
Mechanisms of COVID-19 Associated Pulmonary Thrombosis: A Narrative Review


Abstract
:1. Introduction
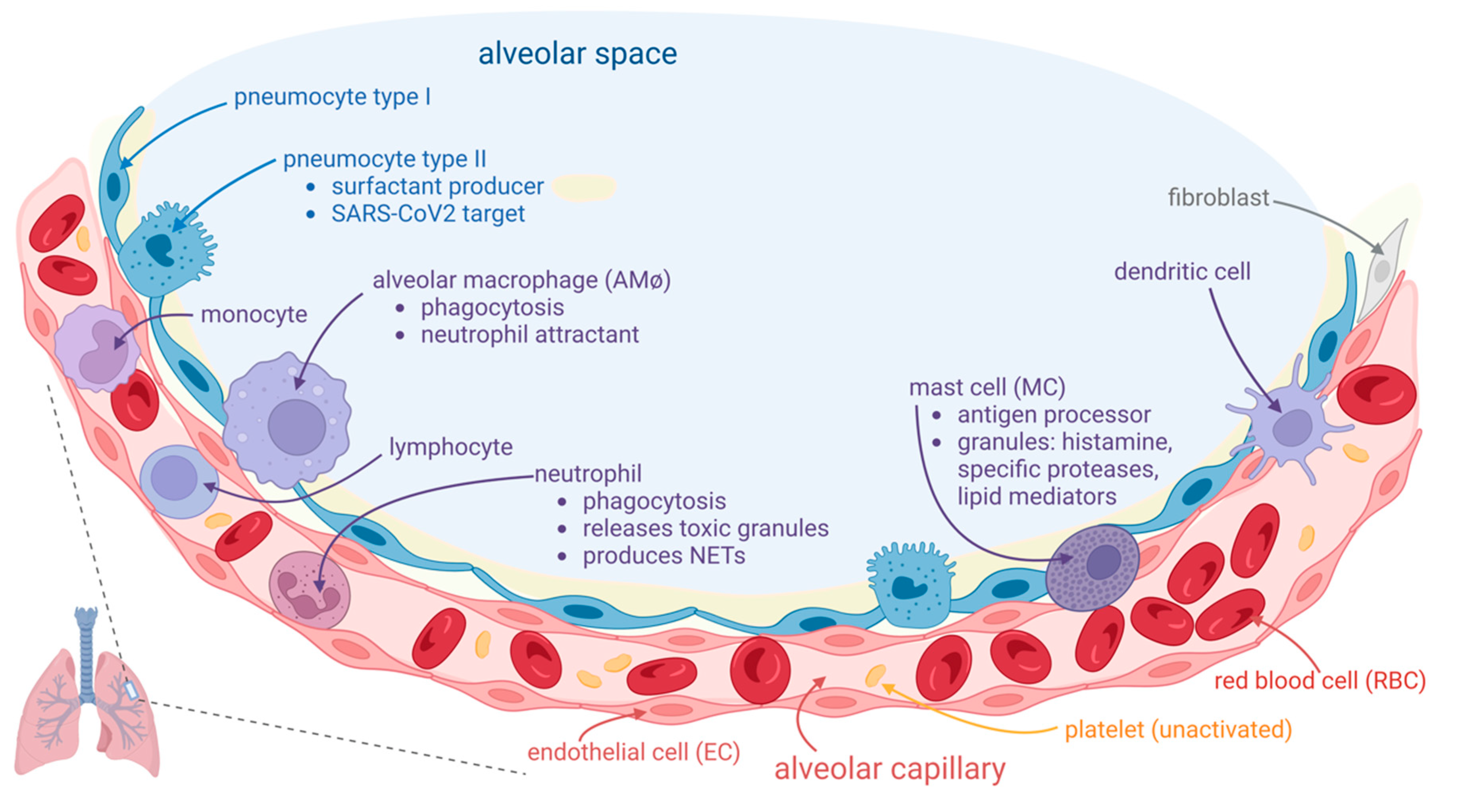
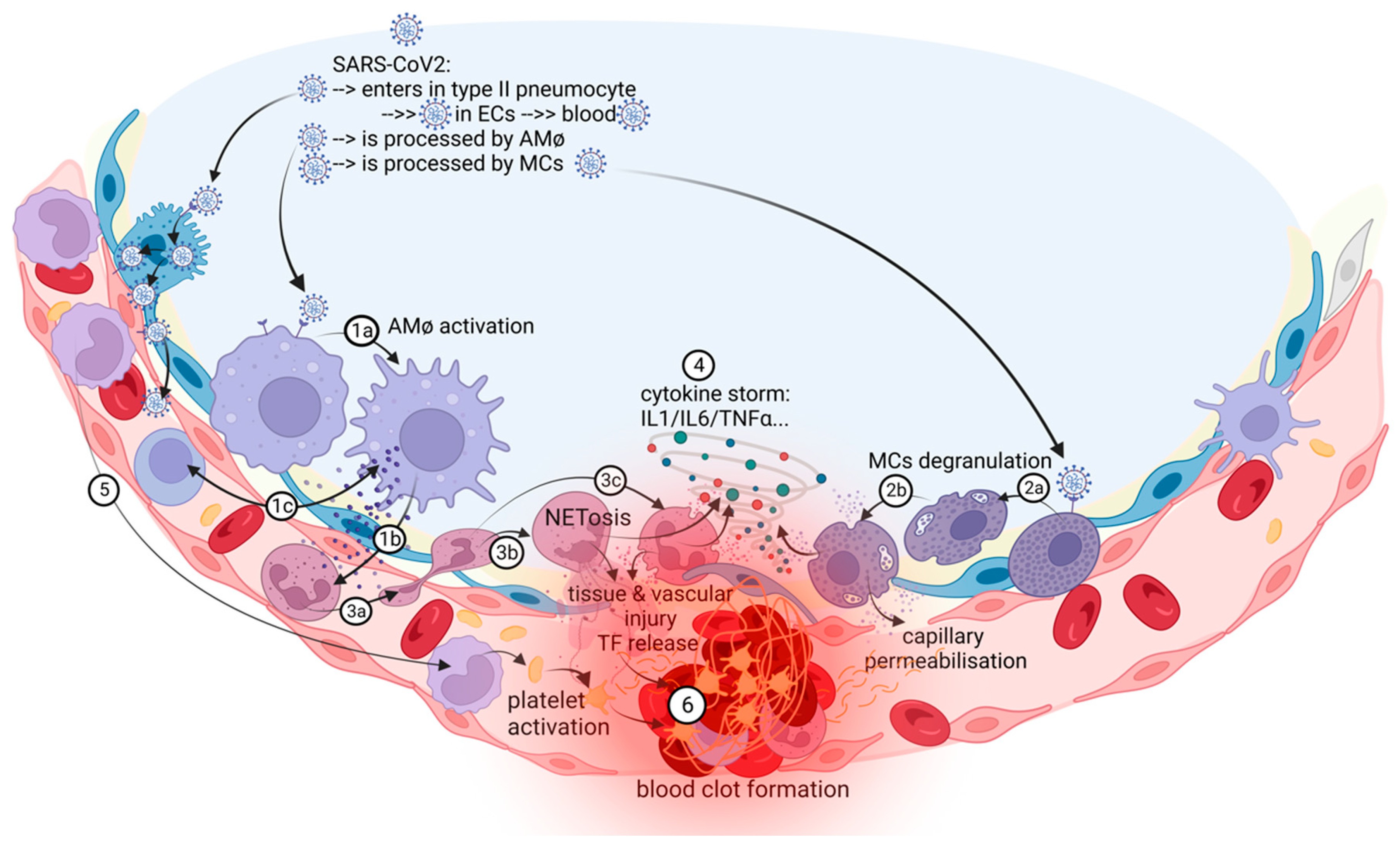
2. COVID-19-Associated Pulmonary Thrombosis Is an In Situ Immunothrombosis
3. Biological Mechanisms for In Situ Pulmonary Immunothrombosis
3.1. Inflammatory Pathways
3.1.1. Macrophages (AMφs), Monocytes, and T Cells
3.1.2. The NETs–Thrombosis Axis in COVID-19
3.1.3. Mast Cells (MCs), Cytokines, and Chemokines
3.1.4. Complement Pathways
3.2. Coagulation Pathways
3.2.1. ECs and Platelets
3.2.2. vWF
3.2.3. Thrombomodulin and P-Selectin
3.2.4. Intrinsic and Extrinsic Coagulation Pathways
3.2.5. Fibrinolytic Disbalance and the Central Role of PAI-1
3.2.6. SARS-CoV-2–RBCs Axis
4. Chest CT Imaging Data Supporting the Role of Pulmonary Immunothrombosis
5. The Role of Venous Thromboembolism
6. The Failure of Anticoagulation Treatment in Immunothrombosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manolis, A.S.; Manolis, T.A.; Manolis, A.A.; Papatheou, D.; Melita, H. COVID-19 Infection: Viral Macro- and Micro-Vascular Coagulopathy and Thromboembolism/Prophylactic and Therapeutic Management. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Loo, J.; Spittle, D.A.; Newnham, M. COVID-19, immunothrombosis and venous thromboembolism: Biological mechanisms. Thorax 2021, 76, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, T.; Wood, M.K.; Hughes, D.M.; Talor, M.V.; Ma, Z.; Schneider, J.; Skinner, J.T.; Asady, B.; Goerlich, E.; Halushka, M.K.; et al. Endothelial thrombomodulin downregulation caused by hypoxia contributes to severe infiltration and coagulopathy in COVID-19 patient lungs. eBioMedicine 2022, 75, 103812. [Google Scholar] [CrossRef]
- Mueller-Peltzer, K.; Krauss, T.; Benndorf, M.; Lang, C.N.; Bamberg, F.; Bode, C.; Duerschmied, D.; Staudacher, D.L.; Zotzmann, V. Pulmonary artery thrombi are co-located with opacifications in SARS-CoV2 induced ARDS. Respir. Med. 2020, 172, 106135. [Google Scholar] [CrossRef]
- Oba, S.; Hosoya, T.; Amamiya, M.; Mitsumura, T.; Kawata, D.; Sasaki, H.; Kamiya, M.; Yamamoto, A.; Ando, T.; Shimada, S.; et al. Arterial and Venous Thrombosis Complicated in COVID-19: A Retrospective Single Center Analysis in Japan. Front. Cardiovasc. Med. 2021, 8, 767074. [Google Scholar] [CrossRef]
- Suh, Y.J.; Hong, H.; Ohana, M.; Bompard, F.; Revel, M.P.; Valle, C.; Gervaise, A.; Poissy, J.; Susen, S.; Hékimian, G.; et al. Pulmonary Embolism and Deep Vein Thrombosis in COVID-19: A Systematic Review and Meta-Analysis. Radiology 2021, 298, E70–E80. [Google Scholar] [CrossRef]
- Delli Muti, N.; Finocchi, F.; Tossetta, G.; Salvio, G.; Cutini, M.; Marzioni, D.; Balercia, G. Could SARS-CoV-2 infection affect male fertility and sexuality? Apmis 2022, 130, 243–252. [Google Scholar] [CrossRef]
- Marshall, M. How COVID-19 can damage the brain. Nature 2020, 585, 342–343. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Delli Muti, N.; Balercia, G.; Ciavattini, A.; Giannubilo, S.R.; Marzioni, D. Preeclampsia and severe acute respiratory syndrome coronavirus 2 infection: A systematic review. J. Hypertens. 2022, 40, 1629–1638. [Google Scholar] [CrossRef]
- Portier, I.; Campbell, R.A.; Denorme, F. Mechanisms of immunothrombosis in COVID-19. Curr. Opin. Hematol. 2021, 28, 445–453. [Google Scholar] [CrossRef]
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; van de Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-associated coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649. [Google Scholar] [CrossRef]
- Payus, A.O.; Lin, C.L.S.; Ibrahim, A. The poorly understood yet potent risk of pulmonary artery thrombosis in-situ in Post-Acute COVID-19 syndrome. J. Cardiothorac. Surg. 2023, 18, 42. [Google Scholar] [CrossRef]
