
Smooth Muscle Cells of Dystrophic (mdx) Mice Are More Susceptible to Hypoxia; The Protective Effect of Reducing Ca2+ Influx



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Vascular Smooth Muscle Cells
2.3. Evaluation of Isolated VSMC
2.4. Ca2+ Selective Microelectrodes and Resting [Ca2+]i Measurements
2.5. Acute Hypoxia Protocol
2.6. Vascular Smooth Muscle Cell Viability
2.7. Solutions
2.8. Statistical Analysis
3. Results
3.1. Aberrant Resting Membrane Potential and [Ca2+]i in Mdx Vascular Smooth Cells
3.2. Hypoxia-Induced Muscle Depolarization and [Ca2+]i Overload
3.3. Reduction of Extracellular Ca2+ Prevented Hypoxia-Induced [Ca2+]i Overload
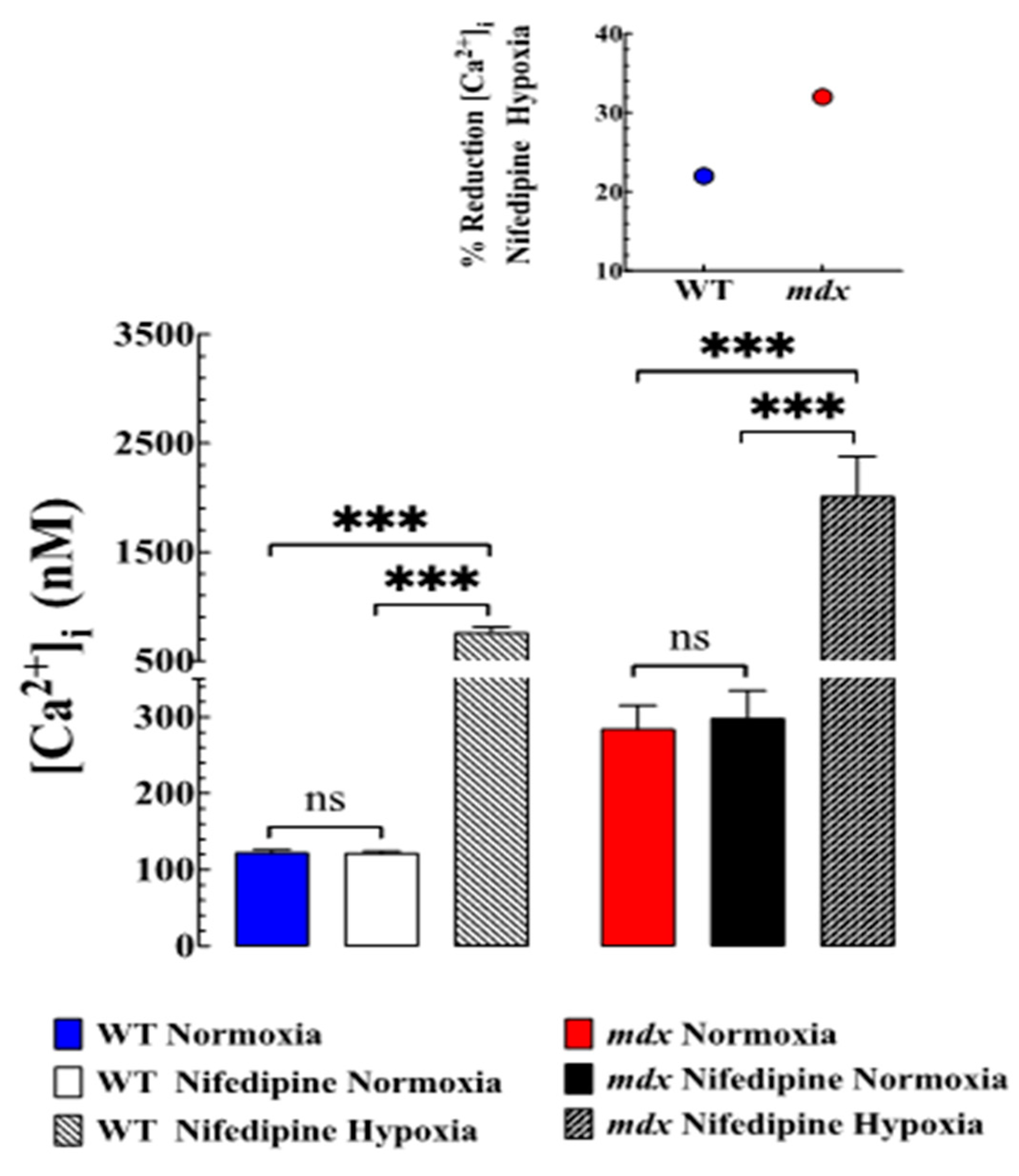
3.4. Nifedipine Did Not Modify Resting [Ca2+]i but Prevented Hypoxia-Induced Elevation of [Ca2+]i
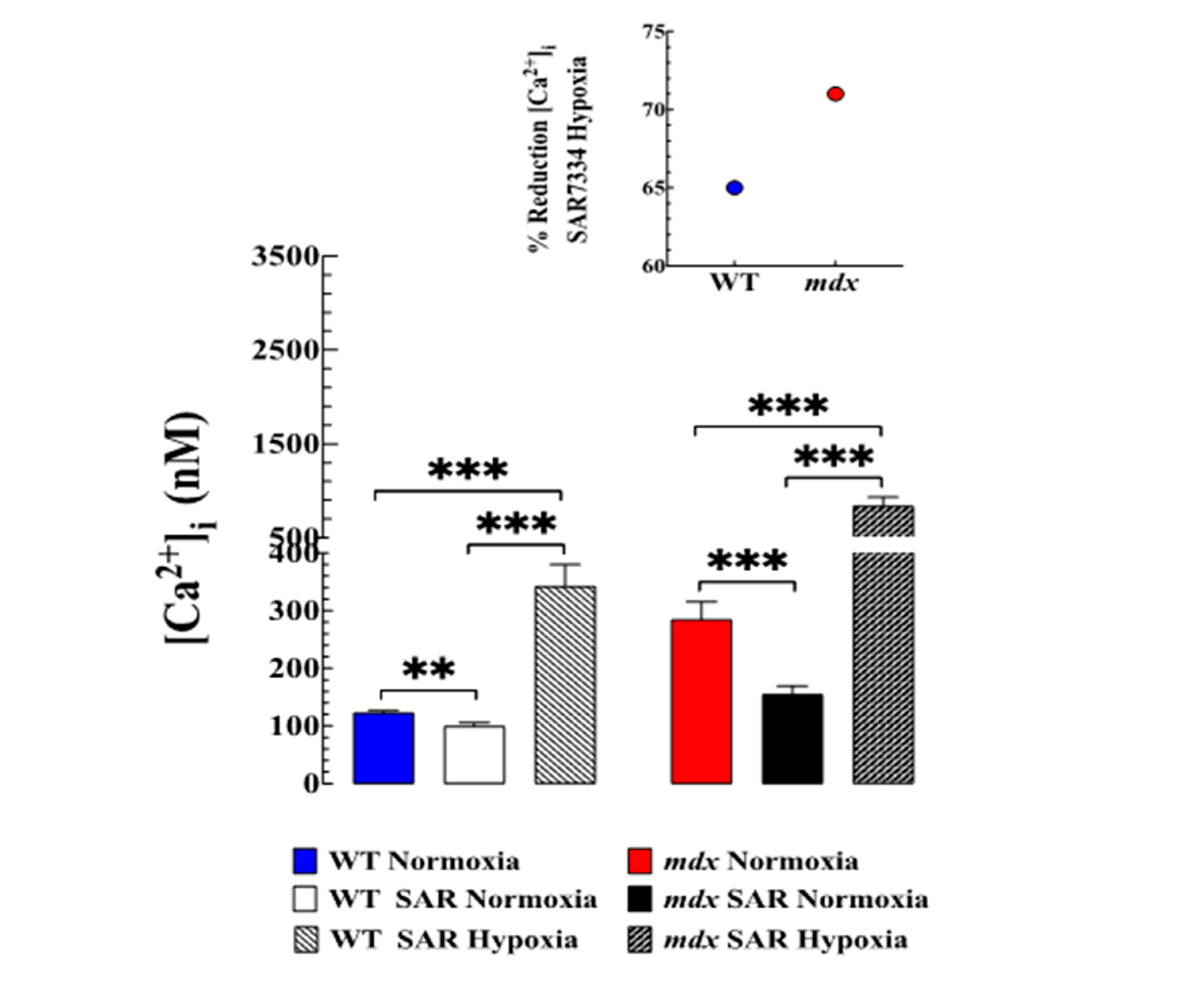
3.5. Contribution of TRPC-Mediated Ca2+ Entry to Hypoxia-Induced Increase in [Ca2+]i
3.6. SAR7334 Improved VSMC Viability Subjected to Hypoxia
4. Discussion
4.1. Study Limitations
4.2. Clinical Consideration
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, F.; Perez, C.F.; Liu, M.; Widrick, J.; Barton, E.R.; Allen, P.D.; Adams, J.A.; Lopez, J.R. Whole Body Periodic Acceleration Is an Effective Therapy to Ameliorate Muscular Dystrophy in mdx Mice. PLoS