adipocytes of Male Wistar Rats. Acta Physiol. 2014, 211, 409–420. [Google Scholar] [CrossRef]
- Ferreira, G.N.; Rossi-Valentim, R.; Buzelle, S.L.; Paula-Gomes, S.; Zanon, N.M.; Garófalo, M.A.R.; Frasson, D.; Navegantes, L.C.C.; Chaves, V.E.; Kettelhut, I.C. Differential Regulation of Glyceroneogenesis by Glucocorticoids in Epididymal and Retroperitoneal White Adipose Tissue from Rats. Endocrine 2017, 57, 287–297. [Google Scholar] [CrossRef]
- Minetto, M.A.; Qaisar, R.; Agoni, V.; Motta, G.; Longa, E.; Miotti, D.; Pellegrino, M.A.; Bottinelli, R. Quantitative and Qualitative Adaptations of Muscle Fibers to Glucocorticoids. Muscle Nerve 2015, 52, 631–639. [Google Scholar] [CrossRef]
- Alev, K.; Vain, A.; Aru, M.; Pehme, A.; Purge, P.; Kaasik, P.; Seene, T. Glucocorticoid-Induced Changes in Rat Skeletal Muscle Biomechanical and Viscoelastic Properties: Aspects of Aging. J. Manipulative Physiol. Ther. 2018, 41, 19–24. [Google Scholar] [CrossRef]
- Wu, T.; Jiang, J.; Yang, L.; Li, H.; Zhang, W.; Chen, Y.; Zhao, B.; Kong, B.; Lu, P.; Zhao, Z.; et al. Timing of Glucocorticoid Administration Determines Severity of Lipid Metabolism and Behavioral Effects in Rats. Chronobiol. Int. 2017, 34, 78–92. [Google Scholar] [CrossRef]
- Chanson, P.; Salenave, S. Metabolic Syndrome in Cushing’s Syndrome. Neuroendocrinology 2010, 92, 96–101. [Google Scholar] [CrossRef]
- Pellegrinelli, V.; Carobbio, S.; Vidal-Puig, A. Adipose Tissue Plasticity: How Fat Depots Respond Differently to Pathophysiological Cues. Diabetologia 2016, 59, 1075–1088. [Google Scholar] [CrossRef] [Green Version]
- Lynes, M.D.; Tseng, Y.H. Deciphering Adipose Tissue Heterogeneity. Ann. N. Y. Acad. Sci. 2018, 1411, 5–20. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef]
- Chusyd, D.E.; Wang, D.; Huffman, D.M.; Nagy, T.R. Relationships between Rodent White Adipose Fat Pads and Human White Adipose Fat Depots. Front. Nutr. 2016, 3, 10. [Google Scholar] [CrossRef]
- Sengupta, P. The Laboratory Rat: Relating Its Age With Human’s. Int. J. Prev. Med. 2013, 4, 624. [Google Scholar]
- Swarbrick, M.; Zhou, H.; Seibel, M. Local and Systemic Effects of Glucocorticoids on Metabolism: New Lessons from Animal Models. Eur. J. Endocrinol. 2021, 185, R113–R129. [Google Scholar] [CrossRef]
- Velasco, A.; Huerta, I.; G-Granda, T.; Cachero, T.G.; Menéndez, E.; Marin, B. Circadian Rhythms of Plasma Corticosterone at Different Times after Induction of Diabetes. Responses to Corticoadrenal Stimulation in Light and Dark Phases. Life Sci. 1993, 52, 965–974. [Google Scholar] [CrossRef]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the Concentration of Low-Density Lipoprotein Cholesterol in Plasma, without Use of the Preparative Ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef]
- Wallace, T.M.; Levy, J.C.; Matthews, D.R. Use and Abuse of HOMA Modeling. Diabetes Care 2004, 27, 1487–1495. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Batista, J.L.; Neves, R.X.; Peres, S.B.; Yamashita, A.S.; Shida, C.S.; Farmer, S.R.; Seelaender, M. Heterogeneous Time-Dependent Response of Adipose Tissue during the Development of Cancer Cachexia. J. Endocrinol. 2012, 215, 363–373. [Google Scholar] [CrossRef]