- Cuevas Vilaplana, A.; Roldan Torres, I.; Vizuete Del Rio, J. Myocarditis and in situ thrombosis in the right ventricle in a COVID-19 patient. Hipertens. Riesgo Vasc. 2021, 38, 148–150. [Google Scholar] [CrossRef]
- Khismatullin, R.R.; Ponomareva, A.A.; Nagaswami, C.; Ivaeva, R.A.; Montone, K.T.; Weisel, J.W.; Litvinov, R.I. Pathology of lung-specific thrombosis and inflammation in COVID-19. J. Thromb. Haemost. 2021, 19, 3062–3072. [Google Scholar] [CrossRef]
- Quartuccio, L.; Sonaglia, A.; Casarotto, L.; McGonagle, D.; Di Loreto, C.; Pegolo, E. Clinical, laboratory and immunohistochemical characterization of in situ pulmonary arterial thrombosis in fatal COVID-19. Thromb. Res. 2022, 219, 95–101. [Google Scholar] [CrossRef]
- Agrati, C.; Sacchi, A.; Tartaglia, E.; Vergori, A.; Gagliardini, R.; Scarabello, A.; Bibas, M. The Role of P-Selectin in COVID-19 Coagulopathy: An Updated Review. Int. J. Mol. Sci. 2021, 22, 7942. [Google Scholar] [CrossRef]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef]
- Lim, M.S.; McRae, S. COVID-19 and immunothrombosis: Pathophysiology and therapeutic implications. Crit. Rev. Oncol. Hematol. 2021, 168, 103529. [Google Scholar] [CrossRef]
- Thachil, J.; Srivastava, A. SARS-2 Coronavirus-Associated Hemostatic Lung Abnormality in COVID-19: Is It Pulmonary Thrombosis or Pulmonary Embolism? Semin. Thromb. Hemost. 2020, 46, 777–780. [Google Scholar] [CrossRef]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Chen, F.; Bai, L.; Yi, Q.; Peng, Y. In situ pulmonary thrombosis in patients with COVID-19 pneumonia: Different phenotypes may exist. Thromb. Res. 2020, 196, 541–542. [Google Scholar] [CrossRef] [PubMed]
- Martín Giménez, V.M.; Inserra, F.; Tajer, C.D.; Mariani, J.; Ferder, L.; Reiter, R.J.; Manucha, W. Lungs as target of COVID-19 infection: Protective common molecular mechanisms of vitamin D and melatonin as a new potential synergistic treatment. Life Sci. 2020, 254, 117808. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Pezzuto, F.; Fortarezza, F.; Hofman, P.; Kern, I.; Panizo, A.; von der Thüsen, J.; Timofeev, S.; Gorkiewicz, G.; Lunardi, F. Pulmonary pathology and COVID-19: Lessons from autopsy. The experience of European Pulmonary Pathologists. Virchows Arch. 2020, 477, 359–372. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef]
- Duarte-Neto, A.N.; Monteiro, R.A.A.; da Silva, L.F.F.; Malheiros, D.; de Oliveira, E.P.; Theodoro-Filho, J.; Pinho, J.R.R.; Gomes-Gouvêa, M.S.; Salles, A.P.M.; de Oliveira, I.R.S.; et al. Pulmonary and systemic involvement in COVID-19 patients assessed with ultrasound-guided minimally invasive autopsy. Histopathology 2020, 77, 186–197. [Google Scholar] [CrossRef]
- Cacciola, R.; Gentilini Cacciola, E.; Vecchio, V.; Cacciola, E. Cellular and molecular mechanisms in COVID-19 coagulopathy: Role of inflammation and endotheliopathy. J. Thromb. Thrombolysis 2022, 53, 282–290. [Google Scholar] [CrossRef]
- Golubeva, M.G. Role of P-Selectin in the Development of Hemostasis Disorders in COVID-19. Biol. Bull. Rev. 2022, 12, 406–413. [Google Scholar] [CrossRef]
- Vivan, M.A.; Rigatti, B.; da Cunha, S.V.; Frison, G.C.; Antoniazzi, L.Q.; de Oliveira, P.H.K.; Oliveira, J.P.S.; Fontanari, C.; Seligman, B.G.S.; Seligman, R. Pulmonary embolism in patients with COVID-19 and D-dimer diagnostic value: A retrospective study. Braz. J. Infect. Dis. 2022, 26, 102702. [Google Scholar] [CrossRef]
- Močibob, L.; Šušak, F.; Šitum, M.; Višković, K.; Papić, N.; Vince, A. COVID-19 and Pulmonary Thrombosis-An Unresolved Clinical Puzzle: A Single-Center Cohort Study. J. Clin. Med. 2022, 11, 7049. [Google Scholar] [CrossRef]
