
Two-Time Multiplexed Targeted Next-Generation Sequencing Might Help the Implementation of Germline Screening Tools for Myelodysplastic Syndromes/Hematologic Neoplasms
 , , , , ,
, , , , , 
Abstract
:1. Introduction
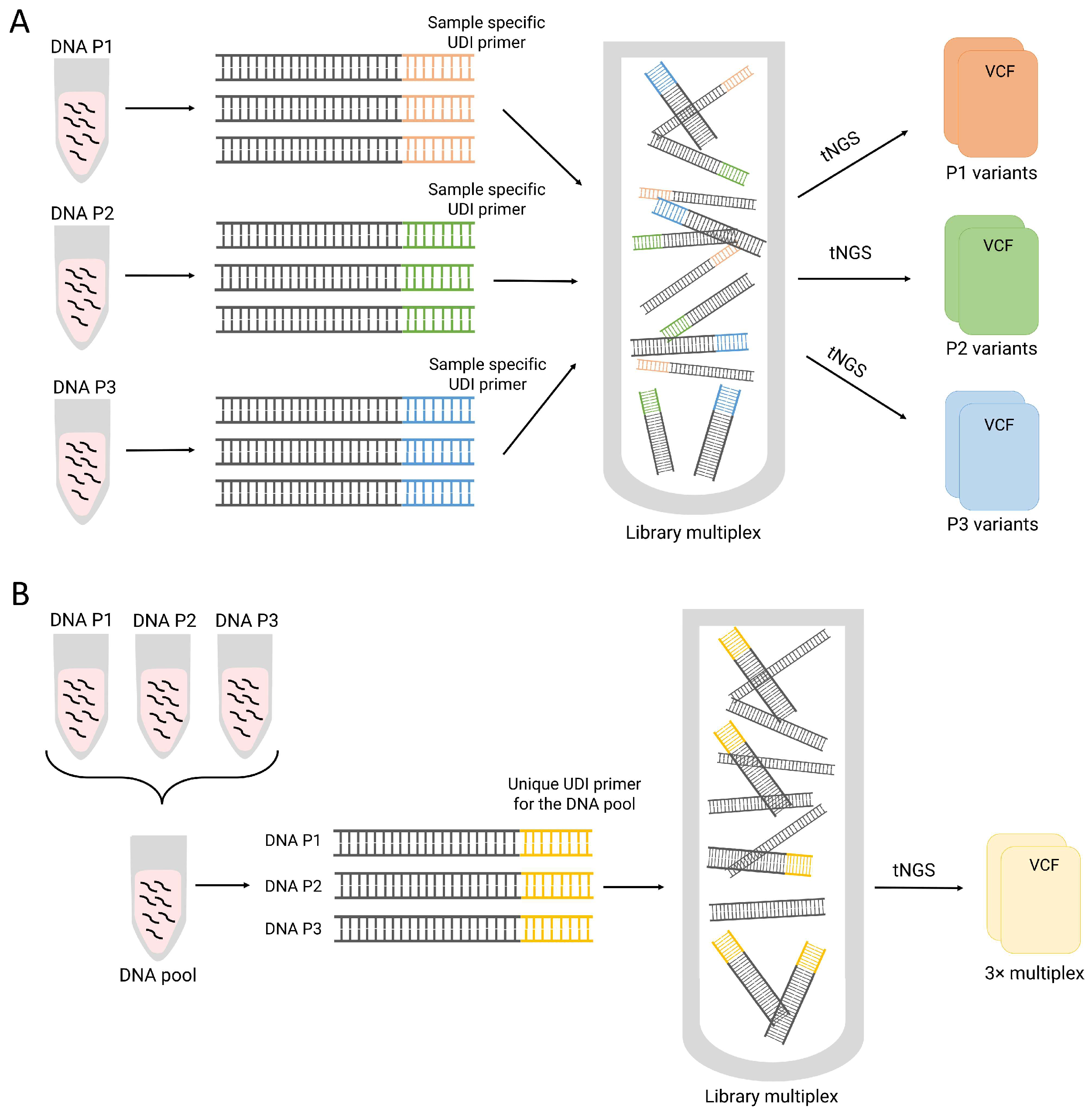
2. Materials and Methods
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mahmoodpour, M.; Kiasari, B.A.; Karimi, M.; Abroshan, A.; Shamshirian, D.; Hosseinalizadeh, H.; Delavari, A.; Mirzei, H. Paper-Based Biosensors as Point-of-Care Diagnostic Devices for the Detection of Cancers: A Review of Innovative Techniques and Clinical Applications. Front. Oncol. 2023, 13, 1131435. [Google Scholar] [CrossRef] [PubMed]
- Nybakken, G.E.; Bagg, A. The Genetic Basis and Expanding Role of Molecular Analysis in the Diagnosis, Prognosis, and Therapeutic Design for Myelodysplastic Syndromes. J. Mol. Diagn. 2014, 16, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef]
- Valent, P. ICUS, IDUS, CHIP and CCUS: Diagnostic Criteria, Separation from MDS and Clinical Implications. Pathobiology 2019, 86, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Pich, O.; Reyes-Salazar, I.; Gonzalez-Perez, A.; Lopez-Bigas, N. Discovering the Drivers of Clonal Hematopoiesis. Nat. Commun. 2022, 13, 4267. [Google Scholar] [CrossRef] [PubMed]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer Therapy Shapes the Fitness Landscape of Clonal Hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; Roboz, G.J. Clonal Hematopoiesis and Therapy Related MDS/AML. Best Pract. Res. Clin. Haematol. 2019, 32, 13–23. [Google Scholar] [CrossRef]
- Furutani, E.; Shimamura, A. Germline Genetic Predisposition to Hematologic Malignancy. J. Clin. Oncol. 2017, 35, 1018–1028. [Google Scholar] [CrossRef]
- Babushok, D.V.; Bessler, M.; Olson, T.S. Genetic Predisposition to Myelodysplastic Syndrome and Acute Myeloid Leukemia in Children and Young Adults. Leuk. Lymphoma 2016, 57, 520–536. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Bagg, A.; Hasserjian, R.P.; DiNardo, C.D.; Godley, L.A.; Iacobucci, I.; Jaiswal, S.; Malcovati, L.; Vannucchi, A.M.; Patel, K.P.; et al. Genomic Profiling for Clinical Decision Making in Myeloid Neoplasms and Acute Leukemia. Blood 2022, 140, 2228–2247. [Google Scholar] [CrossRef]
- Klco, J.M.; Mullighan, C.G. Advances in Germline Predisposition to Acute Leukaemias and Myeloid Neoplasms. Nat. Rev. Cancer 2021, 21, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Churpek, J.E. Familial Myelodysplastic Syndrome/Acute Myeloid Leukemia. Best. Pract. Res. Clin. Haematol. 2017, 30, 287–289. [Google Scholar] [CrossRef]
