
Eph Receptors in Cancer



Abstract
:1. Introduction of Eph Receptors
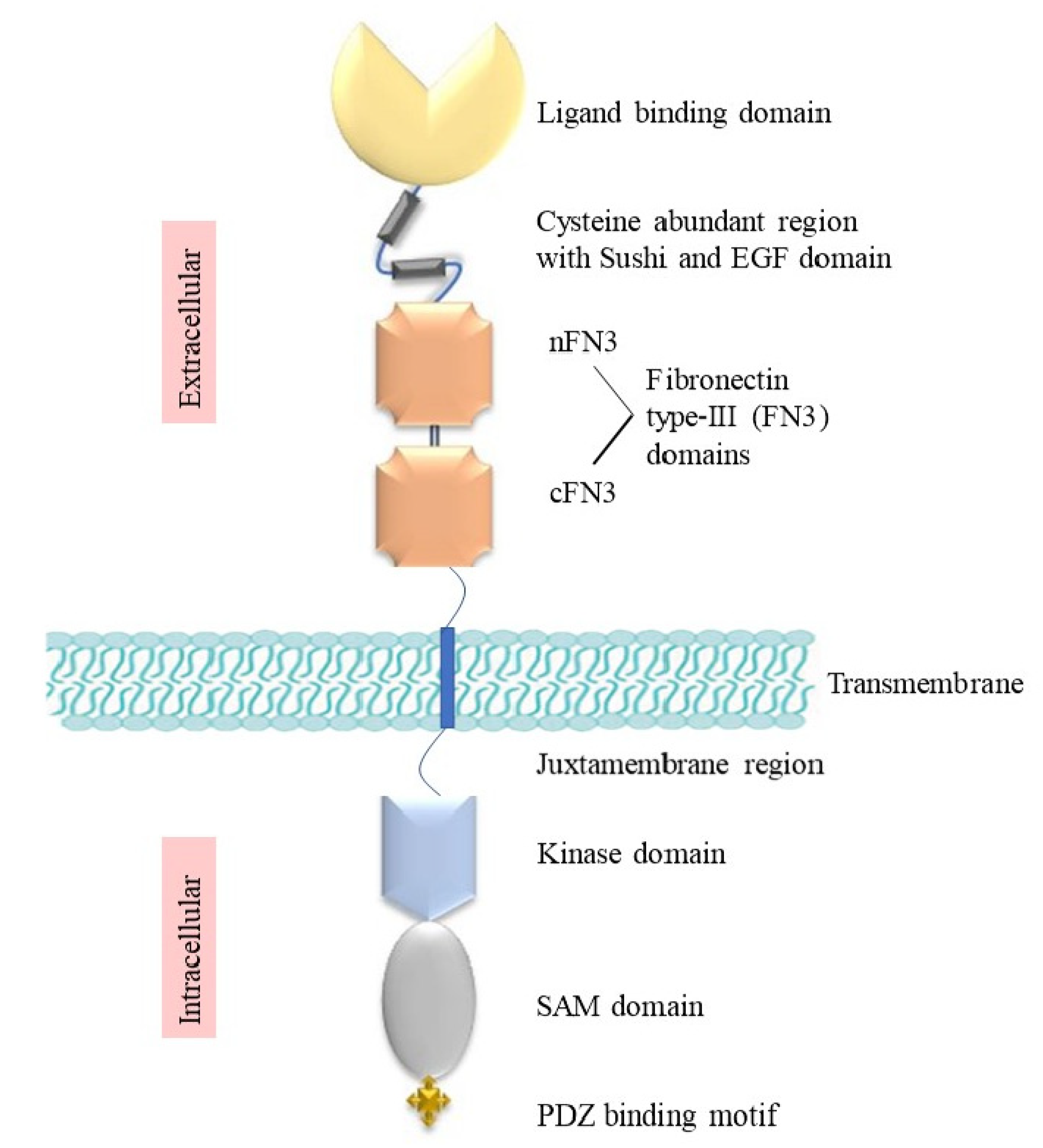
2. Structure and Signalling of Eph Receptors
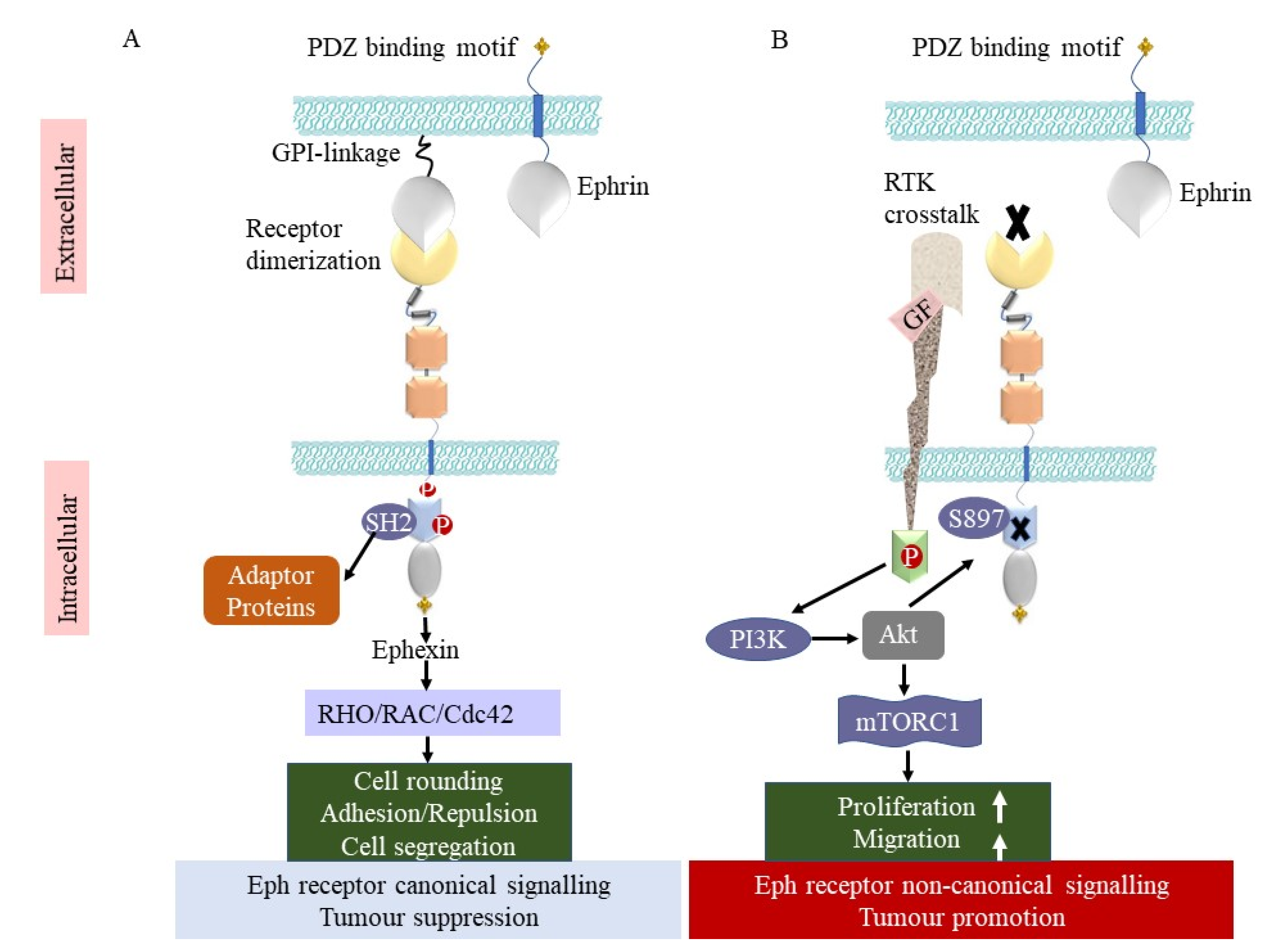
3. Bidirectional Signalling
4. Normal Function of Eph Receptors in Development and Adult Tissues
5. Ephs in Cancer
5.1. Tumour-Promoting Function

5.2. Tumour-Suppressing Function

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Cancer Type | Upregulated/ Downregulated | Tumour Promoting/Tumour Suppressing | mRNA/ Protein | Prognosis | Reference |
|---|---|---|---|---|---|---|
| EphA1 | Colorectal | Downregulated | Tumour suppressing | Both | Poor survival | [48] |
| EphA2 | Breast Pancreatic | Upregulated Upregulated | Tumour promoting Tumour promoting | Both mRNA | Poor survival Poor survival | [55,56,57] |
| EphA3 | Brain Colorectal Prostate | Upregulated Upregulated Upregulated | Tumour promoting Tumour promoting Tumour promoting | Both Protein Protein | Poor survival Poor survival Poor survival | [42,58] |
| EphA4 | Lung Breast | Upregulated Upregulated | Tumour suppressing Tumour promoting | Both mRNA | Increased survival Poor survival | [56,59] |
| EphA7 | Breast | Upregulated | Tumour promoting | mRNA | Poor survival | [56] |
| EphA10 | Breast | Upregulated | Tumour promoting | Protein | Poor survival | [60] |
| EphB2 | Breast Colorectal | Upregulated Downregulated | Tumour promoting Tumour suppressing | Protein mRNA | Poor survival Poor survival | [44,53] |
| EphB3 | Colorectal | Downregulated | Tumour suppressing | Both | Poor survival | [53,61] |
| EphB4 | Colorectal Breast | Downregulated Upregulated | Tumour suppressing Tumour promoting | Protein Both | Poor survival Poor survival | [44,51,56] |
| EphB6 | Breast | Upregulated | Tumour promoting | mRNA | Poor survival | [56] |
| Ephrin-B2 | Breast | Upregulated | Tumour suppressing | Protein | Increased survival | [50] |
5.3. Mutations
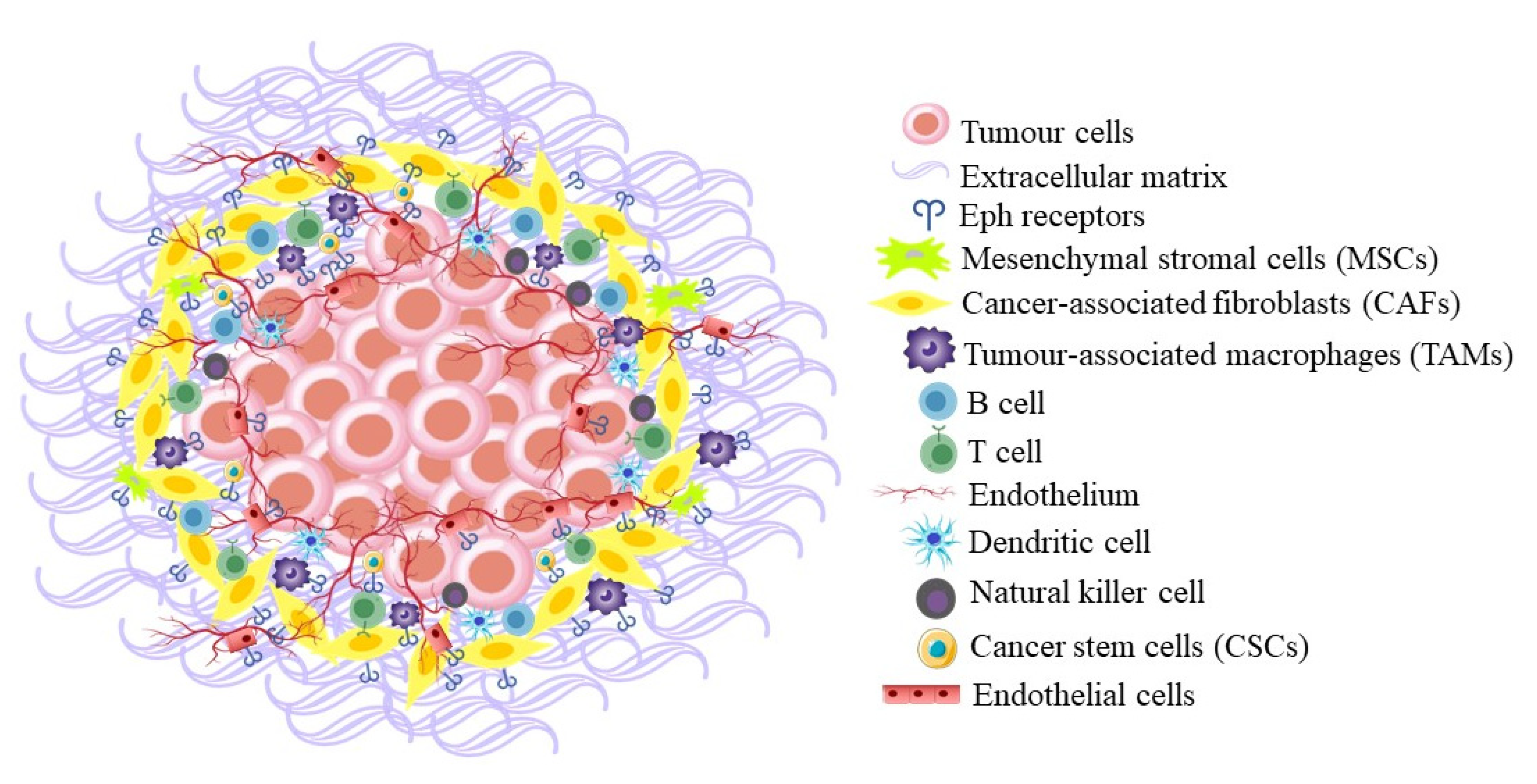
5.4. Roles in the Tumour Microenvironment
5.5. Mechanisms of Drug Resistance Mediated by Eph Receptors
5.6. Eph Receptors as Therapeutic Targets in Specific Cancer Types
5.6.1. Lung Cancer
5.6.2. Brain Cancer
5.6.3. Gastrointestinal Cancers
5.6.4. Breast Cancer
5.6.5. Prostate Cancer
5.6.6. Melanoma
6. EphA10 and EphB6 (Pseudokinases)
7. Therapeutic Strategies to Target Eph Receptors
7.1. Small-Molecule Inhibitors
7.2. Kinase Inhibitors
| Drug | Type | Target | Mechanism of Action (MOA) | Clinical Trial | Clinicaltrials.gov Identifier, Reference |
|---|---|---|---|---|---|
| Azurin | Small-MW inhibitor | EphB2 | Inhibits ligand binding, tyrosine phosphorylation | N/A | [107] |
| 76D10 | Small-MW inhibitor | EphA4 | Inhibits ligand binding and tyrosine phosphorylation | N/A | [106] |
| Compound 1 | Small-MW inhibitor | EphA4 | Inhibits ligand binding, suppresses Akt phosphorylation, and induces apoptosis | N/A | [108] |
| NVP-BHG712 | Kinase inhibitor | EphB4 | inhibits EphB4 autophosphorylation and VEGF-driven vessel formation | N/A | [114] |
| Dasatinib | Kinase inhibitor | EphA2 | Inhibits EphA2 phosphorylation, Cbl binding, internalisation, and degradation | Phase IV | NCT04155411 [115,118] |
| Compound 4a | Kinase inhibitor | EphA2 | Blocks ATP access to the kinase and decreases cell viability of GBM cells | N/A | [117] |
| ALW-II-41-27 | Kinase inhibitor | EphA2 | Blocks ATP binding to the kinase domain | N/A | [76] |
| SWL | Peptide | EphA2 | Induces EphA2 phosphorylation and blocks Erk/Akt pathways | N/A | [119] |
| SWL dimer | Peptide | EphA2 | Induces EphA2 phosphorylation | N/A | [119] |
| SNEW | Peptide | EphB2 | Blocks ephrin-B2 binding to EphB2 | N/A | [120] |
| TNYL-RAW | Peptide | EphB4 | Blocks ephrin-B2 binding to EphB4 | N/A | [121] |
| DS-8895a | Antibody | EphA2 | Antagonist, increases ADCC | Phase I | NCT02252211, NCT02004717 [122,123] |
| IG25 | Antibody | EphA2 | Induces EphA2 degradation | N/A | [124] |
| IG28 | Antibody | EphA2 | Inhibits ephrin-A1 binding to EphA2 | N/A | [124] |
| EphA10/CD3 | Antibody | EphA10 | Induces tumour cell lysis and promotes T-cell activation | N/A | [125] |
| 2H9 | Antibody | EphB2 | Induces internalisation of EphB2 | N/A | [126] |
| 131 and 47 | Antibody | EphB4 | Induces EphB4 degradation, inhibits tumour vasculature | N/A | [127] |
| IIIA4/KB004/Ifabotuzumab | Antibody | EphA3 | Induces receptor phosphorylation and internalisation, and ADCC | Phase I | NCT03374943, [128,129,130] |
| IC1/MEDI-547 | ADC | EphA2 | Induces EphA2 phosphorylation, internalisation, and degradation | Phase I | NCT00796055, [131,132] |
| IIIA4-USAN | ADC | EphA3 | Inhibits GBM cell viability/tumour growth | N/A | [43] |
| IIIA4-177Lu | Radio-labelled antibody | EphA3 | Inhibits GBM cell viability/tumour growth | N/A | [43] |
| PF-06647263 | ADC | Ephrin-A4 | Binds to ephrin-A4-expressing cells and induces DNA cleavage and apoptosis | Phase I | NCT02078752, [133,134] |
7.3. Peptides
7.4. Antibodies
7.5. Antibody–Drug Conjugates
8. Opportunities and Challenges for Therapeutic Targeting of Ephs and Ephrins