- Rodbell, M. Metabolism of Isolated Fat Cells. I. Effects of Hormones on Glucose. J. Biol. Chem. 1964, 239, 375–380. [Google Scholar] [CrossRef]
- Di Girolamo, M.; Mendlinger, S.; Fertig, J.W. A Simple Method to Determine Fat Cell Size and Number in Four Mammalian Species. Am. J. Physiol. 1971, 221, 850–858. [Google Scholar] [CrossRef]
- De Oliveira Caminhotto, R.; Andreotti, S.; Komino, A.C.M.; de Fatima Silva, F.; Sertié, R.A.L.; Christoffolete, M.A.; Reis, G.B.; Lima, F.B. Physiological Concentrations of β-Hydroxybutyrate Do Not Promote Adipocyte Browning. Life Sci. 2019, 232, 116683. [Google Scholar] [CrossRef]
- Sertié, R.A.L.; Andreotti, S.; Proença, A.R.G.; Campaña, A.B.; Lima, F.B. Fat Gain with Physical Detraining Is Correlated with Increased Glucose Transport and Oxidation in Periepididymal White Adipose Tissue in Rats. Braz. J. Med. Biol. Res. 2015, 48, 650–653. [Google Scholar] [CrossRef]
- Sertie, R.A.L.; Curi, R.; Oliveira, A.C.; Andreotti, S.; Caminhotto, R.O.; de Lima, T.M.; Proença, A.R.G.; Reis, G.B.; Lima, F.B. The Mechanisms Involved in the Increased Adiposity Induced by Interruption of Regular Physical Exercise Practice. Life Sci. 2019, 222, 103–111. [Google Scholar] [CrossRef]
- Liu, D.; Ahmet, A.; Ward, L.; Krishnamoorthy, P.; Mandelcorn, E.D.; Leigh, R.; Brown, J.P.; Cohen, A.; Kim, H. A Practical Guide to the Monitoring and Management of the Complications of Systemic Corticosteroid Therapy. Allergy Asthma Clin. Immunol. 2013, 9, 30. [Google Scholar] [CrossRef]
- Fadel, R.; Morrison, A.R.; Vahia, A.; Smith, Z.R.; Chaudhry, Z.; Bhargava, P.; Miller, J.; Kenney, R.M.; Alangaden, G.; Ramesh, M.S.; et al. Early Short-Course Corticosteroids in Hospitalized Patients with COVID-19. Clin. Infect. Dis. 2020, 71, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Paragliola, R.M.; Papi, G.; Pontecorvi, A.; Corsello, S.M. Treatment with Synthetic Glucocorticoids and the Hypothalamus-Pituitary-Adrenal Axis. Int. J. Mol. Sci. 2017, 18, 2201. [Google Scholar] [CrossRef] [PubMed]
- McKay, L.I.; Cidlowski, J.A. Corticosteroids. In Holland-Frei Cancer Medicine, 5th ed.; Bast, R.J., Kufe, D., Pollock, R., Eds.; Decker: Hamilton, ON, Canada, 2000; Chapter 54. [Google Scholar]
- Scaroni, C.; Zilio, M.; Foti, M.; Boscaro, M. Glucose Metabolism Abnormalities in Cushing Syndrome: From Molecular Basis to Clinical Management. Endocr. Rev. 2017, 38, 189–219. [Google Scholar] [CrossRef] [PubMed]
- Mir, N.; Chin, S.A.; Riddell, M.C.; Beaudry, J.L. Genomic and Non-Genomic Actions of Glucocorticoids on Adipose Tissue Lipid Metabolism. Int. J. Mol. Sci. 2021, 22, 8503. [Google Scholar] [CrossRef]
- Zhao, L.F.; Iwasaki, Y.; Zhe, W.; Nishiyama, M.; Taguchi, T.; Tsugita, M.; Kambayashi, M.; Hashimoto, K.; Terada, Y. Hormonal Regulation of Acetyl-CoA Carboxylase Isoenzyme Gene Transcription. Endocr. J. 2010, 57, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Dolinsky, V.W.; Douglas, D.N.; Lehner, R.; Vance, D.E. Regulation of the Enzymes of Hepatic Microsomal Triacylglycerol Lipolysis and Re-Esterification by the Glucocorticoid Dexamethasone. Biochem. J. 2004, 378, 967–974. [Google Scholar] [CrossRef]
