
Pancreatic Cancer Organoids: An Emerging Platform for Precision Medicine?




Abstract
:1. Introduction
2. Current Status
3. Organoids
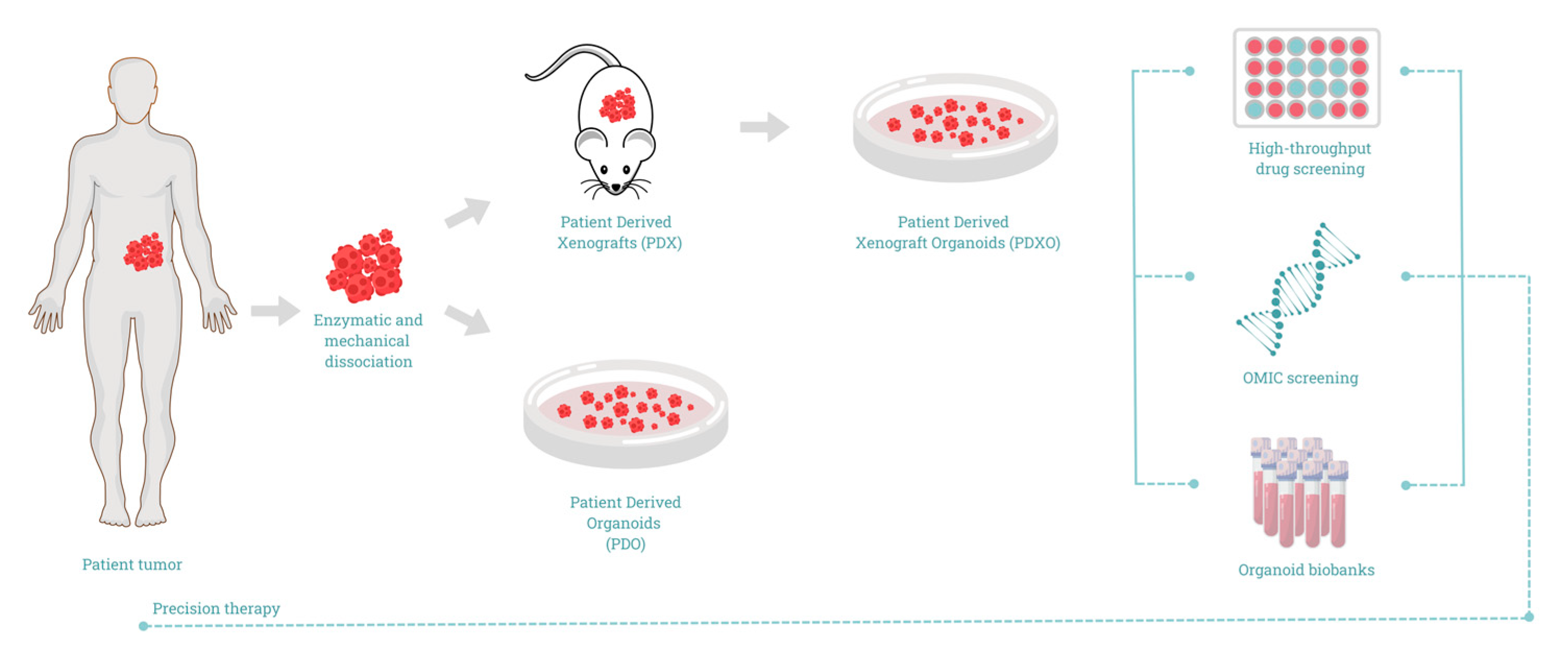
Methods of PDAC Organoids Development
4. Organoids and Tumoroids in the Battle against Pancreatic Cancer
5. Tumor Organoids in Precision Medicine for PDAC
6. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer Burden Statistics and Trends across Europe|ECIS. Available online: https://ecis.jrc.ec.europa.eu/ (accessed on 19 December 2022).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ben, Q.; Xu, M.; Ning, X.; Liu, J.; Hong, S.; Huang, W.; Zhang, H.; Li, Z. Diabetes mellitus and risk of pancreatic cancer: A meta-analysis of cohort studies. Eur. J. Cancer 2011, 47, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, C.; Lucenteforte, E.; Silverman, D.T.; Petersen, G.; Bracci, P.M.; Ji, B.T.; Negri, E.; Li, D.; Risch, H.A.; Olson, S.H.; et al. Cigarette smoking and pancreatic cancer: An analysis from the International Pancreatic Cancer Case-Control Consortium (Panc4). Ann. Oncol. 2012, 23, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Duell, E.; Lucenteforte, E.; Olson, S.; Bracci, P.; Li, D.; Risch, H.; Silverman, D.; Ji, B.; Gallinger, S.; Holly, E.; et al. Pancreatitis and pancreatic cancer risk: A pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann. Oncol. 2012, 23, 2964–2970. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Kenner, B.J.; Chari, S.T.; Maitra, A.M.; Srivastava, S.P.; Cleeter, D.F.; Go, V.L.W.; Rothschild, L.J.M.; Goldberg, A.E.B. Early Detection of Pancreatic Cancer-a Defined Future Using Lessons From Other Cancers: A White Paper. Pancreas 2016, 45, 1073–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, L.; Bakir, B.; Chatterji, P.; Dantes, Z.; Reichert, M.; Rustgi, A.K. Pancreas 3D Organoids: Current and Future Aspects as a Research Platform for Personalized Medicine in Pancreatic Cancer. Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Blackford, A.L.; Molin, M.D.; Wolfgang, C.L.; Goggins, M. Time to progression of pancreatic ductal adenocarcinoma from low-to-high tumour stages. Gut 2015, 64, 1783–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamposioras, K.; Tsimplouli, C.; Verbeke, C.; Anthoney, A.; Daoukopoulou, A.; Papandreou, C.N.; Sakellaridis, N.; Vassilopoulos, G.; Potamianos, S.P.; Liakouli, V.; et al. Silencing of caveolin-1 in fibroblasts as opposed to epithelial tumor cells results in increased tumor growth rate and chemoresistance in a human pancreatic cancer model. Int. J. Oncol. 2019, 54, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Hennig, A.; Wolf, L.; Jahnke, B.; Polster, H.; Seidlitz, T.; Werner, K.; Aust, D.E.; Hampe, J.; Distler, M.; Weitz, J.; et al. CFTR Expression Analysis for Subtyping of Human Pancreatic Cancer Organoids. Stem Cells Int. 2019, 2019, 1024614. [Google Scholar] [CrossRef] [Green Version]