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Giaginis, C.; Tsourouflis, G.; Zizi-Serbetzoglou, A.; Kouraklis, G.; Chatzopoulou, E.; Dimakopoulou, K.; Theocharis, S.E. Clinical significance of ephrin (eph)-A1, -A2, -a4, -a5 and -a7 receptors in pancreatic ductal adenocarcinoma. Pathol. Oncol. Res. 2010, 16, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Kandouz, M. The Eph/Ephrin family in cancer metastasis: Communication at the service of invasion. Cancer Metastasis Rev. 2012, 31, 353–373. [Google Scholar] [CrossRef] [PubMed]
- Himanen, J.P.; Yermekbayeva, L.; Janes, P.W.; Walker, J.R.; Xu, K.; Atapattu, L.; Rajashankar, K.R.; Mensinga, A.; Lackmann, M.; Nikolov, D.B.; et al. Architecture of Eph receptor clusters. Proc. Natl. Acad. Sci. USA 2010, 107, 10860–10865. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.-Y.; Patel, O.; Janes, P.W.; Murphy, J.M.; Lucet, I.S. Eph receptor signalling: From catalytic to non-catalytic functions. Oncogene 2019, 38, 6567–6584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, A.; John, M.; Ruan, Z.; Kannan, N. Coupled regulation by the juxtamembrane and sterile α motif (SAM) linker is a hallmark of ephrin tyrosine kinase evolution. J. Biol. Chem. 2018, 293, 5102–5116. [Google Scholar] [CrossRef] [Green Version]
- Noren, N.K.; Pasquale, E.B. Eph receptor–ephrin bidirectional signals that target Ras and Rho proteins. Cell. Signal. 2004, 16, 655–666. [Google Scholar] [CrossRef]
- Lisabeth, E.M.; Falivelli, G.; Pasquale, E.B. Eph receptor signaling and ephrins. Cold Spring Harb. Perspect. Biol. 2013, 5, a009159. [Google Scholar] [CrossRef] [Green Version]
- Xi, H.-Q.; Wu, X.-S.; Wei, B.; Chen, L. Eph receptors and ephrins as targets for cancer therapy. J. Cell. Mol. Med. 2012, 16, 2894–2909. [Google Scholar] [CrossRef]
- Shamah, S.M.; Lin, M.Z.; Goldberg, J.L.; Estrach, S.; Sahin, M.; Hu, L.; Bazalakova, M.; Neve, R.L.; Corfas, G.; Debant, A.; et al. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell 2001, 105, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Nievergall, E.; Lackmann, M.; Janes, P.W. Eph-dependent cell-cell adhesion and segregation in development and cancer. Cell. Mol. Life Sci. 2012, 69, 1813–1842. [Google Scholar] [CrossRef]
- Han, D.C.; Shen, T.L.; Miao, H.; Wang, B.; Guan, J.L. EphB1 associates with Grb7 and regulates cell migration. J. Biol. Chem. 2002, 277, 45655–45661. [Google Scholar] [CrossRef] [Green Version]
- Borthakur, S.; Lee, H.; Kim, S.; Wang, B.C.; Buck, M. Binding and function of phosphotyrosines of the Ephrin A2 (EphA2) receptor using synthetic sterile α motif (SAM) domains. J. Biol. Chem. 2014, 289, 19694–19703. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shang, Y.; Li, J.; Chen, W.; Li, G.; Wan, J.; Liu, W.; Zhang, M. Specific Eph receptor-cytoplasmic effector signaling mediated by SAM-SAM domain interactions. eLife 2018, 7, e35677. [Google Scholar] [CrossRef]
- Singh, D.R.; Cao, Q.; King, C.; Salotto, M.; Ahmed, F.; Zhou, X.Y.; Pasquale, E.B.; Hristova, K. Unliganded EphA3 dimerization promoted by the SAM domain. Biochem. J. 2015, 471, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.R.; Ahmed, F.; Paul, M.D.; Gedam, M.; Pasquale, E.B.; Hristova, K. The SAM domain inhibits EphA2 interactions in the plasma membrane. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 31–38. [Google Scholar] [CrossRef]
- Romero, G.; von Zastrow, M.; Friedman, P.A. Role of PDZ proteins in regulating trafficking, signaling, and function of GPCRs: Means, motif, and opportunity. Adv. Pharmacol. 2011, 62, 279–314. [Google Scholar] [CrossRef] [Green Version]
- Hock, B.; Böhme, B.; Karn, T.; Yamamoto, T.; Kaibuchi, K.; Holtrich, U.; Holland, S.; Pawson, T.; Rübsamen-Waigmann, H.; Strebhardt, K. PDZ-domain-mediated interaction of the Eph-related receptor tyrosine kinase EphB3 and the ras-binding protein AF6 depends on the kinase activity of the receptor. Proc. Natl. Acad. Sci. USA 1998, 95, 9779–9784. [Google Scholar] [CrossRef] [Green Version]
- Torres, R.; Firestein, B.L.; Dong, H.; Staudinger, J.; Olson, E.N.; Huganir, R.L.; Bredt, D.S.; Gale, N.W.; Yancopoulos, G.D. PDZ Proteins Bind, Cluster, and Synaptically Colocalize with Eph Receptors and Their Ephrin Ligands. Neuron 1998, 21, 1453–1463. [Google Scholar] [CrossRef] [Green Version]
- Brückner, K.; Labrador, J.P.; Scheiffele, P.; Herb, A.; Seeburg, P.H.; Klein, R. EphrinB Ligands Recruit GRIP Family PDZ Adaptor Proteins into Raft Membrane Microdomains. Neuron 1999, 22, 511–524. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, A.R.; Buck, M. Structural and Functional Insights into the Transmembrane Domain Association of Eph Receptors. Int. J. Mol. Sci. 2021, 22, 8593. [Google Scholar] [CrossRef] [PubMed]