- Bowles, N.P.; Karatsoreos, I.N.; Li, X.; Vemuri, V.K.; Wood, J.A.; Li, Z.; Tamashiro, K.L.K.; Schwartz, G.J.; Makriyannis, A.M.; Kunos, G.; et al. A Peripheral Endocannabinoid Mechanism Contributes to Glucocorticoid-Mediated Metabolic Syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 285–290. [Google Scholar] [CrossRef]
- Christ-Crain, M.; Kola, B.; Lolli, F.; Fekete, C.; Seboek, D.; Wittmann, G.; Feltrin, D.; Igreja, S.C.; Ajodha, S.; Harvey-White, J.; et al. AMP-Activated Protein Kinase Mediates Glucocorticoid-Induced Metabolic Changes: A Novel Mechanism in Cushing’s Syndrome. FASEB J. 2008, 22, 1672–1683. [Google Scholar] [CrossRef]
- Lee, S.W.; Son, J.Y.; Kim, J.M.; Hwang, S.S.; Han, J.S.; Heo, N.J. Body Fat Distribution Is More Predictive of All-Cause Mortality than Overall Adiposity. Diabetes Obes. Metab. 2018, 20, 141–147. [Google Scholar] [CrossRef]
- Schoettl, T.; Fischer, I.P.; Ussar, S. Heterogeneity of Adipose Tissue in Development and Metabolic Function. J. Exp. Biol. 2018, 221, jeb162958. [Google Scholar] [CrossRef]
- Pleasure, D.E.; Walsh, G.O.; Engel, W.K. Atrophy of Skeletal Muscle in Patients With Cushing’s Syndrome. Arch. Neurol. 1970, 22, 118–125. [Google Scholar] [CrossRef]
- Ferraù, F.; Korbonits, M. Metabolic Comorbidities in Cushing’s Syndrome. Eur. J. Endocrinol. 2015, 173, M133–M157. [Google Scholar] [CrossRef]
- Fox, C.S.; Massaro, J.M.; Hoffmann, U.; Pou, K.M.; Maurovich-Horvat, P.; Liu, C.Y.; Vasan, R.S.; Murabito, J.M.; Meigs, J.B.; Cupples, L.A.; et al. Abdominal Visceral and Subcutaneous Adipose Tissue Compartments. Circulation 2007, 116, 39–48. [Google Scholar] [CrossRef]
- Palmer, B.F.; Clegg, D.J. The Sexual Dimorphism of Obesity. Mol. Cell. Endocrinol. 2015, 402, 113–119. [Google Scholar] [CrossRef]
- Smith, S.R.; Lovejoy, J.C.; Greenway, F.; Ryan, D.; De Jonge, L.; De La Bretonne, J.; Volafova, J.; Bray, G.A. Contributions of Total Body Fat, Abdominal Subcutaneous Adipose Tissue Compartments, and Visceral Adipose Tissue to the Metabolic Complications of Obesity. Metab. -Clin. Exp. 2001, 50, 425–435. [Google Scholar] [CrossRef]
- Lee, M.J.; Pramyothin, P.; Karastergiou, K.; Fried, S.K. Deconstructing the Roles of Glucocorticoids in Adipose Tissue Biology and the Development of Central Obesity. Biochim. Biophys. Acta -Mol. Basis Dis. 2014, 1842, 473–481. [Google Scholar] [CrossRef]
- Andersson, D.P.; Hogling, D.E.; Thorell, A.; Toft, E.; Qvisth, V.; Näslund, E.; Thörne, A.; Wirén, M.; Löfgren, P.; Hoffstedt, J.; et al. Changes in Subcutaneous Fat Cell Volume and Insulin Sensitivity after Weight Loss. Diabetes Care 2014, 37, 1831–1836. [Google Scholar] [CrossRef]
- Hoffstedt, J.; Arner, E.; Wahrenberg, H.; Andersson, D.P.; Qvisth, V.; Löfgren, P.; Rydén, M.; Thörne, A.; Wirén, M.; Palmér, M.; et al. Regional Impact of Adipose Tissue Morphology on the Metabolic Profile in Morbid Obesity. Diabetologia 2010, 53, 2496–2503. [Google Scholar] [CrossRef]