- Baker, L.A.; Tiriac, H.; Clevers, H.; Tuveson, D.A. Modeling pancreatic cancer with organoids. Trends Cancer 2016, 2, 176–190. [Google Scholar] [CrossRef] [Green Version]
- Ehlen, L.; Arndt, J.; Treue, D.; Bischoff, P.; Loch, F.N.; Hahn, E.; Kotsch, K.; Klauschen, F.; Beyer, K.; Margonis, G.A.; et al. Novel methods for in vitro modeling of pancreatic cancer reveal important aspects for successful primary cell culture. BMC Cancer 2020, 20, 417. [Google Scholar] [CrossRef]
- Mimeault, M.; Brand, R.E.; Sasson, A.A.; Batra, S.K. Recent advances on the molecular mechanisms involved in pancreatic cancer progression and therapies. Pancreas 2005, 31, 301–316. [Google Scholar] [CrossRef]
- Wijnen, R.; Pecoraro, C.; Carbone, D.; Fiuji, H.; Avan, A.; Peters, G.J.; Giovannetti, E.; Diana, P. Cyclin Dependent Kinase-1 (CDK-1) Inhibition as a Novel Therapeutic Strategy against Pancreatic Ductal Adenocarcinoma (PDAC). Cancers 2021, 13, 4389. [Google Scholar] [CrossRef]
- Pecoraro, C.; Parrino, B.; Cascioferro, S.; Puerta, A.; Avan, A.; Peters, G.J.; Diana, P.; Giovannetti, E.; Carbone, D. A New Oxadiazole-Based Topsentin Derivative Modulates Cyclin-Dependent Kinase 1 Expression and Exerts Cytotoxic Effects on Pancreatic Cancer Cells. Molecules 2021, 27, 19. [Google Scholar] [CrossRef]
- Ding, L.; Billadeau, D.D. Glycogen Synthase Kinase-3β: A novel therapeutic target for pancreatic cancer. Expert Opin. Ther. Targets 2020, 24, 417. [Google Scholar] [CrossRef] [PubMed]
- Edderkaoui, M.; Chheda, C.; Soufi, B.; Zayou, F.; Hu, R.W.; Ramanujan, V.K.; Pan, X.; Boros, L.G.; Tajbakhsh, J.; Madhav, A.; et al. An Inhibitor of GSK3B and HDACs Kills Pancreatic Cancer Cells and Slows Pancreatic Tumor Growth and Metastasis in Mice. Gastroenterology 2018, 155, 1985. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.; Seoane, J.; Le Tourneau, C.; Siu, L.; Marais, R.; Michiels, S.; Soria, J.; Campbell, P.; Normanno, N.; Scarpa, A.; et al. The European Society for Medical Oncology (ESMO) Precision Medicine Glossary. Ann. Oncol. 2018, 29, 30–35. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Delord, J.-P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.-A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Keepers, Y.P.; Pizao, P.E.; Peters, G.J.; van Ark-Otte, J.; Winograd, B.; Pinedo, H.M. Comparison of the sulforhodamine B protein and tetrazolium (MTT) assays for in vitro chemosensitivity testing. Eur. J. Cancer 1991, 27, 897–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Calvo, I.; Weber, C.R.; Ray, M.; Brown, M.; Kirby, K.; Nandi, R.K.; Long, T.M.; Sparrow, S.M.; Ugolkov, A.; Qiang, W.; et al. Human Organoids Share Structural and Genetic Features with Primary Pancreatic Adenocarcinoma Tumors. Mol. Cancer Res. 2019, 17, 70–83. [Google Scholar] [CrossRef] [Green Version]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef]