- Menges, C.W.; McCance, D.J. Constitutive activation of the Raf-MAPK pathway causes negative feedback inhibition of Ras-PI3K-AKT and cellular arrest through the EphA2 receptor. Oncogene 2008, 27, 2934–2940. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Li, D.Q.; Mukherjee, A.; Guo, H.; Petty, A.; Cutter, J.; Basilion, J.P.; Sedor, J.; Wu, J.; Danielpour, D.; et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell 2009, 16, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.Y.; Fernandez, C.; Richter, M.; Xiao, Z.; Valencia, F.; Tice, D.A.; Pasquale, E.B. Crosstalk of the EphA2 receptor with a serine/threonine phosphatase suppresses the Akt-mTORC1 pathway in cancer cells. Cell Signal 2011, 23, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soskis, M.J.; Ho, H.-Y.H.; Bloodgood, B.L.; Robichaux, M.A.; Malik, A.N.; Ataman, B.; Rubin, A.A.; Zieg, J.; Zhang, C.; Shokat, K.M.; et al. A chemical genetic approach reveals distinct EphB signaling mechanisms during brain development. Nat. Neurosci. 2012, 15, 1645–1654. [Google Scholar] [CrossRef]
- Flanagan, J.G. Neural map specification by gradients. Curr. Opin. Neurobiol. 2006, 16, 59–66. [Google Scholar] [CrossRef]
- Janes, P.W.; Slape, C.I.; Farnsworth, R.H.; Atapattu, L.; Scott, A.M.; Vail, M.E. EphA3 biology and cancer. Growth Factors 2014, 32, 176–189. [Google Scholar] [CrossRef]
- Chen, J.; Song, W.; Amato, K. Eph receptor tyrosine kinases in cancer stem cells. Cytokine Growth Factor Rev. 2015, 26, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X. The Expression Profile and Prognostic Values of EPHA Family Members in Breast Cancer. Front. Oncol. 2021, 11, 619949. [Google Scholar] [CrossRef]
- Nikas, I.; Giaginis, C.; Petrouska, K.; Alexandrou, P.; Michail, A.; Sarantis, P.; Tsourouflis, G.; Danas, E.; Pergaris, A.; Politis, P.K.; et al. EPHA2, EPHA4, and EPHA7 Expression in Triple-Negative Breast Cancer. Diagnostics 2022, 12, 366. [Google Scholar] [CrossRef]
- Arvanitis, D.; Davy, A. Eph/ephrin signaling: Networks. Genes Dev. 2008, 22, 416–429. [Google Scholar] [CrossRef] [Green Version]
- Janes, P.W.; Vail, M.E.; Ernst, M.; Scott, A.M. Eph Receptors in the Immunosuppressive Tumor Microenvironment. Cancer Res. 2021, 81, 801–805. [Google Scholar] [CrossRef]
- Irie, F.; Yamaguchi, Y. Eph Receptor Signaling and Spine Morphology. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Oxford, UK, 2009; pp. 1141–1145. [Google Scholar]
- Frisén, J.; Holmberg, J.; Barbacid, M. Ephrins and their Eph receptors: Multitalented directors of embryonic development. EMBO J. 1999, 18, 5159–5165. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.U.; Chen, Z.-F.; Anderson, D.J. Molecular Distinction and Angiogenic Interaction between Embryonic Arteries and Veins Revealed by ephrin-B2 and Its Receptor Eph-B4. Cell 1998, 93, 741–753. [Google Scholar] [CrossRef] [Green Version]
- Adams, R.H.; Wilkinson, G.A.; Weiss, C.; Diella, F.; Gale, N.W.; Deutsch, U.; Risau, W.; Klein, R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: Demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999, 13, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Boyd, A.W.; Bartlett, P.F.; Lackmann, M. Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug Discov. 2014, 13, 39–62. [Google Scholar] [CrossRef]
- To, C.; Farnsworth, R.H.; Vail, M.E.; Chheang, C.; Gargett, C.E.; Murone, C.; Llerena, C.; Major, A.T.; Scott, A.M.; Janes, P.W.; et al. Hypoxia-Controlled EphA3 Marks a Human Endometrium-Derived Multipotent Mesenchymal Stromal Cell that Supports Vascular Growth. PLoS ONE 2014, 9, e112106. [Google Scholar] [CrossRef] [Green Version]
- Boyd, A.W.; Ward, L.D.; Wicks, I.P.; Simpson, R.J.; Salvaris, E.; Wilks, A.; Welch, K.; Loudovaris, M.; Rockman, S.; Busmanis, I. Isolation and characterization of a novel receptor-type protein tyrosine kinase (hek) from a human pre-B cell line. J. Biol. Chem. 1992, 267, 3262–3267. [Google Scholar] [CrossRef]
- Shiuan, E.; Chen, J. Eph Receptor Tyrosine Kinases in Tumor Immunity. Cancer Res. 2016, 76, 6452–6457. [Google Scholar] [CrossRef] [Green Version]
- Janes, P.W.; Vail, M.E.; Gan, H.K.; Scott, A.M. Antibody Targeting of Eph Receptors in Cancer. Pharmaceuticals 2020, 13, 88. [Google Scholar] [CrossRef]
- Vail, M.E.; Murone, C.; Tan, A.; Hii, L.; Abebe, D.; Janes, P.W.; Lee, F.-T.; Baer, M.; Palath, V.; Bebbington, C.; et al. Targeting EphA3 Inhibits Cancer Growth by Disrupting the Tumor Stromal Microenvironment. Cancer Res. 2014, 74, 4470–4481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offenhäuser, C.; Al-Ejeh, F.; Puttick, S.; Ensbey, K.S.; Bruce, Z.C.; Jamieson, P.R.; Smith, F.M.; Stringer, B.W.; Carrington, B.; Fuchs, A.V.; et al. EphA3 Pay-Loaded Antibody Therapeutics for the Treatment of Glioblastoma. Cancers 2018, 10, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Suo, Z.; Risberg, B.; Karlsson, M.G.; Villman, K.; Nesland, J.M. Expression of Ephb2 and Ephb4 in breast carcinoma. Pathol. Oncol. Res. 2004, 10, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.R.; Singh, J.; Xia, G.; Krasnoperov, V.; Hassanieh, L.; Ley, E.J.; Scehnet, J.; Kumar, N.G.; Hawes, D.; Press, M.F.; et al. Receptor tyrosine kinase EphB4 is a survival factor in breast cancer. Am. J. Pathol. 2006, 169, 279–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brantley-Sieders, D.M.; Zhuang, G.; Hicks, D.; Fang, W.B.; Hwang, Y.; Cates, J.M.; Coffman, K.; Jackson, D.; Bruckheimer, E.; Muraoka-Cook, R.S.; et al. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J. Clin. Investig. 2008, 118, 64–78. [Google Scholar] [CrossRef] [Green Version]