- Bahceci, M.; Gokalp, D.; Bahceci, S.; Tuzcu, A.; Atmaca, S.; Arikan, S. The Correlation between Adiposity and Adiponectin, TNF-Alpha, IL-6 and High Sensitivity CRP Protein Levels. J. Endocrinol. Investig. 2007, 30, 210–214. [Google Scholar] [CrossRef]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between Adipocyte Size and Adipokine Expression and Secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Hannesian, J.P.O.; Marco, C.C.; Mckee, L.J.; Bauer, T.L.; et al. Serum Immunoreactive-Leptin Concentrations in Normal-Weight and Obese Humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; He, J.; Jiang, H.; Zu, L.; Zhai, W.; Pu, S.; Xu, G. Direct Effect of Glucocorticoids on Lipolysis in Adipocytes. Mol. Endocrinol. 2009, 23, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Arnaldi, G.; Scandali, V.M.; Trementino, L.; Cardinaletti, M.; Appolloni, G.; Boscaro, M. Pathophysiology of Dyslipidemia in Cushing’s Syndrome. Neuroendocrinology 2010, 92, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.A.; Lee, D.P. Enzymes of Triacylglycerol Synthesis and Their Regulation. Prog. Lipid Res. 2004, 43, 134–176. [Google Scholar] [CrossRef]
- Proença, A.R.G.; Sertié, R.A.L.; Oliveira, A.C.; Campaña, A.B.; Caminhotto, R.O.; Chimin, P.; Lima, F.B. New Concepts in White Adipose Tissue Physiology. Braz. J. Med. Biol. Res. 2014, 47, 192–205. [Google Scholar] [CrossRef] [Green Version]
- Lafontan, M. Advances in Adipose Tissue Metabolism. Int. J. Obes. 2008, 32, S39–S51. [Google Scholar] [CrossRef]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients 2015, 7, 9453–9474. [Google Scholar] [CrossRef]
- Nye, C.; Kim, J.; Kalhan, S.C.; Hanson, R.W. Reassessing Triglyceride Synthesis in Adipose Tissue. Trends Endocrinol. Metab. 2008, 19, 356–361. [Google Scholar] [CrossRef]
- Nye, C.K.; Hanson, R.W.; Kalhan, S.C. Glyceroneogenesis Is the Dominant Pathway for Triglyceride Glycerol Synthesis in Vivo in the Rat. J. Biol. Chem. 2008, 283, 27565–27574. [Google Scholar] [CrossRef]
- Newsholme, E.A.; Dimitriadis, G. Integration of Biochemical and Physiologic Effects of Insulin on Glucose Metabolism. Exp. Clin. Endocrinol. Diabetes 2001, 109, 122–134. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Maratou, E. Insulin Effects in Muscle and Adipose Tissue. Diabetes Res. Clin. Pract. 2011, 93, 52–59. [Google Scholar] [CrossRef]
- Kobayashi, N.; Ueki, K.; Okazaki, Y.; Iwane, A.; Kubota, N.; Ohsugi, M. Blockade of Class IB Phosphoinositide-3 Kinase Ameliorates Obesity-Induced in Fl Ammation and Insulin Resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 5753–5758. [Google Scholar] [CrossRef]
- Shimizu, I.; Walsh, K. The Whitening of Brown Fat and Its Implications for Weight Management in Obesity. Curr. Obes. Rep. 2015, 4, 224–229. [Google Scholar] [CrossRef]
- Hill, B.G. Insights into an Adipocyte Whitening Program. Adipocyte 2015, 4, 75–80. [Google Scholar] [CrossRef]
- Bel, J.S.; Tai, T.C.; Khaper, N.; Lees, S.J. Chronic Glucocorticoid Exposure Causes Brown Adipose Tissue Whitening, Alters Whole-Body Glucose Metabolism and Increases Tissue Uncoupling Protein-1. Physiol. Rep. 2022, 10, e15292. [Google Scholar] [CrossRef]