- Schrag, D.; Garewal, H.S.; Burstein, H.J.; Samson, D.J.; Von Hoff, D.D.; Somerfield, M.R. American Society of Clinical Oncology Technology Assessment: Chemotherapy sensitivity and resistance assays. J. Clin. Oncol. 2004, 22, 3631–3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samson, D.J.; Seidenfeld, J.; Ziegler, K.; Aronson, N. Chemotherapy sensitivity and resistance assays: A systematic review. J. Clin. Oncol. 2004, 22, 3618–3630. [Google Scholar] [CrossRef]
- Cortazar, P.; Johnson, B.E. Review of the efficacy of individualized chemotherapy selected by in vitro drug sensitivity testing for patients with cancer. J. Clin. Oncol. 1999, 17, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, P.E.; Cree, I.A.; Kurbacher, C.M.; Hartmann, D.M.; Linder, D.; Harel, G.; Gleiberman, I.; Caruso, P.A.; Ricks, S.H.; Untch, M.; et al. Chemosensitivity Testing of Human Tumors Using a Microplate Adenosine Triphosphate Luminescence Assay: Clinical Correlation for Cisplatin Resistance of Ovarian Carcinoma. Cancer Res. 1995, 55, 5276–5282. [Google Scholar]
- Cree, I.A.; Kurbacher, C.M.; Untch, M.; Sutherland, L.A.; Hunter, E.M.; Mc Subedi, A.; James, E.A.; Dewar, J.A.; Preece, P.E.; Andreotti, P.E.; et al. Correlation of the clinical response to chemotherapy in breast cancer with ex vivo chemosensitivity. Anticancer. Drugs 1996, 7, 630–635. [Google Scholar] [CrossRef]
- Kurbacher, C.M.; Cree, I.A.; Bruckner, H.W.; Brenne, U.; Kurbacher, J.A.; Müller, K.; Ackermann, T.; Gilster, T.J.; Wilhelm, L.M.; Engel, H.; et al. Use of an ex vivo ATP luminescence assay to direct chemotherapy for recurrent ovarian cancer. Anticancer. Drugs 1998, 9, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Sereti, E.; Karagianellou, T.; Kotsoni, I.; Magouliotis, D.; Kamposioras, K.; Ulukaya, E.; Sakellaridis, N.; Zacharoulis, D.; Dimas, K. Patient Derived Xenografts (PDX) for personalized treatment of pancreatic cancer: Emerging allies in the war on a devastating cancer? J. Proteom. 2018, 188, 107–118. [Google Scholar] [CrossRef]
- Sutherland, R.M. Importance of Critical Metabolites and Cellular Interactions in the Biology of Microregions of Tumors. Cancer 1986, 58, 1668–1680. [Google Scholar] [CrossRef]
- Lee, J.; Cuddihy, M.J.; Kotov, N.A. Three-Dimensional Cell Culture Matrices: State of the Art. Tissue Eng. Part B Rev. 2008, 14, 61–86. [Google Scholar] [CrossRef] [Green Version]
- Kunz-Schughart, L.A.; Kreutz, M.; Knuechel, R. Multicellular spheroids: A three-dimensional in vitro culture system to study tumour biology. Int. J. Exp. Pathol. 1998, 79, 1. [Google Scholar] [CrossRef]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almany, L.; Seliktar, D. Biosynthetic hydrogel scaffolds made from fibrinogen and polyethylene glycol for 3D cell cultures. Biomaterials 2005, 26, 2467–2477. [Google Scholar] [CrossRef]
- Friedrich, J.; Ebner, R.; Kunz-Schughart, L. Experimental anti-tumor therapy in 3-D: Spheroids—Old hat or new challenge? Int. J. Radiat. Biol. 2007, 83, 849–871. [Google Scholar] [CrossRef]