- Baliakas, P.; Tesi, B.; Wartiovaara-Kautto, U.; Stray-Pedersen, A.; Friis, L.S.; Dybedal, I.; Hovland, R.; Jahnukainen, K.; Raaschou-Jensen, K.; Ljungman, P.; et al. Nordic Guidelines for Germline Predisposition to Myeloid Neoplasms in Adults: Recommendations for Genetic Diagnosis, Clinical Management and Follow-Up. Hemasphere 2019, 3, e321. [Google Scholar] [CrossRef] [PubMed]
- Schlegelberger, B.; Mecucci, C.; Wlodarski, M. Review of Guidelines for the Identification and Clinical Care of Patients with Genetic Predisposition for Hematological Malignancies. Fam. Cancer 2021, 20, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Feurstein, S.K.; Trottier, A.M.; Estrada-Merly, N.; Pozsgai, M.J.; McNeely, K.E.; Drazer, M.W.; Ruhle, B.; Sadera, K.; Koppayi, A.L.; Scott, B.L.; et al. Germline Predisposition Variants Occur in Myelodysplastic Syndrome Patients of All Ages. Blood 2022, 140, 2533–2548. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Zawit, M.; Durrani, J.; Shen, W.; Bahaj, W.; Kewan, T.; Ponvilawan, B.; Mori, M.; Meggendorfer, M.; Gurnari, C.; et al. Significance of Hereditary Gene Alterations for the Pathogenesis of Adult Bone Marrow Failure versus Myeloid Neoplasia. Leukemia 2022, 36, 2827–2834. [Google Scholar] [CrossRef]
- Calvete, O.; Mestre, J.; Durmaz, A.; Gurnari, C.; Maciejewski, J.P.; Solé, F. Are the Current Guidelines for Identification of Myelodysplastic Syndrome with Germline Predisposition Strong Enough? Br. J. Haematol. 2023, 201, e5–e11. [Google Scholar] [CrossRef]
- Bersanelli, M.; Travaglino, E.; Meggendorfer, M.; Matteuzzi, T.; Sala, C.; Mosca, E.; Chiereghin, C.; di Nanni, N.; Gnocchi, M.; Zampini, M.; et al. Classification and Personalized Prognostic Assessment on the Basis of Clinical and Genomic Features in Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1223–1233. [Google Scholar] [CrossRef]
- Tawana, K.; Brown, A.L.; Churpek, J.E. Integrating Germline Variant Assessment into Routine Clinical Practice for Myelodysplastic Syndrome and Acute Myeloid Leukaemia: Current Strategies and Challenges. Br. J. Haematol. 2022, 196, 1293–1310. [Google Scholar] [CrossRef]