ONE 2014, 9, e106590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.R.; Kolster, J.; Zhang, R.; Adams, J. Increased constitutive nitric oxide production by whole body periodic acceleration ameliorates alterations in cardiomyocytes associated with utrophin/dystrophin deficiency. J. Mol. Cell. Cardiol. 2017, 108, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.R.; Uryash, A.; Faury, G.; Estève, E.; Adams, J.A. Contribution of TRPC Channels to Intracellular Ca2+ Dyshomeostasis in Smooth Muscle From mdx Mice. Front. Physiol. 2020, 11, 126. [Google Scholar] [CrossRef]
- Lopez, J.R.; Kolster, J.; Uryash, A.; Estève, E.; Altamirano, F.; Adams, J.A. Dysregulation of Intracellular Ca2+ in Dystrophic Cortical and Hippocampal Neurons. Mol. Neurobiol. 2016, 55, 603–618. [Google Scholar] [CrossRef]
- Gomez-Merino, E.; Bach, J.R. Duchenne muscular dystrophy: Prolongation of life by noninvasive ventilation and mechanically assisted coughing. Am. J. Phys. Med. Rehabil. 2002, 81, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Mhandire, D.Z.; Burns, D.P.; Roger, A.L.; O’Halloran, K.D.; ElMallah, M.K. Breathing in Duchenne muscular dystrophy: Translation to therapy. J. Physiol. 2022, 600, 3465–3482. [Google Scholar] [CrossRef]