- Costa, E.C.; Moreira, A.F.; De Melo-Diogo, D.M.; Gaspar, V.M.; Carvalho, M.P.; Correia, I.J. 3D tumor spheroids: An overview on the tools and techniques used for their analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef]
- Yeon, S.-E.; No, D.Y.; Lee, S.-H.; Nam, S.W.; Oh, I.-H.; Lee, J.; Kuh, H.-J. Application of concave microwells to pancreatic tumor spheroids enabling anticancer drug evaluation in a clinically relevant drug resistance model. PLoS ONE 2013, 8, e73345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Ootani, A.; Li, X.; Sangiorgi, E.; Ho, Q.T.; Ueno, H.; Toda, S.; Sugihara, H.; Fujimoto, K.; Weissman, I.L.; Capecchi, M.R.; et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med. 2009, 15, 701–706. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.M.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.J.; Castellanos, J.A.; Nagathihalli, N.S.; Merchant, N.B.; Skala, M.C. Optical Imaging of Drug-Induced Metabolism Changes in Murine and Human Pancreatic Cancer Organoids Reveals Heterogeneous Drug Response. Pancreas 2016, 45, 863–869. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.I.; Jeon, A.R.; Kim, M.K.; Lee, Y.S.; Im, J.E.; Koh, J.W.; Han, S.S.; Kong, S.Y.; Yoon, K.A.; Koh, Y.H.; et al. Development of Patient-Derived Preclinical Platform for Metastatic Pancreatic Cancer: PDOX and a Subsequent Organoid Model System Using Percutaneous Biopsy Samples. Front. Oncol. 2019, 9, 875. [Google Scholar] [CrossRef] [Green Version]
- Huch, M.; Bonfanti, P.; Boj, S.F.; Sato, T.; Loomans, C.J.M.; Van De Wetering, M.; Sojoodi, M.; Li, V.S.W.; Schuijers, J.; Gracanin, A.; et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013, 32, 2708–2721. [Google Scholar] [CrossRef] [Green Version]
- Longo, D.L.; Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic Adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [Green Version]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef] [PubMed]
- Marjon, K.; Cameron, M.J.; Quang, P.; Clasquin, M.F.; Mandley, E.; Kunii, K.; McVay, M.; Choe, S.; Kernytsky, A.; Gross, S.; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016, 15, 574–587. [Google Scholar] [CrossRef] [Green Version]
- Seppälä, T.T.; Zimmerman, J.W.; Sereni, E.; Plenker, D.; Suri, R.M.; Rozich, N.; Blair, A.; Thomas, D.L.I.B.; Teinor, J.B.; Javed, A.; et al. Patient-derived Organoid Pharmacotyping is a Clinically Tractable Strategy for Precision Medicine in Pancreatic Cancer. Ann. Surg. 2020, 272, 427–435. [Google Scholar] [CrossRef]
- Farshadi, E.A.; Chang, J.; Sampadi, B.; Doukas, M.; Land, F.V.; van der Sijde, F.; Vietsch, E.E.; Pothof, J.; Koerkamp, B.G.; van Eijck, C.H. Organoids Derived from Neoadjuvant FOLFIRINOX Patients Recapitulate Therapy Resistance in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2021, 27, 6602–6612. [Google Scholar] [CrossRef] [PubMed]