- Mousovich-Neto, F.; Matos, M.S.; Costa, A.C.R.; de Melo Reis, R.A.; Atella, G.C.; Miranda-Alves, L.; Carvalho, D.P.; Ketzer, L.A.; Corrêa da Costa, V.M. Brown Adipose Tissue Remodelling Induced by Corticosterone in Male Wistar Rats. Exp. Physiol. 2019, 104, 514–528. [Google Scholar] [CrossRef]
- Poggioli, R.; Ueta, C.B.; Drigo, R.A.E.; Castillo, M.; Fonseca, T.L.; Bianco, A.C. Dexamethasone Reduces Energy Expenditure and Increases Susceptibility to Diet-Induced Obesity in Mice. Obesity 2013, 21, 415–420. [Google Scholar] [CrossRef]
- Scotney, H.; Symonds, M.E.; Law, J.; Budge, H.; Sharkey, D.; Manolopoulos, K.N. Glucocorticoids Modulate Human Brown Adipose Tissue Thermogenesis in Vivo. Metabolism 2017, 70, 125–132. [Google Scholar] [CrossRef]
- Van Den Beukel, J.C.; Boon, M.R.; Steenbergen, J.; Rensen, P.C.N.; Meijer, O.C.; Themmen, A.P.N.; Grefhorst, A. Cold Exposure Partially Corrects Disturbances in Lipid Metabolism in a Male Mouse Model of Glucocorticoid Excess. Endocrinology 2015, 156, 4115–4128. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. The Biochemistry of an Inefficient Tissue: Brown Adipose Tissue. Essays Biochem. 1985, 20, 110–164. [Google Scholar]
- Murano, I.; Barbatelli, G.; Giordano, A.; Cinti, S. Noradrenergic Parenchymal Nerve Fiber Branching after Cold Acclimatisation Correlates with Brown Adipocyte Density in Mouse Adipose Organ. J. Anat. 2009, 214, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Soumano, K.; Desbiens, S.; Rabelo, R.; Bakopanos, E.; Camirand, A.; Silva, J.E. Glucocorticoids Inhibit the Transcriptional Response of the Uncoupling Protein-1 Gene to Adrenergic Stimulation in a Brown Adipose Cell Line. Mol. Cell. Endocrinol. 2000, 165, 7–15. [Google Scholar] [CrossRef]
- McNeill, B.T.; Morton, N.M.; Stimson, R.H. Substrate Utilization by Brown Adipose Tissue: What’s Hot and What’s Not? Front. Endocrinol. 2020, 11, 571659. [Google Scholar] [CrossRef] [PubMed]
- Berbeé, J.F.P.; Boon, M.R.; Khedoe, P.P.S.J.; Bartelt, A.; Schlein, C.; Worthmann, A.; Kooijman, S.; Hoeke, G.; Mol, I.M.; John, C.; et al. Brown Fat Activation Reduces Hypercholesterolaemia and Protects from Atherosclerosis Development. Nat. Commun. 2015, 6, 6356. [Google Scholar] [CrossRef]
- Sanchez-Gurmaches, J.; Tang, Y.; Jespersen, N.Z.; Wallace, M.; Martinez Calejman, C.; Gujja, S.; Li, H.; Edwards, Y.J.K.; Wolfrum, C.; Metallo, C.M.; et al. Brown Fat AKT2 Is a Cold-Induced Kinase That Stimulates ChREBP-Mediated De Novo Lipogenesis to Optimize Fuel Storage and Thermogenesis. Cell Metab. 2018, 27, 195–209.e6. [Google Scholar] [CrossRef]
- Petersen, C.; Nielsen, M.D.; Andersen, E.S.; Basse, A.L.; Isidor, M.S.; Markussen, L.K.; Viuff, B.M.; Lambert, I.H.; Hansen, J.B.; Pedersen, S.F. MCT1 and MCT4 Expression and Lactate Flux Activity Increase during White and Brown Adipogenesis and Impact Adipocyte Metabolism. Sci. Rep. 2017, 7, 13101. [Google Scholar] [CrossRef] [Green Version]