- Jalde, F.C.; Beckman, M.O.; Svensson, A.M.; Bell, M.; Skold, M.; Strand, F.; Nyren, S.; Kistner, A. Widespread Parenchymal Abnormalities and Pulmonary Embolism on Contrast-Enhanced CT Predict Disease Severity and Mortality in Hospitalized COVID-19 Patients. Front. Med. 2021, 8, 666723. [Google Scholar] [CrossRef]
- Masselli, G.; Almberger, M.; Tortora, A.; Capoccia, L.; Dolciami, M.; D’Aprile, M.R.; Valentini, C.; Avventurieri, G.; Bracci, S.; Ricci, P. Role of CT angiography in detecting acute pulmonary embolism associated with COVID-19 pneumonia. Radiol. Med. 2021, 126, 1553–1560. [Google Scholar] [CrossRef]
- Smilowitz, N.R.; Kunichoff, D.; Garshick, M.; Shah, B.; Pillinger, M.; Hochman, J.S.; Berger, J.S. C-reactive protein and clinical outcomes in patients with COVID-19. Eur. Heart J. 2021, 42, 2270–2279. [Google Scholar] [CrossRef]
- Sultana, G.N.N.; Srivastava, A.; Akhtaar, K.; Singh, P.P.; Islam, M.A.; Mishra, R.K.; Chaubey, G. Studying C-reactive protein and D-dimer levels in blood may prevent severe complications: A study in Bangladeshi COVID-19 patients. Front. Genet. 2022, 13, 966595. [Google Scholar] [CrossRef]
- Gorog, D.A.; Storey, R.F.; Gurbel, P.A.; Tantry, U.S.; Berger, J.S.; Chan, M.Y.; Duerschmied, D.; Smyth, S.S.; Parker, W.A.E.; Ajjan, R.A.; et al. Current and novel biomarkers of thrombotic risk in COVID-19: A Consensus Statement from the International COVID-19 Thrombosis Biomarkers Colloquium. Nat. Rev. Cardiol. 2022, 19, 475–495. [Google Scholar] [CrossRef]
- Lucijanić, M.; Stojić, J.; Atić, A.; Čikara, T.; Osmani, B.; Barišić-Jaman, M.; Andrilović, A.; Bistrović, P.; Zrilić Vrkljan, A.; Lagančić, M.; et al. Clinical and prognostic significance of C-reactive protein to albumin ratio in hospitalized coronavirus disease 2019 (COVID-19) patients. Wien. Klin. Wochenschr. 2022, 134, 377–384. [Google Scholar] [CrossRef]
- Fay, W.P. Linking inflammation and thrombosis: Role of C-reactive protein. World J. Cardiol. 2010, 2, 365–369. [Google Scholar] [CrossRef]
- Galland, J.; Thoreau, B.; Delrue, M.; Neuwirth, M.; Stepanian, A.; Chauvin, A.; Dellal, A.; Nallet, O.; Roriz, M.; Devaux, M.; et al. White blood count, D-dimers, and ferritin levels as predictive factors of pulmonary embolism suspected upon admission in noncritically ill COVID-19 patients: The French multicenter CLOTVID retrospective study. Eur. J. Haematol. 2021, 107, 190–201. [Google Scholar] [CrossRef]
- Kaushal, K.; Kaur, H.; Sarma, P.; Bhattacharyya, A.; Sharma, D.J.; Prajapat, M.; Pathak, M.; Kothari, A.; Kumar, S.; Rana, S.; et al. Serum ferritin as a predictive biomarker in COVID-19. A systematic review, meta-analysis and meta-regression analysis. J. Crit. Care 2022, 67, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, N.; Comish, P.B.; Tang, D.; Kang, R. Inflammasome-Dependent Coagulation Activation in Sepsis. Front. Immunol. 2021, 12, 641750. [Google Scholar] [CrossRef] [PubMed]
- Tuculeanu, G.; Barbu, E.C.; Lazar, M.; Chitu-Tisu, C.E.; Moisa, E.; Negoita, S.I.; Ion, D.A. Coagulation Disorders in Sepsis and COVID-19—Two Sides of the Same Coin? A Review of Inflammation–Coagulation Crosstalk in Bacterial Sepsis and COVID-19. J. Clin. Med. 2023, 12, 601. [Google Scholar] [PubMed]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Savla, S.R.; Prabhavalkar, K.S.; Bhatt, L.K. Cytokine storm associated coagulation complications in COVID-19 patients: Pathogenesis and Management. Expert Rev. Anti-Infect Ther. 2021, 19, 1397–1413. [Google Scholar] [CrossRef]
- Bain, C.C.; Lucas, C.D.; Rossi, A.G. Pulmonary macrophages and SARS-Cov2 infection. Int. Rev. Cell Mol. Biol. 2022, 367, 1–28. [Google Scholar] [CrossRef]
- Faggioli, P.M.; Mumoli, N.; Mazzone, A. Iloprost in COVID-19: The Rationale of Therapeutic Benefit. Front. Cardiovasc. Med. 2021, 8, 649499. [Google Scholar] [CrossRef]