- Grossman, J.E.; Muthuswamy, L.; Huang, L.; Akshinthala, D.; Perea, S.; Gonzalez, R.S.; Tsai, L.L.; Cohen, J.; Bockorny, B.; Bullock, A.J.; et al. Organoid Sensitivity Correlates with Therapeutic Response in Patients with Pancreatic Cancer. Clin. Cancer Res. 2022, 28, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Sharick, J.T.; Walsh, C.M.; Sprackling, C.M.; Pasch, C.A.; Pham, D.L.; Esbona, K.; Choudhary, A.; Garcia-Valera, R.; Burkard, M.E.; McGregor, S.M.; et al. Metabolic Heterogeneity in Patient Tumor-Derived Organoids by Primary Site and Drug Treatment. Front. Oncol. 2020, 10, 553. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, L.; Vähä-Koskela, M.; Juusola, M.; Mustonen, H.; Wennerberg, K.; Hagström, J.; Puolakkainen, P.; Seppänen, H. Pancreatic Cancer Organoids in the Field of Precision Medicine: A Review of Literature and Experience on Drug Sensitivity Testing with Multiple Readouts and Synergy Scoring. Cancers 2022, 14, 525. [Google Scholar] [CrossRef]
- Koike, H.; Iwasawa, K.; Ouchi, R.; Maezawa, M.; Giesbrecht, K.; Saiki, N.; Ferguson, A.; Kimura, M.; Thompson, W.L.; Wells, J.M.; et al. Modelling human hepato-biliary-pancreatic organogenesis from the foregut-midgut boundary. Nature 2019, 574, 112–116. [Google Scholar] [CrossRef]
- Tiriac, H.; French, R.; Lowy, A.M. Isolation and Characterization of Patient-derived Pancreatic Ductal Adenocarcinoma Organoid Models. J. Vis. Exp. 2020, 2020, e60364. [Google Scholar] [CrossRef]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef]
- Grebenyuk, S.; Ranga, A. Engineering Organoid Vascularization. Front. Bioeng. Biotechnol. 2019, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, S.; Winter, P.S.; Navia, A.W.; Williams, H.L.; DenAdel, A.; Lowder, K.E.; Galvez-Reyes, J.; Kalekar, R.L.; Mulugeta, N.; Kapner, K.S.; et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 2021, 184, 6119–6137.e26. [Google Scholar] [CrossRef] [PubMed]
- de la Peña, D.O.; Trabulo, S.M.D.; Collin, E.; Liu, Y.; Sharma, S.; Tatari, M.; Behrens, D.; Erkan, M.; Lawlor, R.T.; Scarpa, A.; et al. Bioengineered 3D models of human pancreatic cancer recapitulate in vivo tumour biology. Nat. Commun. 2021, 12, 5623. [Google Scholar] [CrossRef]
- Park, S.E.; Georgescu, A.; Huh, D. Organoids-on-a-chip. Science 2019, 364, 960–965. [Google Scholar] [CrossRef]
- Ponz-Sarvise, M.; Corbo, V.; Tiriac, H.; Engle, D.D.; Frese, K.K.; Oni, T.E.; Hwang, C.-I.; Öhlund, D.; Chio, I.I.C.; Baker, L.A.; et al. Identification of Resistance Pathways Specific to Malignancy Using Organoid Models of Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 6742–6755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachir, E.H.; Poiraud, C.; Paget, S.; Stoup, N.; El Moghrabi, S.; Duchêne, B.; Jouy, N.; Bongiovanni, A.; Tardivel, M.; Weiswald, L.; et al. A new pancreatic adenocarcinoma-derived organoid model of acquired chemoresistance to FOLFIRINOX: First insight of the underlying mechanisms. Biol. Cell 2022, 114, 32–55. [Google Scholar] [CrossRef]