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Starý, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; van den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, Clinical Characteristics, and Prognosis of GATA2-Related Myelodysplastic Syndromes in Children and Adolescents. Blood 2016, 127, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Calvete, O.; Mestre, J.; Jerez, A.; Solé, F. The Secondary Myelodysplastic Neoplasms (MDS) Jigsaw. Cancers 2023, 15, 1483. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.L.; Shimamura, A. Genetic Predisposition to MDS: Clinical Features and Clonal Evolution. Blood 2019, 133, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Palomino-Echeverría, S.; Vázquez, I.; Alfonso-Piérola, A.; José Larrayoz, M.; Aguilera-Díaz, A.; Ariceta, B.; Daniela Urribarri, A.; Mañú, A.; Blasco-Iturri, Z.; Prósper, F.; et al. Predisposición a Neoplasias Mieloides: El Nuevo Desafío En La Consulta de Hematología. Genética Médica Genómica 2020, 4, 33–46. [Google Scholar]
- Keel, S.B.; Scott, A.; Sanchez-Bonilla, M.; Ho, P.A.; Gulsuner, S.; Pritchard, C.C.; Abkowitz, J.L.; King, M.-C.; Walsh, T.; Shimamura, A. Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients. Haematologica 2016, 101, 1343–1350. [Google Scholar] [CrossRef]
- Babushok, D.V.; Bessler, M. Genetic Predisposition Syndromes: When Should They Be Considered in the Work-up of MDS? Best. Pract. Res. Clin. Haematol. 2015, 28, 55–68. [Google Scholar] [CrossRef]


{kind=link}
| Filtered Variants | Candidate Variants | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient | Variants | tNGS | 3× M | (%) | 4× M | (%) | tNGS | 3× M | 4× M |
| 1 | 232 | 41 | 24 | 58.54 | 20 | 48.78 | 1 | 1 | 1 |
| 2 | 229 | 30 | 20 | 66.67 | 14 | 46.67 | 0 | 0 | 0 |
| 3 | 187 | 30 | 18 | 60.00 | 13 | 43.33 | 5 | 5 | 5 |
| Total (different variants) | 350 | 75 | 40 | 53.33 | 30 | 40.00 | 6 | 6 | 6 |
| VAF Range | Patient | tNGS | 3× M | (%) | 4× M | (%) |
|---|---|---|---|---|---|---|
| (<10%) | 1 | 30 | 13 | 43.33 | 13 | 43.33 |
| 2 | 24 | 14 | 58.33 | 13 | 54.17 | |
| 3 | 17 | 6 | 35.29 | 4 | 23.53 | |
| TOTAL | 71 | 33 | 46.48 | 20 | 28.17 | |
| (10–30%) | 1 | 0 | 0 | NA | 0 | NA |
| 2 | 0 | 0 | NA | 0 | NA | |
| 3 | 1 | 1 | 100.00 | 1 | 100.00 | |
| TOTAL | 1 | 1 | 100.00 | 1 | 100.00 | |
| (>30%) | 1 | 11 | 11 | 100.00 | 11 | 100.00 |
| 2 | 6 | 6 | 100.00 | 6 | 100.00 | |
| 3 | 12 | 11 | 91.67 | 11 | 91.67 | |
| TOTAL | 29 | 28 | 96.55 | 28 | 96.55 |
| Patient | 3× Multiplex | 4× Multiplex | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Variant | 1 | 2 | 3 | Expected VAF | Observed VAF | VAF Difference | Expected VAF | Observed VAF | VAF Difference |