- Barbe, F.; Quera-Salva, M.; McCann, C.; Gajdos, P.; Raphael, J.; de Lattre, J.; Agusti, A. Sleep-related respiratory disturbances in patients with Duchenne muscular dystrophy. Eur. Respir. J. 1994, 7, 1403–1408. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.E.M.; Calverley, P.M.A.; Edwards, R.H.T. Hypoxemia during Sleep in Duchenne Muscular Dystrophy. Am. Rev. Respir. Dis. 1988, 137, 884–888. [Google Scholar] [CrossRef]
- Mauro, M.A.L.; D’Angelo, M.G.; Aliverti, A. Sleep Disordered Breathing in Duchenne Muscular Dystrophy. Curr. Neurol. Neurosci. Rep. 2017, 17, 44. [Google Scholar] [CrossRef]
- Townsend, D. Diastolic dysfunction precedes hypoxia-induced mortality in dystrophic mice. Physiol. Rep. 2015, 3, e12513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelter, Z.; Straková, J.; Yellamilli, A.; Fischer, K.; Sharpe, K.M.; Townsend, D. Hypoxia-induced cardiac injury in dystrophic mice. Am. J. Physiol. Circ. Physiol. 2016, 310, H938–H948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metz, R.P.; Patterson, J.L.; Wilson, E. Vascular Smooth Muscle Cells: Isolation, Culture, and Characterization. Methods Mol. Biol. 2012, 843, 169–176. [Google Scholar] [CrossRef] [PubMed]
- AVMA. Guidelines for the Euthanasia of Animals. Available online: https://www.avma.org/KB/Policies/Documents/euthanasia.pdf2020 (accessed on 1 June 2020).