- Kumar, K.; Chow, C.R.; Ebine, K.; Arslan, A.D.; Kwok, B.; Bentrem, D.J.; Eckerdt, F.D.; Platanias, L.C.; Munshi, H.G. Differential Regulation of ZEB1 and EMT by MAPK-interacting protein kinases (MNKs) and eIF4E in Pancreatic Cancer. Mol. Cancer Res. 2016, 14, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Navarro-Serer, B.; Jeong, Y.J.; Chianchiano, P.; Xia, L.; Luchini, C.; Veronese, N.; Dowiak, C.; Ng, T.; Trujillo, M.A.; et al. Pattern of Invasion in Human Pancreatic Cancer Organoids Is Associated with Loss of SMAD4 and Clinical Outcome. Cancer Res. 2020, 80, 2804–2817. [Google Scholar] [CrossRef]
- Wang, E.; Xiang, K.; Zhang, Y.; Wang, X.-F. Patient-derived organoids (PDOs) and PDO-derived xenografts (PDOXs): New opportunities in establishing faithful pre-clinical cancer models. J. Natl. Cancer Cent. 2022, 2, 263–276. [Google Scholar] [CrossRef]


{kind=link}
| Reference [No] | Sample | Matrix | Culture Medium | Transplantation of Organoids | Success of Organoid Culture | Passage | Targeted Treatment on Organoids | Storage |
|---|---|---|---|---|---|---|---|---|
| Boj et al. 2015 [44] | Fresh tumors from surgical resection and FNA biopsies | GFR Matrigel (embedded) | AdDMEM/F12 supplemented with: (1) Glutamax 1X, (2) Hepes 1X, (3) Noggin 10% v/v or recombinant protein 0.1 μg/mL, (4) Gastrin 10 nM, (5) Nicotinamide 10 mM, (6) R-spondin-1 10% v/v, (7) EGF 50 ng/mL, (8) FGF-10 100 ng/mL, (9) N-acetyl-L-cysteine 1 mM, (10) B27 supplement, (11) Wnt3a 50% v/v, (12) primocin 1 mg/mL, (13) Penicillin-streptomycin 1X, (14) A83-01 0.5 μΜ | PDOX creation by orthotopical injection of human PDAC organoids to Nu/Nu mice, PanIN like lesions | 75% | Indefinite passaging capability | Not mentioned | Organoids can be cryo-preserved |
| Huang et al. 2015 [45] | Fresh tumors from surgical resection | Bed of Matrigel | Pancreatic Progenitor and Tumor Organoid Media (PTOM) consisted of DMEM with (1) 1% B27, (2) 50 ug/mL ascorbic acid, (3) 20 μg/mL insulin, (4) 0.25 μg/mL hydrocortisone, (5) 100 ng/mL FGF2, (6) 100 nM all-trans retinoic acid, (7) 10 μΜ Υ267632. At day 8 in 3D culture, culture medium was replaced with fresh pancreatic organoid maintenance medium (POMM, which contains 1% B27 and 50 μg/mL ascorbic acid) with 5% Matrigel. Culture media was replaced every 4 days. Pancreatic Organoid Differentation Media 1 (PODM 1) consisted of DMEM supplemented with (1) 1% B27, (2) 300 μΜ 2-phospho ascorbic acid, (3) 100 ng/ml FGF7, (4) 10 ng/mL EGF, (5) 1 μΜ A8301 and (6) 1 μΜ DBZ. Pancreatic Organoid Differentation Medium 2 (PODM 2) consisted of DMEM supplemented with (1) 1% B27, (2) 300 μΜ 2-phospho ascorbic acid, (3) 100 ng/mL FGF7 and (4) 10 ng/mL EGF. | 0.3–0.5 million cells were injected into the mammary glands of female NOD/SCID mice | 85% (17/20 samples) | At day 16, organoids are treated with collagenase for 2 hours and then additionaly with trypsin for another 10–30 min. The cells are collected and re-seeded in 3D culture. | Drugs targeting epigenetic regulators were tested, i.e A366 and UNC1999, alone or in combination with Gemcitabine | Tumor organoids could be freezed-thawed and still re-establish cultures that preserve phase and hematoxylin and eosin stain (H&E) morphology across passages |