- Hottz, E.D.; Martins-Gonçalves, R.; Palhinha, L.; Azevedo-Quintanilha, I.G.; de Campos, M.M.; Sacramento, C.Q.; Temerozo, J.R.; Soares, V.C.; Dias, S.S.G.; Teixeira, L.; et al. Platelet-monocyte interaction amplifies thromboinflammation through tissue factor signaling in COVID-19. Blood Adv. 2022, 6, 5085–5099. [Google Scholar] [CrossRef]
- Szturmowicz, M.; Demkow, U. Neutrophil Extracellular Traps (NETs) in Severe SARS-CoV-2 Lung Disease. Int. J. Mol. Sci. 2021, 22, 8854. [Google Scholar] [CrossRef]
- Zhou, Y.; Tao, W.; Shen, F.; Du, W.; Xu, Z.; Liu, Z. The Emerging Role of Neutrophil Extracellular Traps in Arterial, Venous and Cancer-Associated Thrombosis. Front. Cardiovasc. Med. 2021, 8, 786387. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Al-Hussaniy, H.A.; Al-Harcan, N.A.H.; Alexiou, A.; Batiha, G.E. Neutrophil Extracellular Traps (NETs) and Covid-19: A new frontiers for therapeutic modality. Int. Immunopharmacol. 2022, 104, 108516. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Ackermann, M.; Anders, H.-J.; Bilyy, R.; Bowlin, G.L.; Daniel, C.; De Lorenzo, R.; Egeblad, M.; Henneck, T.; Hidalgo, A.; Hoffmann, M.; et al. Patients with COVID-19: In the dark-NETs of neutrophils. Cell Death Differ. 2021, 28, 3125–3139. [Google Scholar] [CrossRef]
- Pastorek, M.; Dúbrava, M.; Celec, P. On the Origin of Neutrophil Extracellular Traps in COVID-19. Front. Immunol. 2022, 13, 821007. [Google Scholar] [CrossRef]
- Teluguakula, N. Neutrophils Set Extracellular Traps to Injure Lungs in Coronavirus Disease 2019. J. Infect. Dis. 2021, 223, 1503–1505. [Google Scholar] [CrossRef]
- Ouwendijk, W.J.D.; Raadsen, M.P.; van Kampen, J.J.A.; Verdijk, R.M.; von der Thusen, J.H.; Guo, L.; Hoek, R.A.S.; van den Akker, J.P.C.; Endeman, H.; Langerak, T.; et al. High Levels of Neutrophil Extracellular Traps Persist in the Lower Respiratory Tract of Critically Ill Patients with Coronavirus Disease 2019. J. Infect. Dis. 2021, 223, 1512–1521. [Google Scholar] [CrossRef]
- Sung, P.-S.; Yang, S.-P.; Peng, Y.-C.; Sun, C.-P.; Tao, M.-H.; Hsieh, S.-L. CLEC5A and TLR2 are critical in SARS-CoV-2-induced NET formation and lung inflammation. J. Biomed. Sci. 2022, 29, 52. [Google Scholar] [CrossRef]
- Lam, H.Y.; Tergaonkar, V.; Kumar, A.P.; Ahn, K.S. Mast cells: Therapeutic targets for COVID-19 and beyond. IUBMB Life 2021, 73, 1278–1292. [Google Scholar] [CrossRef]
- Galli, S.J.; Gaudenzio, N.; Tsai, M. Mast Cells in Inflammation and Disease: Recent Progress and Ongoing Concerns. Annu. Rev. Immunol. 2020, 38, 49–77. [Google Scholar] [CrossRef]
- Cildir, G.; Pant, H.; Lopez, A.F.; Tergaonkar, V. The transcriptional program, functional heterogeneity, and clinical targeting of mast cells. J. Exp. Med. 2017, 214, 2491–2506. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Valent, P.; Akin, C. Mast Cells, Mastocytosis, and Related Disorders. N. Engl. J. Med. 2015, 373, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Gebremeskel, S.; Schanin, J.; Coyle, K.M.; Butuci, M.; Luu, T.; Brock, E.C.; Xu, A.; Wong, A.; Leung, J.; Korver, W.; et al. Mast Cell and Eosinophil Activation Are Associated with COVID-19 and TLR-Mediated Viral Inflammation: Implications for an Anti-Siglec-8 Antibody. Front. Immunol. 2021, 12, 650331. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.S.; Portales-Cervantes, L.; Leong, E. Mast Cell Responses to Viruses and Pathogen Products. Int. J. Mol. Sci. 2019, 20, 4241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budnevsky, A.V.; Avdeev, S.N.; Kosanovic, D.; Shishkina, V.V.; Filin, A.A.; Esaulenko, D.I.; Ovsyannikov, E.S.; Samoylenko, T.V.; Redkin, A.N.; Suvorova, O.A.; et al. Role of mast cells in the pathogenesis of severe lung damage in COVID-19 patients. Respir. Res. 2022, 23, 371. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Caraffa, A.; Tetè, G.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Ronconi, G. Mast cells activated by SARS-CoV-2 release histamine which increases IL-1 levels causing cytokine storm and inflammatory reaction in COVID-19. J. Biol. Regul. Homeost. Agents 2020, 34, 1629–1632. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Ronconi, G.; Toniato, E. IL-1 induces throboxane-A2 (TxA2) in COVID-19 causing inflammation and micro-thrombi: Inhibitory effect of the IL-1 receptor antagonist (IL-1Ra). J. Biol. Regul. Homeost. Agents 2020, 34, 1623–1627. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Wismans, L.V.; Lopuhaä, B.; de Koning, W.; Moeniralam, H.; van Oosterhout, M.; Ambarus, C.; Hofman, F.N.; Kuiken, T.; Endeman, H.; Mustafa, D.A.M.; et al. Increase of mast cells in COVID-19 pneumonia may contribute to pulmonary fibrosis and thrombosis. Histopathology 2023, 82, 407–419. [Google Scholar] [CrossRef]
- Wu, M.L.; Liu, F.L.; Sun, J.; Li, X.; He, X.Y.; Zheng, H.Y.; Zhou, Y.H.; Yan, Q.; Chen, L.; Yu, G.Y.; et al. SARS-CoV-2-triggered mast cell rapid degranulation induces alveolar epithelial inflammation and lung injury. Signal Transduct. Target. Ther. 2021, 6, 428. [Google Scholar] [CrossRef]
- Liu, S.; Suzuki, Y.; Takemasa, E.; Watanabe, R.; Mogi, M. Mast cells promote viral entry of SARS-CoV-2 via formation of chymase/spike protein complex. Eur. J. Pharmacol. 2022, 930, 175169. [Google Scholar] [CrossRef]
- Krysko, O.; Bourne, J.H.; Kondakova, E.; Galova, E.A.; Whitworth, K.; Newby, M.L.; Bachert, C.; Hill, H.; Crispin, M.; Stamataki, Z.; et al. Severity of SARS-CoV-2 infection is associated with high numbers of alveolar mast cells and their degranulation. Front. Immunol. 2022, 13, 968981. [Google Scholar] [CrossRef]
- Motta Junior, J.D.S.; Miggiolaro, A.; Nagashima, S.; de Paula, C.B.V.; Baena, C.P.; Scharfstein, J.; de Noronha, L. Mast Cells in Alveolar Septa of COVID-19 Patients: A Pathogenic Pathway That May Link Interstitial Edema to Immunothrombosis. Front. Immunol. 2020, 11, 574862. [Google Scholar] [CrossRef]
- Gasparello, J.; d’Aversa, E.; Breveglieri, G.; Borgatti, M.; Finotti, A.; Gambari, R. In vitro induction of interleukin-8 by SARS-CoV-2 Spike protein is inhibited in bronchial epithelial IB3-1 cells by a miR-93-5p agomiR. Int. Immunopharmacol. 2021, 101, 108201. [Google Scholar] [CrossRef]
- Halova, I.; Draberova, L.; Draber, P. Mast cell chemotaxis—Chemoattractants and signaling pathways. Front. Immunol. 2012, 3, 119. [Google Scholar] [CrossRef] [Green Version]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef]
- Hafezi, B.; Chan, L.; Knapp, J.P.; Karimi, N.; Alizadeh, K.; Mehrani, Y.; Bridle, B.W.; Karimi, K. Cytokine Storm Syndrome in SARS-CoV-2 Infections: A Functional Role of Mast Cells. Cells 2021, 10, 1761. [Google Scholar] [CrossRef]
- Theoharides, T.C. Potential association of mast cells with coronavirus disease 2019. Ann. Allergy Asthma Immunol. 2021, 126, 217–218. [Google Scholar] [CrossRef]
- Setyo Nugroho, G.M.; Marhana, I.A.; Kusumastuti, E.H.; Semedi, B.P.; Maimunah, U.; Lefi, A.; Suyanto, E.; Rosyid, A.N.; Wahyu, D.; Wiratama, P.A.; et al. Interleukin-6 (IL-6) expression of lung tissue in COVID-19 patient severity through core biopsy post mortem. Ann. Med. Surg. 2022, 82, 104648. [Google Scholar] [CrossRef]
- Harapan, H.; Fajar, J.K.; Supriono, S.; Soegiarto, G.; Wulandari, L.; Seratin, F.; Prayudi, N.G.; Dewi, D.P.; Monica Elsina, M.T.; Atamou, L.; et al. The prevalence, predictors and outcomes of acute liver injury among patients with COVID-19: A systematic review and meta-analysis. Rev. Med. Virol. 2022, 32, e2304. [Google Scholar] [CrossRef]