- Eltit, J.M.; Ding, X.; Pessah, I.N.; Allen, P.D.; Lopez, J.R. Nonspecific sarcolemmal cation channels are critical for the pathogenesis of malignant hyperthermia. FASEB J. 2013, 27, 991–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ervasti, J.M.; Campbell, K. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Tran, T.; Stelmack, G.L.; McNeill, K.; Gosens, R.; Mutawe, M.M.; Unruh, H.; Gerthoffer, W.T.; Halayko, A.J. Expression of the dystrophin-glycoprotein complex is a marker for human airway smooth muscle phenotype maturation. Am. J. Physiol. Cell. Mol. Physiol. 2008, 294, L57–L68. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, K.M.; McDonald, C.M.; Ingman, E.; Haas, J. Symptoms of upper gastrointestinal dysfunction in Duchenne muscular dys-trophy: Case-control study. Arch. Phys. Med. Rehabil. 1990, 71, 742–744. [Google Scholar]
- Miike, T.; Sugino, S.; Ohtani, Y.; Taku, K.; Yoshioka, K. Vascular endothelial cell injury and platelet embolism in Duchenne muscular dystrophy at the preclinical stage. J. Neurol. Sci. 1987, 82, 67–80. [Google Scholar] [CrossRef]
- Brown, I.A.; Diederich, L.; Good, M.; DeLalio, L.; Murphy, S.; Cortese-Krott, M.M.; Hall, J.L.; Le, T.H.; Isakson, B.E. Vascular Smooth Muscle Remodeling in Conductive and Resistance Arteries in Hypertension. Arter. Thromb. Vasc. Biol. 2018, 38, 1969–1985. [Google Scholar] [CrossRef]
- Robin, G.; López, J.R.; Espinal, G.M.; Hulsizer, S.; Hagerman, P.J.; Pessah, I.N. Calcium dysregulation and Cdk5-ATM pathway involved in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2017, 26, 2649–2666. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef] [PubMed]
- Danialou, G.; Comtois, A.S.; Dudley, R.; Karpati, G.; Vincent, G.; Rosiers, C.D.; Petrof, B.J. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 2001, 15, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.R.; Briceno, L.E.; Sanchez, V.; Horvart, D. Myoplasmic (Ca2+) in Duchenne muscular dystrophy patients. Acta Cient. Venez. 1987, 38, 503–504. [Google Scholar] [PubMed]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicotera, P.; Orrenius, S. The role of calcium in apoptosis. Cell Calcium 1998, 23, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Uryash, A.; Mijares, A.; Esteve, E.; Adams, J.A.; Lopez, J.R. Cardioprotective Effect of Whole Body Periodic Acceleration in Dystrophic Phenotype mdx Rodent. Front. Physiol. 2021, 12, 658042. [Google Scholar] [CrossRef]
- Yuan, X.J.; Goldman, W.F.; Tod, M.L.; Rubin, L.J.; Blaustein, M.P. Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am. J. Physiol. Cell. Mol. Physiol. 1993, 264 Pt 1, L116–L123. [Google Scholar] [CrossRef]
- Sung, J.Y.; Choi, H.C. Nifedipine inhibits vascular smooth muscle cell proliferation and reactive oxygen species production through AMP-activated protein kinase signaling pathway. Vasc. Pharmacol. 2012, 56, 1–8. [Google Scholar] [CrossRef]
- Altamirano, F.; Valladares, D.; Henríquez-Olguín, C.; Casas, M.; López, J.R.; Allen, P.D.; Jaimovich, E. Nifedipine Treatment Reduces Resting Calcium Concentration, Oxidative and Apoptotic Gene Expression, and Improves Muscle Function in Dystrophic mdx Mice. PLoS ONE 2013, 8, e81222. [Google Scholar] [CrossRef] [Green Version]
- Hess, P.; Lansman, J.B.; Tsien, R.W. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature 1984, 311 (Suppl. S1), 538–544. [Google Scholar] [CrossRef]