- Duxbury, M.S.; Ito, H.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. EphA2: A determinant of malignant cellular behavior and a potential therapeutic target in pancreatic adenocarcinoma. Oncogene 2004, 23, 1448–1456. [Google Scholar] [CrossRef] [Green Version]
- Herath, N.I.; Doecke, J.; Spanevello, M.D.; Leggett, B.A.; Boyd, A.W. Epigenetic silencing of EphA1 expression in colorectal cancer is correlated with poor survival. Br. J. Cancer 2009, 100, 1095–1102. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef] [Green Version]
- Magic, Z.; Sandström, J.; Perez-Tenorio, G.; Magic, Z.; Sandström, J.; Perez-Tenorio, G.; Magic, Z.; Sandström, J.; Perez-Tenorio, G.; Magic, Z.; et al. Ephrin-B2 inhibits cell proliferation and motility in vitro and predicts longer metastasis-free survival in breast cancer. Int. J. Oncol. 2019, 55, 1275–1286. [Google Scholar] [CrossRef] [Green Version]
- Davalos, V.; Dopeso, H.; Castaño, J.; Wilson, A.J.; Vilardell, F.; Romero-Gimenez, J.; Espín, E.; Armengol, M.; Capella, G.; Mariadason, J.M.; et al. EPHB4 and Survival of Colorectal Cancer Patients. Cancer Res. 2006, 66, 8943–8948. [Google Scholar] [CrossRef] [Green Version]
- Dopeso, H.; Mateo-Lozano, S.; Mazzolini, R.; Rodrigues, P.; Lagares-Tena, L.; Ceron, J.; Romero, J.; Esteves, M.; Landolfi, S.; Hernández-Losa, J.; et al. The Receptor Tyrosine Kinase EPHB4 Has Tumor Suppressor Activities in Intestinal Tumorigenesis. Cancer Res. 2009, 69, 7430–7438. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Bacani, J.; Begthel, H.; Jonkeer, S.; Gregorieff, A.; van de Born, M.; Malats, N.; Sancho, E.; Boon, E.; Pawson, T.; et al. EphB receptor activity suppresses colorectal cancer progression. Nature 2005, 435, 1126–1130. [Google Scholar] [CrossRef]
- Cortina, C.; Palomo-Ponce, S.; Iglesias, M.; Fernández-Masip, J.L.; Vivancos, A.; Whissell, G.; Humà, M.; Peiró, N.; Gallego, L.; Jonkheer, S.; et al. EphB–ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nat. Genet. 2007, 39, 1376–1383. [Google Scholar] [CrossRef]
- Brantley-Sieders, D.M.; Fang, W.B.; Hicks, D.J.; Zhuang, G.; Shyr, Y.; Chen, J. Impaired tumor microenvironment in EphA2-deficient mice inhibits tumor angiogenesis and metastatic progression. Fed. Am. Soc. Exp. Biol. J. 2005, 19, 1884–1886. [Google Scholar] [CrossRef]
- Brantley-Sieders, D.M.; Jiang, A.; Sarma, K.; Badu-Nkansah, A.; Walter, D.L.; Shyr, Y.; Chen, J. Eph/ephrin profiling in human breast cancer reveals significant associations between expression level and clinical outcome. PLoS ONE 2011, 6, e24426. [Google Scholar] [CrossRef] [Green Version]
- Markosyan, N.; Li, J.; Sun, Y.H.; Richman, L.P.; Lin, J.H.; Yan, F.; Quinones, L.; Sela, Y.; Yamazoe, T.; Gordon, N.; et al. Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2). J. Clin. Investig. 2019, 129, 3594–3609. [Google Scholar] [CrossRef] [Green Version]
- Day, B.W.; Stringer, B.W.; Al-Ejeh, F.; Ting, M.J.; Wilson, J.; Ensbey, K.S.; Jamieson, P.R.; Bruce, Z.C.; Lim, Y.C.; Offenhäuser, C.; et al. EphA3 Maintains Tumorigenicity and Is a Therapeutic Target in Glioblastoma Multiforme. Cancer Cell 2013, 23, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Saintigny, P.; Peng, S.; Zhang, L.; Sen, B.; Wistuba, I.I.; Lippman, S.M.; Girard, L.; Minna, J.D.; Heymach, J.V.; Johnson, F.M. Global Evaluation of Eph Receptors and Ephrins in Lung Adenocarcinomas Identifies EphA4 as an Inhibitor of Cell Migration and Invasion. Mol. Cancer Ther. 2012, 11, 2021–2032. [Google Scholar] [CrossRef] [Green Version]
- Nagano, K.; Maeda, Y.; Kanasaki, S.-i.; Watanabe, T.; Yamashita, T.; Inoue, M.; Higashisaka, K.; Yoshioka, Y.; Abe, Y.; Mukai, Y.; et al. Ephrin receptor A10 is a promising drug target potentially useful for breast cancers including triple negative breast cancers. J. Control. Release 2014, 189, 72–79. [Google Scholar] [CrossRef]
- Xuan, Z.; Huang, J.; Gao, L.; Wang, Y.; Wang, J.; Sun, Y. Receptor Tyrosine Kinase EphB3: A Prognostic Indicator in Colorectal Carcinoma. Pathol. Oncol. Res. 2020, 26, 541–549. [Google Scholar] [CrossRef]
- Guo, H.; Miao, H.; Gerber, L.; Singh, J.; Denning, M.F.; Gilliam, A.C.; Wang, B. Disruption of EphA2 Receptor Tyrosine Kinase Leads to Increased Susceptibility to Carcinogenesis in Mouse Skin. Cancer Res. 2006, 66, 7050–7058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisabeth, E.M.; Fernandez, C.; Pasquale, E.B. Cancer somatic mutations disrupt functions of the EphA3 receptor tyrosine kinase through multiple mechanisms. Biochemistry 2012, 51, 1464–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, G.; Song, W.; Amato, K.; Hwang, Y.; Lee, K.; Boothby, M.; Ye, F.; Guo, Y.; Shyr, Y.; Lin, L.; et al. Effects of Cancer-Associated EPHA3 Mutations on Lung Cancer. J. Natl. Cancer Inst. 2012, 104, 1183–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Fröse, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.-H.; Cha, J.-H.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Wang, Y.-N.; Hsu, J.L.; Ren, G.; Hung, M.-C. Juxtacrine Signaling Inhibits Antitumor Immunity by Upregulating PD-L1 Expression. Cancer Res. 2018, 78, 3761–3768. [Google Scholar] [CrossRef] [Green Version]