| Walsh et al. 2016 [46] | Biopsy from a patient with poorly differentiated pancreatic ductal adenocarcinoma | Matrigel (embedded at 1:2 ratio) | RPMI supllemented with (1) 10% FBS, (2) 1% Penicillin/Streptomycin, (3) 10 ng EGFR. Culture medium was replaced every 3 days. | Not reported | Not reported | Not mentioned | Organoids were cultured for 3 days and treated with (1) DMSO (control), (2) Gemcitabine (25 μg/mL), (3) AZD1480 (100 nM), (4) AZD6244 (4 μM), (5) XL147 (25 nM) and (6) combination of gemcitabine plus (a) AZD1480, (b) AZD1480 + AZD6244, (c) AZD1480 + AZD6244 + XL147 | Not reported |
| Romero-Calvo et al. 2019 [22] | Biopsies (n = 10) from PDAC patients and PDX derived organoid cultures | GFR Matrigel (embedded) | Complete media [Intesticult (Stemcell Technologies, 6005), (1) A83-01 (0.5 mmol/L), (2) fibroblast growth factor 10 (FGF10, 100 ng/mL), (3) gastrin I (10 nmol/L), (3) N-acetyl-L-cysteine (10 mmol/L), (4) nicotinamide (10 mmol/L), (5) B27 1X, (6) primocin (1 mg/mL), and (7) Y-27632 (10.5 mmol/L) | Not reported | 100% | Organoids were passaged via mechanical dissociation with TrypLE Express and passage was performed weekly with a 1:2 ratio. | Chemotherapeutic drugs tested: (1) gemcitabine (3 nmol/L, 10 nmol/L, 30 nmol/L) (2) FOLFIRINOX (10 nmol/L) (3) Abraxane (10 nm/L) | Not reported |
| Neal et al. 2018 [47] | 100 patients representing 19 distinct tissue sites and 28 unique disease subtypes | ALI method | ADMEM/F12 supplemented with 50% Wnt3a, RSPO1, Noggin-conditioned media (L-WRN) with HEPES (1 mM), Glutamax (1X), Nicotinamide (10 mM), N-Acetylcysteine (1 mM), B-27 without vitamin A (1X), A83-01 (0.5 mM), Pen-Strep Glutamine (1X), Gastrin (10 nM), SB-202190 (10 mM), and EGF (50 ng/mL). The transwell containing tumor tissue and collagen was placed into an outer 60 mm cell culture dish containing 1.0 mL of medium. | Not reported | 73% (at 1-month culture across tumor histologies) | After continued growth (>4 passages, >100 days) some PDOs did not maintain the complex tissue architecture exhibiting a simple, cystic morphology. PDOs could be xenografted into immunocompromised mice and re-derived as organoids. | Not reported | 80% (cryo-recovered and serially re-propagated every few weeks) |
| Choi et al. 2019 [48] | Biopsies of liver metastasis from patients with PDAC were used to develop PDOX in female Hsd:athymic nude-Foxn1 mice aged 5–8 weeks. Then, first passage cells isolated from F1 PDX tissues were used to develop organoids. | GFR Matrigel | Advanced DMEM/F12 with (1) B27, (2) N-acetylcysteine, (3) EGF, (4) FGF-10, (5) R-spondin 1 and (6) Noggin. Medium was replaced every 3 days. | Not reported | Not mentioned | The researchers used first passage cells isolated from F1 PDOX tissues | Organoids were treated with (1) DMSO (control), (2) Gemcitabine-HCL and (3) Albumin-bound paclitaxel for 7 days | Not mentioned |