- Jurevičius, J.; Muckus, K.; Mačianskiene, R.; Chmel-Dunaj, G.; Jureviius, J. Influence of action potential duration and resting potential on effects of quinidine, lidocaine and ethmozin on in guinea-pig papillary muscles. J. Mol. Cell. Cardiol. 1991, 23, 103–114. [Google Scholar] [CrossRef]
- Maier, T.; Follmann, M.; Hessler, G.; Kleemann, H.-W.; Hachtel, S.; Fuchs, B.; Weissmann, N.; Linz, W.; Schmidt, T.; Löhn, M.; et al. Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels. Br. J. Pharmacol. 2015, 172, 3650–3660. [Google Scholar] [CrossRef] [PubMed]
- Vandebrouck, C.; Martin, D.; Schoor, M.C.-V.; Debaix, H.; Gailly, P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 2002, 158, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.; Chan, W.Y.; Leung, P.C.; Kwan, H.Y.; Liu, C.; Huang, Y.; Michel, V.; Yew, D.T.; Yao, X. Expression of TRPC homologs in endothelial cells and smooth muscle layers of human arteries. Histochem. Cell Biol. 2004, 122, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Inoue, R.; Jensen, L.J.; Shi, J.; Morita, H.; Nishida, M.; Honda, A.; Ito, Y. Transient Receptor Potential Channels in Cardiovascular Function and Disease. Circ. Res. 2006, 99, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yotsukura, M.; Miyagawa, M.; Tsua, T.; Ishihara, T.; Ishikawa, K. Pulmonary hypertension in progressive muscular dystrophy of the Duchenne type. Jpn. Circ. J. 1988, 52, 321–326. [Google Scholar] [CrossRef] [Green Version]
- AbuZahra, H.M.; Rajendran, P.; Ismail, M.B. Zerumbone Exhibit Protective Effect against Zearalenone Induced Toxicity via Ameliorating Inflammation and Oxidative Stress Induced Apoptosis. Antioxidants 2021, 10, 1593. [Google Scholar] [CrossRef]
- Brookes, P.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Baydun, A.; Gilgoff, I.; Prentice, W.; Carlson, M.; Fischer, D.A. Decline in Respiratory Function and Experience with Long-Term Assisted Ventilation in Advanced Duchenne’s Muscular Dystrophy. Chest 1990, 97, 884–889. [Google Scholar] [CrossRef]
- Melacini, P.; Vianello, A.; Villanova, C.; Fanin, M.; Miorin, M.; Angelini, C.; Volta, S.D. Cardiac and respiratory involvement in advanced stage Duchenne muscular dystrophy. Neuromuscul. Disord. 1996, 6, 367–376. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uryash, A.; Mijares, A.; Estève, E.; Adams, J.A.; Lopez, J.R. Smooth Muscle Cells of Dystrophic (mdx) Mice Are More Susceptible to Hypoxia; The Protective Effect of Reducing Ca2+ Influx. Biomedicines 2023, 11, 623. https://doi.org/10.3390/biomedicines11020623
Uryash A, Mijares A, Estève E, Adams JA, Lopez JR. Smooth Muscle Cells of Dystrophic (mdx) Mice Are More Susceptible to Hypoxia; The Protective Effect of Reducing Ca2+ Influx. Biomedicines. 2023; 11(2):623. https://doi.org/10.3390/biomedicines11020623
Chicago/Turabian StyleUryash, Arkady, Alfredo Mijares, Eric Estève, Jose A. Adams, and Jose R. Lopez. 2023. "Smooth Muscle Cells of Dystrophic (mdx) Mice Are More Susceptible to Hypoxia; The Protective Effect of Reducing Ca2+ Influx" Biomedicines 11, no. 2: 623. https://doi.org/10.3390/biomedicines11020623
APA StyleUryash, A., Mijares, A., Estève, E., Adams, J. A., & Lopez, J. R. (2023). Smooth Muscle Cells of Dystrophic (mdx) Mice Are More Susceptible to Hypoxia; The Protective Effect of Reducing Ca2+ Influx. Biomedicines, 11(2), 623. https://doi.org/10.3390/biomedicines11020623