- Cioce, M.; Fazio, V.M. EphA2 and EGFR: Friends in Life, Partners in Crime, Can EphA2 Be a Predictive Biomarker of Response to Anti-EGFR Agents? Cancers 2021, 13, 700. [Google Scholar] [CrossRef]
- Lu, M.; Miller, K.D.; Gokmen-Polar, Y.; Jeng, M.-H.; Kinch, M.S. EphA2 Overexpression Decreases Estrogen Dependence and Tamoxifen Sensitivity1. Cancer Res. 2003, 63, 3425–3429. [Google Scholar]
- Zhuang, G.; Brantley-Sieders, D.M.; Vaught, D.; Yu, J.; Xie, L.; Wells, S.; Jackson, D.; Muraoka-Cook, R.; Arteaga, C.; Chen, J. Elevation of Receptor Tyrosine Kinase EphA2 Mediates Resistance to Trastuzumab Therapy. Cancer Res. 2010, 70, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Miao, B.; Ji, Z.; Tan, L.; Taylor, M.; Zhang, J.; Choi, H.G.; Frederick, D.T.; Kumar, R.; Wargo, J.A.; Flaherty, K.T.; et al. EPHA2 Is a Mediator of Vemurafenib Resistance and a Novel Therapeutic Target in Melanoma. Cancer Discov. 2015, 5, 274–287. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Shen, Y.; Ni, J.; Ou, Y.; Ou, Y.; Liu, H. EphA4 promotes cell proliferation and cell adhesion–mediated drug resistance via the AKT pathway in multiple myeloma. Tumor Biol. 2017, 39, 1010428317694298. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Xu, N.; Zhang, J.F.; Xu, L.L.; Zhou, X.; Huang, B.T.; Li, Y.L.; Liu, X.L. EphB4/ephrinB2 Contributes to Imatinib Resistance in Chronic Myeloid Leukemia Involved in Cytoskeletal Proteins. Int. J. Med. Sci. 2016, 13, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yang, Y.; Tang, S.; Tang, H.; Yang, G.; Xu, Q.; Wu, J. EphB4 inhibitor overcome the acquired resistance to cisplatin in melanomas xenograft model. J. Pharmacol. Sci. 2015, 129, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Wang, Q.; Liu, H.; Ye, M.; Wu, X.; Guo, L. EPHA3 regulates the multidrug resistance of small cell lung cancer via the PI3K/BMX/STAT3 signaling pathway. Tumor Biol. 2016, 37, 11959–11971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brannan, J.M.; Sen, B.; Saigal, B.; Prudkin, L.; Behrens, C.; Solis, L.; Dong, W.; Bekele, B.N.; Wistuba, I.; Johnson, F.M. EphA2 in the Early Pathogenesis and Progression of Non–Small Cell Lung Cancer. Cancer Prev. Res. 2009, 2, 1039–1049. [Google Scholar] [CrossRef] [Green Version]
- Amato, K.R.; Wang, S.; Hastings, A.K.; Youngblood, V.M.; Santapuram, P.R.; Chen, H.; Cates, J.M.; Colvin, D.C.; Ye, F.; Brantley-Sieders, D.M.; et al. Genetic and pharmacologic inhibition of EPHA2 promotes apoptosis in NSCLC. J. Clin. Investig. 2014, 124, 2037–2049. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.-D.; Li, G.; Feng, Y.-X.; Zhao, J.-S.; Li, J.-J.; Sun, Z.-J.; Shi, S.; Deng, Y.-Z.; Xu, J.-F.; Zhu, Y.-Q.; et al. EphB3 Is Overexpressed in Non–Small-Cell Lung Cancer and Promotes Tumor Metastasis by Enhancing Cell Survival and Migration. Cancer Res. 2011, 71, 1156–1166. [Google Scholar] [CrossRef] [Green Version]
- Barquilla, A.; Pasquale, E.B. Eph Receptors and Ephrins: Therapeutic Opportunities. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 465–487. [Google Scholar] [CrossRef] [Green Version]
- Binda, E.; Visioli, A.; Giani, F.; Lamorte, G.; Copetti, M.; Pitter, K.L.; Huse, J.T.; Cajola, L.; Zanetti, N.; DiMeco, F.; et al. The EphA2 Receptor Drives Self-Renewal and Tumorigenicity in Stem-like Tumor-Propagating Cells from Human Glioblastomas. Cancer Cell 2012, 22, 765–780. [Google Scholar] [CrossRef] [Green Version]
- Miao, H.; Gale, N.W.; Guo, H.; Qian, J.; Petty, A.; Kaspar, J.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.; Hambardzumyan, D.; et al. EphA2 promotes infiltrative invasion of glioma stem cells in vivo through cross-talk with Akt and regulates stem cell properties. Oncogene 2015, 34, 558–567. [Google Scholar] [CrossRef] [Green Version]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Adams, J.; Singh, M.; Hu, A.; Gorelik, M.; Subapanditha, M.K.; Savage, N.; Yang, J.; et al. Cotargeting Ephrin Receptor Tyrosine Kinases A2 and A3 in Cancer Stem Cells Reduces Growth of Recurrent Glioblastoma. Cancer Res. 2018, 78, 5023–5037. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.D.; Rath, P.; Lal, B.; Richard, J.P.; Li, Y.; Goodwin, C.R.; Laterra, J.; Xia, S. EphB2 receptor controls proliferation/migration dichotomy of glioblastoma by interacting with focal adhesion kinase. Oncogene 2012, 31, 5132–5143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmberg, J.; Genander, M.; Halford, M.M.; Annerén, C.; Sondell, M.; Chumley, M.J.; Silvany, R.E.; Henkemeyer, M.; Frisén, J. EphB Receptors Coordinate Migration and Proliferation in the Intestinal Stem Cell Niche. Cell 2006, 125, 1151–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genander, M.; Halford, M.M.; Xu, N.-J.; Eriksson, M.; Yu, Z.; Qiu, Z.; Martling, A.; Greicius, G.; Thakar, S.; Catchpole, T.; et al. Dissociation of EphB2 Signaling Pathways Mediating Progenitor Cell Proliferation and Tumor Suppression. Cell 2009, 139, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Xia, Q.; Wang, J.; Shen, Q.; Zhang, J.; Zhou, X. EphB4 promotes the proliferation, invasion, and angiogenesis of human colorectal cancer. Exp. Mol. Pathol. 2016, 100, 402–408. [Google Scholar] [CrossRef]