- Colafrancesco, S.; Scrivo, R.; Barbati, C.; Conti, F.; Priori, R. Targeting the Immune System for Pulmonary Inflammation and Cardiovascular Complications in COVID-19 Patients. Front. Immunol. 2020, 11, 1439. [Google Scholar] [CrossRef] [PubMed]
- Rad, F.; Dabbagh, A.; Dorgalaleh, A.; Biswas, A. The Relationship between Inflammatory Cytokines and Coagulopathy in Patients with COVID-19. J. Clin. Med. 2021, 10, 2020. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Ghani, S.E.-S.; Hamed, R.M.R.; Eid, R.A.; Ibrahim, A.Y.M.; Abdel-Hamid, H.M.; Abdelrahman, W.; Ibrahim, R.E.; Abdel-Aziz, M.M.; Mohamed, M.S. Serum interleukin 1β and sP-selectin as biomarkers of inflammation and thrombosis, could they be predictors of disease severity in COVID 19 Egyptian patients? (a cross-sectional study). Thromb. J. 2022, 20, 77. [Google Scholar] [CrossRef] [PubMed]
- Gianni, P.; Goldin, M.; Ngu, S.; Zafeiropoulos, S.; Geropoulos, G.; Giannis, D. Complement-mediated microvascular injury and thrombosis in the pathogenesis of severe COVID-19: A review. World J. Exp. Med. 2022, 12, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.M.; Ferrari, M.; Lynch, N.J.; Yaseen, S.; Dudler, T.; Gragerov, S.; Demopulos, G.; Heeney, J.L.; Schwaeble, W.J. Lectin Pathway Mediates Complement Activation by SARS-CoV-2 Proteins. Front. Immunol. 2021, 12, 714511. [Google Scholar] [CrossRef]
- Boussier, J.; Yatim, N.; Marchal, A.; Hadjadj, J.; Charbit, B.; El Sissy, C.; Carlier, N.; Pène, F.; Mouthon, L.; Tharaux, P.L.; et al. Severe COVID-19 is associated with hyperactivation of the alternative complement pathway. J. Allergy Clin. Immunol. 2022, 149, 550–556.e552. [Google Scholar] [CrossRef]
- Niederreiter, J.; Eck, C.; Ries, T.; Hartmann, A.; Märkl, B.; Büttner-Herold, M.; Amann, K.; Daniel, C. Complement Activation via the Lectin and Alternative Pathway in Patients with Severe COVID-19. Front. Immunol. 2022, 13, 182. [Google Scholar] [CrossRef]
- Foley, J.H.; Walton, B.L.; Aleman, M.M.; O’Byrne, A.M.; Lei, V.; Harrasser, M.; Foley, K.A.; Wolberg, A.S.; Conway, E.M. Complement Activation in Arterial and Venous Thrombosis is Mediated by Plasmin. EBioMedicine 2016, 5, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Tomo, S.; Kumar, K.P.; Roy, D.; Sankanagoudar, S.; Purohit, P.; Yadav, D.; Banerjee, M.; Sharma, P.; Misra, S. Complement activation and coagulopathy—An ominous duo in COVID19. Expert Rev. Hematol. 2021, 14, 155–173. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Holter, J.C.; Pischke, S.E.; de Boer, E.; Lind, A.; Jenum, S.; Holten, A.R.; Tonby, K.; Barratt-Due, A.; Sokolova, M.; Schjalm, C.; et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl. Acad. Sci. USA 2020, 117, 25018–25025. [Google Scholar] [CrossRef]
- Peffault de Latour, R.; Bergeron, A.; Lengline, E.; Dupont, T.; Marchal, A.; Galicier, L.; de Castro, N.; Bondeelle, L.; Darmon, M.; Dupin, C.; et al. Complement C5 inhibition in patients with COVID-19—A promising target? Haematologica 2020, 105, 2847–2850. [Google Scholar] [CrossRef]
- Soma, P.; Bester, J. Pathophysiological Changes in Erythrocytes Contributing to Complications of Inflammation and Coagulation in COVID-19. Front. Physiol. 2022, 13, 899629. [Google Scholar] [CrossRef]
- de Andrade, S.A.; de Souza, D.A.; Torres, A.L.; de Lima, C.F.G.; Ebram, M.C.; Celano, R.M.G.; Schattner, M.; Chudzinski-Tavassi, A.M. Pathophysiology of COVID-19: Critical Role of Hemostasis. Front. Cell. Infect. Microbiol. 2022, 12, 896972. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Tafazoli, A.; Anil Kumar, S.; Othman, M. Thrombocytopathy vs Platelet hyper-reactivity in COVID-19: Diverse pathologies, disease outcomes and therapeutic implications. Platelets 2022, 33, 48–53. [Google Scholar] [CrossRef]