- Saggerson, E.D.; McAllister, T.W.J.; Baht, H.S. Lipogenesis in Rat Brown Adipocytes. Effects of Insulin and Noradrenaline, Contributions from Glucose and Lactate as Precursors and Comparisons with White Adipocytes. Biochem. J. 1988, 251, 701–709. [Google Scholar] [CrossRef]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; Sutton, S.W.; Li, X.; Su, J.Y.; Mirzadegan, T.; et al. Lactate Inhibits Lipolysis in Fat Cells through Activation of an Orphan G-Protein-Coupled Receptor, GPR81. J. Biol. Chem. 2009, 284, 2811–2822. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequences (5′-3′) | |
|---|---|---|
| Forward | Reverse | |
| Actb | AGCCTGGATGGCTACGTACA | CCTCTGAACCCTAAGGCCAA |
| Agrp | AGGACTCGTGCAGCCTTACAC | GCAGAGGTGCTAGATCCACAGAA |
| Cartpt | CCGCCTTGGCAGCTCCTT | CCGAGCCCTGGACATCTACT |
| Crh | CCGATAATCTCCATCAGTTTCCTG | TGGATCTCACCTTCCACCTTCTG |
| Gapdh | CCGTTCAGCTCTGGGATGAC | GGGCAGCCCAGAACATCAT |
| Hcrp | AGGGAGAGGCAATCCGGAGAG | GCGGCCTCAGACTCCT |
| Npy | CCCTCAGCCAGAATGCCCAA | CCGCCCGCCATGATGCTAGGTA |
| Lepr | CCAGAAGAAGAGGACCAAATATCAC | ACTTAATTTCCAAAAGCCTGAAACA |
| Ppia | TATCTGCACTGCCAAGACTGAGT | CTTCTTGCTGGTCTTGCCATTCC |
| Pmch | CTTCTACGTTCCTGATGGACTT | ATGCTGGCCTTTTCTTTGTTT |
| Pomc | GCAAGCCAGCAGGTTGCT | ATAGACGTGTGGAGCTGGTGC |
| Tnf | GGTTGTCTTTGAGATCCATGC | TCTCAAAACTCGAGTGACAAGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Fatima Silva, F.; Komino, A.C.M.; Andreotti, S.; Boltes Reis, G.; Caminhotto, R.O.; Landgraf, R.G.; de Souza, G.O.; Sertié, R.A.L.; Collins, S.; Donato, J., Jr.; et al. Dexamethasone-Induced Adipose Tissue Redistribution and Metabolic Changes: Is Gene Expression the Main Factor? An Animal Model of Chronic Hypercortisolism. Biomedicines 2022, 10, 2328. https://doi.org/10.3390/biomedicines10092328
de Fatima Silva F, Komino ACM, Andreotti S, Boltes Reis G, Caminhotto RO, Landgraf RG, de Souza GO, Sertié RAL, Collins S, Donato J Jr., et al. Dexamethasone-Induced Adipose Tissue Redistribution and Metabolic Changes: Is Gene Expression the Main Factor? An Animal Model of Chronic Hypercortisolism. Biomedicines. 2022; 10(9):2328. https://doi.org/10.3390/biomedicines10092328
Chicago/Turabian Stylede Fatima Silva, Flaviane, Ayumi Cristina Medeiros Komino, Sandra Andreotti, Gabriela Boltes Reis, Rennan Oliveira Caminhotto, Richardt Gama Landgraf, Gabriel Orefice de Souza, Rogerio Antonio Laurato Sertié, Sheila Collins, Jose Donato, Jr., and et al. 2022. "Dexamethasone-Induced Adipose Tissue Redistribution and Metabolic Changes: Is Gene Expression the Main Factor? An Animal Model of Chronic Hypercortisolism" Biomedicines 10, no. 9: 2328. https://doi.org/10.3390/biomedicines10092328
APA Stylede Fatima Silva, F., Komino, A. C. M., Andreotti, S., Boltes Reis, G., Caminhotto, R. O., Landgraf, R. G., de Souza, G. O., Sertié, R. A. L., Collins, S., Donato, J., Jr., & Bessa Lima, F. (2022). Dexamethasone-Induced Adipose Tissue Redistribution and Metabolic Changes: Is Gene Expression the Main Factor? An Animal Model of Chronic Hypercortisolism. Biomedicines, 10(9), 2328. https://doi.org/10.3390/biomedicines10092328