- Potla, L.; Boghaert, E.R.; Armellino, D.; Frost, P.; Damle, N.K. Reduced expression of EphrinA1 (EFNA1) inhibits three-dimensional growth of HT29 colon carcinoma cells. Cancer Lett. 2002, 175, 187–195. [Google Scholar] [CrossRef]
- Lu, C.-Y.; Yang, Z.-X.; Zhou, L.; Huang, Z.-Z.; Zhang, H.T.; Li, J.; Tao, K.-S.; Xie, B.-Z. High levels of EphA3 expression are associated with high invasive capacity and poor overall survival in hepatocellular carcinoma. Oncol. Rep. 2013, 30, 2179–2186. [Google Scholar] [CrossRef] [Green Version]
- Psilopatis, I.; Souferi-Chronopoulou, E.; Vrettou, K.; Troungos, C.; Theocharis, S. EPH/Ephrin-Targeting Treatment in Breast Cancer: A New Chapter in Breast Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 15275. [Google Scholar] [CrossRef]
- Anderton, M.; van der Meulen, E.; Blumenthal, M.J.; Schäfer, G. The Role of the Eph Receptor Family in Tumorigenesis. Cancers 2021, 13, 206. [Google Scholar] [CrossRef]
- Kou, C.-T.J.; Kandpal, R.P. Differential Expression Patterns of Eph Receptors and Ephrin Ligands in Human Cancers. BioMed Res. Int. 2018, 2018, 7390104. [Google Scholar] [CrossRef] [Green Version]
- Lisle, J.E.; Mertens-Walker, I.; Rutkowski, R.; Herington, A.C.; Stephenson, S.-A. Eph receptors and their ligands: Promising molecular biomarkers and therapeutic targets in prostate cancer. Biochim. Biophys. Acta Rev. Cancer 2013, 1835, 243–257. [Google Scholar] [CrossRef]
- Fox, B.P.; Tabone, C.J.; Kandpal, R.P. Potential clinical relevance of Eph receptors and ephrin ligands expressed in prostate carcinoma cell lines. Biochem. Biophys. Res. Commun. 2006, 342, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Ashida, S.; Nakagawa, H.; Katagiri, T.; Furihata, M.; Iiizumi, M.; Anazawa, Y.; Tsunoda, T.; Takata, R.; Kasahara, K.; Miki, T.; et al. Molecular Features of the Transition from Prostatic Intraepithelial Neoplasia (PIN) to Prostate Cancer: Genome-wide Gene-expression Profiles of Prostate Cancers and PINs. Cancer Res. 2004, 64, 5963–5972. [Google Scholar] [CrossRef] [Green Version]
- Soler, M.; Mancini, F.; Meca-Cortés, Ó.; Sánchez-Cid, L.; Rubio, N.; López-Fernández, S.; Lozano, J.J.; Blanco, J.; Fernández, P.L.; Thomson, T.M. HER3 is required for the maintenance of neuregulin-dependent and -independent attributes of malignant progression in prostate cancer cells. Int. J. Cancer 2009, 125, 2565–2575. [Google Scholar] [CrossRef]
- Udayakumar, D.; Zhang, G.; Ji, Z.; Njauw, C.N.; Mroz, P.; Tsao, H. Epha2 is a critical oncogene in melanoma. Oncogene 2011, 30, 4921–4929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easty, D.J.; Hill, S.P.; Hsu, M.-Y.; Fallowfield, M.E.; Flørenes, V.A.; Herlyn, M.; Bennett, D.C. Up-regulation of ephrin-a1 during melanoma progression. Int. J. Cancer 1999, 84, 494–501. [Google Scholar] [CrossRef]
- Shin, W.S.; Park, M.K.; Lee, Y.H.; Kim, K.W.; Lee, H.; Lee, S.T. The catalytically defective receptor protein tyrosine kinase EphA10 promotes tumorigenesis in pancreatic cancer cells. Cancer Sci. 2020, 111, 3292–3302. [Google Scholar] [CrossRef]
- Strozen, T.G.; Sharpe, J.C.; Harris, E.D.; Uppalapati, M.; Toosi, B.M. The EphB6 Receptor: Kinase-Dead but Very Much Alive. Int. J. Mol. Sci. 2021, 22, 8211. [Google Scholar] [CrossRef]
- Peng, L.; Tu, P.; Wang, X.; Shi, S.; Zhou, X.; Wang, J. Loss of EphB6 protein expression in human colorectal cancer correlates with poor prognosis. J. Mol. Histol. 2014, 45, 555–563. [Google Scholar] [CrossRef]
- Mateo-Lozano, S.; Bazzocco, S.; Rodrigues, P.; Mazzolini, R.; Andretta, E.; Dopeso, H.; Fernández, Y.; del Llano, E.; Bilic, J.; Suárez-López, L.; et al. Loss of the EPH receptor B6 contributes to colorectal cancer metastasis. Sci. Rep. 2017, 7, 43702. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.Y.; Roy, M.; Horne, C.R.; Sandow, J.J.; Surudoi, M.; Dagley, L.F.; Young, S.N.; Dite, T.; Babon, J.J.; Janes, P.W.; et al. The intracellular domains of the EphB6 and EphA10 receptor tyrosine pseudokinases function as dynamic signalling hubs. Biochem. J. 2021, 478, 3351–3371. [Google Scholar] [CrossRef]
- Sato, S.; Tanaka, H.; Hikichi, Y.; Sunahara, E.; Kaieda, I.; Ishii, T.; Yoshimura, K. Abstract 3951: EphA10 exhibits anti-apoptotic activity through its dominant-negative role for EphA signaling in cancer cells. Cancer Res. 2010, 70, 3951. [Google Scholar] [CrossRef]
- Li, Y.; Jin, L.; Ye, F.; Ma, Q.; Yang, Z.; Liu, D.; Yang, J.; Ma, D.; Gao, Q. Isoform expression patterns of EPHA10 protein mediate breast cancer progression by regulating the E-Cadherin and β-catenin complex. Oncotarget 2017, 8, 30344–30356. [Google Scholar] [CrossRef] [PubMed]
- Noberini, R.; Koolpe, M.; Peddibhotla, S.; Dahl, R.; Su, Y.; Cosford, N.D.P.; Roth, G.P.; Pasquale, E.B. Small Molecules Can Selectively Inhibit Ephrin Binding to the EphA4 and EphA2 Receptors. J. Biol. Chem. 2008, 283, 29461–29472. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, C.; Mohamed, H.I.; Flammini, L.; Barocelli, E.; Incerti, M.; Lodola, A.; Tognolini, M. Lithocholic Acid Is an Eph-ephrin Ligand Interfering with Eph-kinase Activation. PLoS ONE 2011, 6, e18128. [Google Scholar] [CrossRef]