- Guo, L.; Rondina, M.T. The Era of Thromboinflammation: Platelets Are Dynamic Sensors and Effector Cells During Infectious Diseases. Front. Immunol. 2019, 10, 2204. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Tyagi, T.; Antoniak, S. Platelet in thrombo-inflammation: Unraveling new therapeutic targets. Front. Immunol. 2022, 13, 1039843. [Google Scholar] [CrossRef]
- Comer, S.P.; Cullivan, S.; Szklanna, P.B.; Weiss, L.; Cullen, S.; Kelliher, S.; Smolenski, A.; Murphy, C.; Altaie, H.; Curran, J.; et al. COVID-19 induces a hyperactive phenotype in circulating platelets. PLoS Biol. 2021, 19, e3001109. [Google Scholar] [CrossRef]
- Tunjungputri, R.N.; Li, Y.; de Groot, P.G.; Dinarello, C.A.; Smeekens, S.P.; Jaeger, M.; Doppenberg-Oosting, M.; Cruijsen, M.; Lemmers, H.; Toenhake-Dijkstra, H.; et al. The Inter-Relationship of Platelets with Interleukin-1β-Mediated Inflammation in Humans. Thromb. Haemost. 2018, 118, 2112–2125. [Google Scholar] [CrossRef] [PubMed]
- Fard, M.B.; Fard, S.B.; Ramazi, S.; Atashi, A.; Eslamifar, Z. Thrombosis in COVID-19 infection: Role of platelet activation-mediated immunity. Thromb. J. 2021, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- Le, H.T.; Golla, K.; Karimi, R.; Hughes, M.R.; Lakschevitz, F.; Cines, D.B.; Kowalska, M.A.; Poncz, M.; McNagny, K.M.; Häkkinen, L.; et al. Platelet factor 4 (CXCL4/PF4) upregulates matrix metalloproteinase-2 (MMP-2) in gingival fibroblasts. Sci. Rep. 2022, 12, 18636. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Miao, H.; Li, S.; Zhang, P.; Gerber, G.F.; Follmann, D.; Ji, H.; Zeger, S.L.; Chertow, D.S.; Quinn, T.C.; et al. Anti-PF4 antibodies associated with disease severity in COVID-19. Proc. Natl. Acad. Sci. USA 2022, 119, e2213361119. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, D.; Klug, M.; Lazareva, O.; Weidlich, S.; Biasi, M.; Ursu, S.; Warth, S.; Buske, C.; Lukas, M.; Spinner, C.D.; et al. SARS-CoV-2 infection is associated with a pro-thrombotic platelet phenotype. Cell Death Dis. 2021, 12, 50. [Google Scholar] [CrossRef]
- Aloui, C.; Prigent, A.; Sut, C.; Tariket, S.; Hamzeh-Cognasse, H.; Pozzetto, B.; Richard, Y.; Cognasse, F.; Laradi, S.; Garraud, O. The signaling role of CD40 ligand in platelet biology and in platelet component transfusion. Int. J. Mol. Sci. 2014, 15, 22342–22364. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Xie, G.; Xiao, H.; Ding, F.; Bao, W.; Zhang, M. CXCR4 knockdown prevents inflammatory cytokine expression in macrophages by suppressing activation of MAPK and NF-κB signaling pathways. Cell Biosci. 2019, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Gavins, F.N.; Li, G.; Russell, J.; Perretti, M.; Granger, D.N. Microvascular thrombosis and CD40/CD40L signaling. J. Thromb. Haemost. 2011, 9, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Kremer Hovinga, J.A.; George, J.N. Hereditary Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 381, 1653–1662. [Google Scholar] [CrossRef]
- Naß, J.; Terglane, J.; Gerke, V. Weibel Palade Bodies: Unique Secretory Organelles of Endothelial Cells that Control Blood Vessel Homeostasis. Front. Cell Dev. Biol. 2021, 9, 813995. [Google Scholar] [CrossRef]
- Mei, Z.W.; van Wijk, X.M.R.; Pham, H.P.; Marin, M.J. Role of von Willebrand Factor in COVID-19 Associated Coagulopathy. J. Appl. Lab. Med. 2021, 6, 1305–1315. [Google Scholar] [CrossRef]
- Manz, X.D.; Szulcek, R.; Pan, X.; Symersky, P.; Dickhoff, C.; Majolée, J.; Kremer, V.; Michielon, E.; Jordanova, E.S.; Radonic, T.; et al. Epigenetic Modification of the von Willebrand Factor Promoter Drives Platelet Aggregation on the Pulmonary Endothelium in Chronic Thromboembolic Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2022, 205, 806–818. [Google Scholar] [CrossRef]