| Hennig et al. 2019 [11] | Human primary tumor samples from 31 treatment-naïve PDAC patients: 25 specimens from surgical tumor resections and 6 from endoscopic ultrasound- (EUS-) guided fine needle aspiration (FNA) | GFR Matrigel | Advanced DMEM/F12 suplemented with (1) WNT3a 50% v/v, (2) noggin 10% v/v, (3) Rspondin1 10% v/v, (4) B27 1X, (5) nicotinamide 10 mM, (6) Gastrin 1 nM, (7) n-acetyl-L-cysteine 1 mM, (8) primocin 1 mg/mL, (9) mEGF 50 ng/mL, (10) hFGF10 100 ng/mL, (11) A83-01 0.5 μΜ, (12) Ν2 1Χ | Not reported | 71% (68% from surgical resections and 83% from fine needle aspirations) | Not mentioned | Chemotherapeutic drugs tested: (1) Gemcitabine at 1 μΜ, 200 nM, 100 nM, 50 nM, 25 nM, 10 nM and 1 nM, (2) 5-Fuorouracil at 50 μΜ, 25 μΜ, 10 μΜ, 5 μΜ, 1 μΜ, 100 nM and 10 nM, (3) Oxaliplatin at 250 μΜ, 25 μΜ, 10 μΜ, 1 μΜ, 100 nM, 10 nM and 1 nM and (4) Irinotecan at 250 μΜ, 25 μΜ, 10 μΜ, 1 μΜ, 100 nM, 10 nM and 1 nM | Repeated freeze-thaw cycles |
| Supplements | Function |
|---|---|
| 2-phospho ascorbic acid | Maintains cellular differentiation |
| A 83-01 | TGF-b inhibitor |
| B27 supplement | Vitamins and growth factors mix |
| EGF | Mitogen. |
| EGFR | Mitogen |
| FGF-10 | Stimulates cell proliferation |
| FGF-2 | Stimulates cell proliferation |
| FGF-7 | Stimulates cell proliferation |
| Gastrin | TGF-b inhibitor |
| Glutamax | Substitute for L-glutamine |
| HEPES | Provides a buffered pH environment |
| IGF-1 | Mitogen |
| N-acetyl-L-cysteine | Precursor in glutathione synthesis |
| N2 supplement | Vitamins and growth factors mix |
| Nicotinamide | Maintain cell stemness and cystic phenotype |
| Noggin | Inhibits BMP-4 |
| Penicillin/streptomycin | Avoid bacterial contamination |
| PGE2 | Maintain cell stemness and cystic phenotype |
| Primocin | Antibiotic |
| RSPO-1 | Wnt signaling activator |
| SB202190 | p38 MAPK inhibitor |
| Wnt3A conditioned Medium | Wnt signaling activator |
| Y-27632 | ROCK inhibitor to prevent anoikis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sereti, E.; Papapostolou, I.; Dimas, K. Pancreatic Cancer Organoids: An Emerging Platform for Precision Medicine? Biomedicines 2023, 11, 890. https://doi.org/10.3390/biomedicines11030890
Sereti E, Papapostolou I, Dimas K. Pancreatic Cancer Organoids: An Emerging Platform for Precision Medicine? Biomedicines. 2023; 11(3):890. https://doi.org/10.3390/biomedicines11030890
Chicago/Turabian StyleSereti, Evangelia, Irida Papapostolou, and Konstantinos Dimas. 2023. "Pancreatic Cancer Organoids: An Emerging Platform for Precision Medicine?" Biomedicines 11, no. 3: 890. https://doi.org/10.3390/biomedicines11030890
APA StyleSereti, E., Papapostolou, I., & Dimas, K. (2023). Pancreatic Cancer Organoids: An Emerging Platform for Precision Medicine? Biomedicines, 11(3), 890. https://doi.org/10.3390/biomedicines11030890