- Noberini, R.; De, S.K.; Zhang, Z.; Wu, B.; Raveendra-Panickar, D.; Chen, V.; Vazquez, J.; Qin, H.; Song, J.; Cosford, N.D.; et al. A disalicylic acid-furanyl derivative inhibits ephrin binding to a subset of Eph receptors. Chem. Biol. Drug Des. 2011, 78, 667–678. [Google Scholar] [CrossRef]
- Chaudhari, A.; Mahfouz, M.; Fialho, A.M.; Yamada, T.; Granja, A.T.; Zhu, Y.; Hashimoto, W.; Schlarb-Ridley, B.; Cho, W.; Gupta, T.K.D.; et al. Cupredoxin−Cancer Interrelationship: Azurin Binding with EphB2, Interference in EphB2 Tyrosine Phosphorylation, and Inhibition of Cancer Growth. Biochemistry 2007, 46, 1799–1810. [Google Scholar] [CrossRef]
- Takano, H.; Nakamura, T.; Tsuchikawa, T.; Kushibiki, T.; Hontani, K.; Inoko, K.; Takahashi, M.; Sato, S.; Abe, H.; Takeuchi, S.; et al. Inhibition of Eph receptor A4 by 2,5-dimethylpyrrolyl benzoic acid suppresses human pancreatic cancer growing orthotopically in nude mice. Oncotarget 2015, 6, 41063–41076. [Google Scholar] [CrossRef]
- Castelli, R.; Tognolini, M.; Vacondio, F.; Incerti, M.; Pala, D.; Callegari, D.; Bertoni, S.; Giorgio, C.; Hassan-Mohamed, I.; Zanotti, I.; et al. Δ5-Cholenoyl-amino acids as selective and orally available antagonists of the Eph–ephrin system. Eur. J. Med. Chem. 2015, 103, 312–324. [Google Scholar] [CrossRef]
- Su, Q.; Wang, J.; Wu, Q.; Ullah, A.; Ghauri, M.A.; Sarwar, A.; Chen, L.; Liu, F.; Zhang, Y. Sanguinarine combats hypoxia-induced activation of EphB4 and HIF-1α pathways in breast cancer. Phytomedicine 2021, 84, 153503. [Google Scholar] [CrossRef]
- Choi, Y.; Syeda, F.; Walker, J.R.; Finerty, P.J.; Cuerrier, D.; Wojciechowski, A.; Liu, Q.; Dhe-Paganon, S.; Gray, N.S. Discovery and structural analysis of Eph receptor tyrosine kinase inhibitors. Bioorganic Med. Chem. Lett. 2009, 19, 4467–4470. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Choi, S.; Case, A.; Gainer, T.G.; Seyb, K.; Glicksman, M.A.; Lo, D.C.; Stein, R.L.; Cuny, G.D. Structure–activity relationship study of EphB3 receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 6122–6126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlaam, B.; Ducray, R.; Brempt, C.L.-v.d.; Plé, P.; Bardelle, C.; Brooks, N.; Coleman, T.; Cross, D.; Kettle, J.G.; Read, J. Inhibitors of the tyrosine kinase EphB4. Part 4: Discovery and optimization of a benzylic alcohol series. Bioorg. Med. Chem. Lett. 2011, 21, 2207–2211. [Google Scholar] [CrossRef]
- Martiny-Baron, G.; Holzer, P.; Billy, E.; Schnell, C.; Brueggen, J.; Ferretti, M.; Schmiedeberg, N.; Wood, J.M.; Furet, P.; Imbach, P. The small molecule specific EphB4 kinase inhibitor NVP-BHG712 inhibits VEGF driven angiogenesis. Angiogenesis 2010, 13, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Eustace, A.J.; Crown, J.; Clynes, M.; O’Donovan, N. Preclinical evaluation of dasatinib, a potent Src kinase inhibitor, in melanoma cell lines. J. Transl. Med. 2008, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Torres-Adorno, A.M.; Vitrac, H.; Qi, Y.; Tan, L.; Levental, K.R.; Fan, Y.-Y.; Yang, P.; Chapkin, R.S.; Eckhardt, B.L.; Ueno, N.T. Eicosapentaenoic acid in combination with EPHA2 inhibition shows efficacy in preclinical models of triple-negative breast cancer by disrupting cellular cholesterol efflux. Oncogene 2019, 38, 2135–2150. [Google Scholar] [CrossRef]
- Heinzlmeir, S.; Lohse, J.; Treiber, T.; Kudlinzki, D.; Linhard, V.; Gande, S.L.; Sreeramulu, S.; Saxena, K.; Liu, X.; Wilhelm, M.; et al. Chemoproteomics-Aided Medicinal Chemistry for the Discovery of EPHA2 Inhibitors. ChemMedChem 2017, 12, 999–1011. [Google Scholar] [CrossRef] [Green Version]
- Chang, Q.; Jorgensen, C.; Pawson, T.; Hedley, D.W. Effects of dasatinib on EphA2 receptor tyrosine kinase activity and downstream signalling in pancreatic cancer. Br. J. Cancer 2008, 99, 1074–1082. [Google Scholar] [CrossRef] [Green Version]
- Duggineni, S.; Mitra, S.; Lamberto, I.; Han, X.; Xu, Y.; An, J.; Pasquale, E.B.; Huang, Z. Design and Synthesis of Potent Bivalent Peptide Agonists Targeting the EphA2 Receptor. ACS Med. Chem. Lett. 2013, 4, 344–348. [Google Scholar] [CrossRef]
- Chrencik, J.E.; Brooun, A.; Recht, M.I.; Nicola, G.; Davis, L.K.; Abagyan, R.; Widmer, H.; Pasquale, E.B.; Kuhn, P. Three-dimensional Structure of the EphB2 Receptor in Complex with an Antagonistic Peptide Reveals a Novel Mode of Inhibition. J. Biol. Chem. 2007, 282, 36505–36513. [Google Scholar] [CrossRef] [Green Version]
- Duggineni, S.; Mitra, S.; Noberini, R.; Han, X.; Lin, N.; Xu, Y.; Tian, W.; An, J.; Pasquale, E.B.; Huang, Z. Design, synthesis and characterization of novel small molecular inhibitors of ephrin-B2 binding to EphB4. Biochem. Pharmacol. 2013, 85, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, J.; Sue, M.; Yamato, M.; Ichikawa, J.; Ishida, S.; Shibutani, T.; Kitamura, M.; Wada, T.; Agatsuma, T. Novel anti-EPHA2 antibody, DS-8895a for cancer treatment. Cancer Biol. Ther. 2016, 17, 1158–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shitara, K.; Satoh, T.; Iwasa, S.; Yamaguchi, K.; Muro, K.; Komatsu, Y.; Nishina, T.; Esaki, T.; Hasegawa, J.; Kakurai, Y.; et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the afucosylated, humanized anti-EPHA2 antibody DS-8895a: A first-in-human phase I dose escalation and dose expansion study in patients with advanced solid tumors. J. ImmunoTherapy Cancer 2019, 7, 219. [Google Scholar] [CrossRef] [PubMed]