| TET2:p.C1135W | 3.8 | 0 | 0 | 1.27 | 1.90 | 0.63 | 0.95 | ND | ND |
| TET2:p.L1819 * | 4.9 | 0 | 0 | 1.63 | 2.10 | 0.47 | 1.23 | ND | ND |
| TET2:p.P401del | 0 | 0.7 | 0 | 0.24 | 0.50 | 0.26 | 0.18 | ND | ND |
| TET2:p.G613W | 0 | 1.4 | 0 | 0.47 | 1.10 | 0.63 | 0.35 | ND | ND |
| TET2:p.G614E | 0 | 1.3 | 0 | 0.43 | 0.94 | 0.50 | 0.33 | ND | ND |
| TET2:p.L615P | 0 | 1.3 | 0 | 0.43 | 0.93 | 0.49 | 0.33 | ND | ND |
| TET2:p.P616P | 0 | 1.3 | 0 | 0.43 | 0.95 | 0.51 | 0.33 | ND | ND |
| DDX41:p.R400R | 46.4 | 47.8 | 0 | 31.40 | 49.10 | 17.70 | 23.55 | 60.50 | 36.95 † |
| CUX1:p.P1439P | 55.2 | 0 | 49.4 | 34.87 | 35.30 | 0.43 | 26.15 | 55.00 | 28.85 † |
| CUX1:p.S1448del | 3.8 | 1.9 | 2.3 | 2.67 | 3.90 | 1.23 | 2.00 | 3.50 | 1.50 |
| CUX1:p.R558Q | 2.6 | 1.5 | 0 | 1.37 | 0.96 | −0.40 | 1.03 | 2.10 | 1.08 |
| EZH2:p.E401Kfs*22 | 5.1 | 0 | 0 | 1.70 | 1.40 | −0.30 | 1.28 | 1.40 | 0.13 |
| EZH2:p.P132P | 53.6 | 0 | 0 | 17.87 | 16.80 | −1.07 | 13.40 | 11.80 | −1.60 |
| EZH2:p.M1? | 5.1 | 0 | 0 | 1.70 | 1.80 | 0.10 | 1.28 | ND | ND |
| EZH2:p.D185H | 0 | 0 | 99.9 | 33.30 | 25.90 | −7.40 | 24.98 | 18.40 | −6.58 |
| ANKRD26:p.Q20R | 51.6 | 50.7 | 49.7 | 50.67 | 48.10 | −2.57 | 38.00 | 50.20 | 12.20 |
| CBL:p.D460del | 3.7 | 3.9 | 3 | 3.53 | 3.20 | −0.33 | 2.65 | 3.40 | 0.75 |
| TP53:p.S366A | 51.2 | 0 | 0 | 17.07 | 18.90 | 1.83 | 12.80 | 13.00 | 0.2 |
| NF1:p.L234L | 99.9 | 99.8 | 49.3 | 83.00 | 86.30 | 3.30 | 62.25 | 89.50 | 27.25 † |
| CEBPA:p.T230T | 53.5 | 0 | 0 | 17.83 | 18.80 | 0.97 | 13.38 | 14.20 | 0.82 |
| CEBPA:p.P189del | 2.9 | 4 | 4.9 | 3.93 | 4.60 | 0.67 | 2.95 | 5.30 | 2.35 |
| CEBPA:p.G104del | 1.9 | 1.9 | 0 | 1.27 | 1.50 | 0.23 | 0.95 | 1.70 | 0.75 |
| ASXL1:p.Q428Sfs*10 | 6.3 | 0 | 0 | 2.10 | 1.50 | −0.60 | 1.58 | 1.50 | −0.08 |
| ASXL1:p.G645Vfs*58 | 2.8 | 2.4 | 1.7 | 2.30 | 2.60 | 0.30 | 1.73 | 2.80 | 1.08 |
| ASXL1:p.G646Wfs*12 | 1.2 | 1.7 | 35.4 | 12.77 | 11.80 | −0.97 | 9.58 | 7.80 | −1.775 |
| ASXL1:p.T655T | 49.1 | 0 | 0 | 16.37 | 16.00 | −0.37 | 12.28 | 12.50 | 0.23 |
| ASXL1:p.L1325F | 51 | 0 | 0 | 17.00 | 17.20 | 0.20 | 12.75 | 13.10 | 0.35 |
| BCOR:p.D420D | 99.9 | 46.3 | 99.6 | 81.93 | 78.60 | −3.33 | 61.45 | 67.30 | 5.85 |
| BCORL1:p.E1324del | 0.7 | 0.7 | 0 | 0.47 | 0.87 | 0.40 | 0.36 | 0.57 | 0.22 |
| JAK2:p.V617F | 0 | 7.5 | 0 | 2.50 | 1.70 | −0.80 | 1.88 | 1.80 | −0.08 |
| IDH2:p.V109V | 0 | 46.9 | 0 | 15.63 | 16.60 | 0.97 | 11.73 | 11.90 | 0.18 |
| NRAS:p.Q61H | 0 | 0 | 5.6 | 1.87 | 2.10 | 0.23 | 1.40 | ND | ND |
| NRAS:p.G13R | 0 | 0 | 19.5 | 6.50 | 4.00 | −2.50 | 4.88 | 3.40 | −1.48 |
| NRAS:p.G12C | 0 | 0 | 1.8 | 0.60 | 0.76 | 0.16 | 0.45 | ND | ND |
| GATA2:p.P5P | 0 | 46.9 | 0 | 15.63 | 17.90 | 2.27 | 11.73 | 14.50 | 2.78 |
| GATA2:p.R396Gfs*81 | 0 | 0 | 44.3 | 14.77 | 14.60 | −0.17 | 11.08 | 8.80 | −2.28 |
| SRSF2:p.P95H | 0 | 0 | 41.8 | 13.93 | 13.10 | −0.83 | 10.45 | 7.30 | −3.15 |
| SETBP1:p.D868N | 0 | 0 | 43.4 | 14.47 | 12.10 | −2.37 | 10.85 | 9.30 | −1.55 |
| SETBP1:p.H1206H | 0 | 0 | 50.6 | 16.87 | 12.10 | −4.77 | 12.65 | 10.00 | −2.65 |
| ZRSR2:p.P303P | 0 | 0 | 46.7 | 15.57 | 14.60 | −0.97 | 11.68 | 11.70 | 0.02 |
| Average of total negative VAF deviation | −1.75 | −2.12 | |||||||
| Average of total positive VAF deviation | 1.47 | 1.54 | |||||||
| Average VAF of candidate variants | 39.4 | 13.25 | 12.42 | −0.83 | 9.94 | 8.27 | −1.67 | ||
| Average of candidate variants negative VAF deviation | −1.37 | −2.05 | |||||||
| Average of candidate variants positive VAF deviation | 1.83 | 0.20 | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvete, O.; Mestre, J.; Risueño, R.M.; Manzanares, A.; Acha, P.; Xicoy, B.; Solé, F. Two-Time Multiplexed Targeted Next-Generation Sequencing Might Help the Implementation of Germline Screening Tools for Myelodysplastic Syndromes/Hematologic Neoplasms. Biomedicines 2023, 11, 3222. https://doi.org/10.3390/biomedicines11123222
Calvete O, Mestre J, Risueño RM, Manzanares A, Acha P, Xicoy B, Solé F. Two-Time Multiplexed Targeted Next-Generation Sequencing Might Help the Implementation of Germline Screening Tools for Myelodysplastic Syndromes/Hematologic Neoplasms. Biomedicines. 2023; 11(12):3222. https://doi.org/10.3390/biomedicines11123222
Chicago/Turabian StyleCalvete, Oriol, Julia Mestre, Ruth M. Risueño, Ana Manzanares, Pamela Acha, Blanca Xicoy, and Francesc Solé. 2023. "Two-Time Multiplexed Targeted Next-Generation Sequencing Might Help the Implementation of Germline Screening Tools for Myelodysplastic Syndromes/Hematologic Neoplasms" Biomedicines 11, no. 12: 3222. https://doi.org/10.3390/biomedicines11123222
APA StyleCalvete, O., Mestre, J., Risueño, R. M., Manzanares, A., Acha, P., Xicoy, B., & Solé, F. (2023). Two-Time Multiplexed Targeted Next-Generation Sequencing Might Help the Implementation of Germline Screening Tools for Myelodysplastic Syndromes/Hematologic Neoplasms. Biomedicines, 11(12), 3222. https://doi.org/10.3390/biomedicines11123222