- Ansuini, H.; Meola, A.; Gunes, Z.; Paradisi, V.; Pezzanera, M.; Acali, S.; Santini, C.; Luzzago, A.; Mori, F.; Lazzaro, D.; et al. Anti-EphA2 Antibodies with Distinct In Vitro Properties Have Equal In Vivo Efficacy in Pancreatic Cancer. J. Oncol. 2009, 2009, 951917. [Google Scholar] [CrossRef]
- Taki, S.; Kamada, H.; Inoue, M.; Nagano, K.; Mukai, Y.; Higashisaka, K.; Yoshioka, Y.; Tsutsumi, Y.; Tsunoda, S.-i. A Novel Bispecific Antibody against Human CD3 and Ephrin Receptor A10 for Breast Cancer Therapy. PLoS ONE 2015, 10, e0144712. [Google Scholar] [CrossRef] [Green Version]
- Mao, W.; Luis, E.; Ross, S.; Silva, J.; Tan, C.; Crowley, C.; Chui, C.; Franz, G.; Senter, P.; Koeppen, H.; et al. EphB2 as a Therapeutic Antibody Drug Target for the Treatment of Colorectal Cancer. Cancer Res. 2004, 64, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Krasnoperov, V.; Kumar, S.R.; Ley, E.; Li, X.; Scehnet, J.; Liu, R.; Zozulya, S.; Gill, P.S. Novel EphB4 monoclonal antibodies modulate angiogenesis and inhibit tumor growth. Am. J. Pathol. 2010, 176, 2029–2038. [Google Scholar] [CrossRef]
- Palath, V.; Vekhande, R.; Baer, M.; Ching, W.; Tomasevic, N.; Martinez, D.; Yi, C.; Bhaskar, S.; Luehrsen, K.; Williams, J.; et al. A Recombinant Antibody to EphA3 with Pro-Apoptotic and Enhanced ADCC Activity Against Various Hematologic Malignancies Shows Selective Inhibition of Colony Formation from Long-Term Culture-Initiating Cells (LTC-ICs) In Primary Leukemia Samples. Blood 2010, 116, 2897. [Google Scholar] [CrossRef]
- Swords, R.T.; Greenberg, P.L.; Wei, A.H.; Durrant, S.; Advani, A.S.; Hertzberg, M.S.; Lewis, I.D.; Rivera, G.; Gratzinger, D.; Fan, A.C.; et al. KB004, a first in class monoclonal antibody targeting the receptor tyrosine kinase EphA3, in patients with advanced hematologic malignancies: Results from a phase 1 study. Leuk. Res. 2016, 50, 123–131. [Google Scholar] [CrossRef]
- Gan, H.; Cher, L.; Inglis, P.; Lwin, Z.; Lau, E.; Wichmann, C.; Ackermann, U.; Coombs, N.; Remen, K.; Guo, N.; et al. Abstract CT063: Preliminary findings of a Phase I safety and bioimaging trial of KB004 (ifabotuzumab) in patients with glioblastoma. Cancer Res. 2019, 79, CT063. [Google Scholar] [CrossRef]
- Jackson, D.; Gooya, J.; Mao, S.; Kinneer, K.; Xu, L.; Camara, M.; Fazenbaker, C.; Fleming, R.; Swamynathan, S.; Meyer, D.; et al. A Human Antibody–Drug Conjugate Targeting EphA2 Inhibits Tumor Growth in vivo. Cancer Res. 2008, 68, 9367–9374. [Google Scholar] [CrossRef] [Green Version]
- Annunziata, C.M.; Kohn, E.C.; LoRusso, P.; Houston, N.D.; Coleman, R.L.; Buzoianu, M.; Robbie, G.; Lechleider, R. Phase 1, open-label study of MEDI-547 in patients with relapsed or refractory solid tumors. Investig. New Drugs 2013, 31, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damelin, M.; Bankovich, A.; Park, A.; Aguilar, J.; Anderson, W.; Santaguida, M.; Aujay, M.; Fong, S.; Khandke, K.; Pulito, V.; et al. Anti-EFNA4 Calicheamicin Conjugates Effectively Target Triple-Negative Breast and Ovarian Tumor-Initiating Cells to Result in Sustained Tumor Regressions. Clin. Cancer Res. 2015, 21, 4165–4173. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Laguna, I.; Krop, I.; Burris, H.A., III; Hamilton, E.; Braiteh, F.; Weise, A.M.; Abu-Khalaf, M.; Werner, T.L.; Pirie-Shepherd, S.; Zopf, C.J.; et al. First-in-human, phase I study of PF-06647263, an anti-EFNA4 calicheamicin antibody–drug conjugate, in patients with advanced solid tumors. Int. J. Cancer 2019, 145, 1798–1808. [Google Scholar] [CrossRef]
- Cha, J.-H.; Chan, L.-C.; Wang, Y.-N.; Chu, Y.-Y.; Wang, C.-H.; Lee, H.-H.; Xia, W.; Shyu, W.-C.; Liu, S.-P.; Yao, J.; et al. Ephrin receptor A10 monoclonal antibodies and the derived chimeric antigen receptor T cells exert an antitumor response in mouse models of triple-negative breast cancer. J. Biol. Chem. 2022, 298, 101817. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Noberini, R.; Lamberto, I.; Pasquale, E.B. Targeting Eph receptors with peptides and small molecules: Progress and challenges. Semin. Cell Dev. Biol. 2012, 23, 51–57. [Google Scholar] [CrossRef] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arora, S.; Scott, A.M.; Janes, P.W. Eph Receptors in Cancer. Biomedicines 2023, 11, 315. https://doi.org/10.3390/biomedicines11020315
Arora S, Scott AM, Janes PW. Eph Receptors in Cancer. Biomedicines. 2023; 11(2):315. https://doi.org/10.3390/biomedicines11020315
Chicago/Turabian StyleArora, Sakshi, Andrew M. Scott, and Peter W. Janes. 2023. "Eph Receptors in Cancer" Biomedicines 11, no. 2: 315. https://doi.org/10.3390/biomedicines11020315
APA StyleArora, S., Scott, A. M., & Janes, P. W. (2023). Eph Receptors in Cancer. Biomedicines, 11(2), 315. https://doi.org/10.3390/biomedicines11020315