- Manz, X.D.; Bogaard, H.J.; Aman, J. Regulation of VWF (Von Willebrand Factor) in Inflammatory Thrombosis. Arter. Thromb. Vasc. Biol. 2022, 42, 1307–1320. [Google Scholar] [CrossRef]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. eLife 2021, 10, e68563. [Google Scholar] [CrossRef]
- O’Sullivan, J.M.; Gonagle, D.M.; Ward, S.E.; Preston, R.J.S.; O’Donnell, J.S. Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol. 2020, 7, e553–e555. [Google Scholar] [CrossRef]
- Seibert, F.S.; Blazquez-Navarro, A.; Hölzer, B.; Doevelaar, A.A.N.; Nusshag, C.; Merle, U.; Morath, C.; Zgoura, P.; Dittmer, R.; Schneppenheim, S.; et al. Effect of plasma exchange on COVID-19 associated excess of von Willebrand factor and inflammation in critically ill patients. Sci. Rep. 2022, 12, 4801. [Google Scholar] [CrossRef]
- Doevelaar, A.A.N.; Bachmann, M.; Hölzer, B.; Seibert, F.S.; Rohn, B.J.; Bauer, F.; Witzke, O.; Dittmer, U.; Bachmann, M.; Yilmaz, S.; et al. von Willebrand Factor Multimer Formation Contributes to Immunothrombosis in Coronavirus Disease 2019. Crit. Care Med. 2021, 49, e512–e520. [Google Scholar] [CrossRef]
- Mancini, I.; Baronciani, L.; Artoni, A.; Colpani, P.; Biganzoli, M.; Cozzi, G.; Novembrino, C.; Boscolo Anzoletti, M.; De Zan, V.; Pagliari, M.T.; et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J. Thromb. Haemost. 2021, 19, 513–521. [Google Scholar] [CrossRef]
- Fenyves, B.G.; Mehta, A.; MGH COVID-19 Collection & Processing Team; Kays, K.R.; Beakes, C.; Margolin, J.; Goldberg, M.B.; Hacohen, N.; Filbin, M.R. Plasma P-selectin is an early marker of thromboembolism in COVID-19. Am. J. Hematol. 2021, 96, E468–E471. [Google Scholar] [CrossRef]
- Ito, T.; Thachil, J.; Asakura, H.; Levy, J.H.; Iba, T. Thrombomodulin in disseminated intravascular coagulation and other critical conditions—A multi-faceted anticoagulant protein with therapeutic potential. Crit. Care 2019, 23, 280. [Google Scholar] [CrossRef] [Green Version]
- Yamakawa, K.; Murao, S.; Aihara, M. Recombinant Human Soluble Thrombomodulin in Sepsis-Induced Coagulopathy: An Updated Systematic Review and Meta-Analysis. Thromb. Haemost. 2019, 119, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Leucker, T.M.; Osburn, W.O.; Reventun, P.; Smith, K.; Claggett, B.; Kirwan, B.A.; de Brouwer, S.; Williams, M.S.; Gerstenblith, G.; Hager, D.N.; et al. Effect of Crizanlizumab, a P-Selectin Inhibitor, in COVID-19: A Placebo-Controlled, Randomized Trial. JACC Basic Transl. Sci. 2021, 6, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Neri, T.; Nieri, D.; Celi, A. P-selectin blockade in COVID-19-related ARDS. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L1237–L1238. [Google Scholar] [CrossRef] [PubMed]
- Karsli, E.; Sabirli, R.; Altintas, E.; Canacik, O.; Sabirli, G.T.; Kaymaz, B.; Kurt, O.; Koseler, A. Soluble P-selectin as a potential diagnostic and prognostic biomarker for COVID-19 disease: A case-control study. Life Sci. 2021, 277, 119634. [Google Scholar] [CrossRef] [PubMed]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Muller, R.; Rink, G.; Uzun, G.; Bakchoul, T.; Wuchter, P.; Kluter, H.; Bugert, P. Increased plasma level of soluble P-selectin in non-hospitalized COVID-19 convalescent donors. Thromb. Res. 2022, 216, 120–124. [Google Scholar] [CrossRef]
- Wang, S.S.Y.; Chee, K.; Wong, S.W.; Tan, G.B.; Ang, H.; Leung, B.P.; Tan, C.W.; Ramanathan, K.; Dalan, R.; Cheung, C.; et al. Increased Platelet Activation demonstrated by Elevated CD36 and P-Selectin Expression in 1-Year Post-Recovered COVID-19 Patients. Semin. Thromb. Hemost. 2023, 1–4. [Google Scholar] [CrossRef]
- FitzGerald, E.S.; Chen, Y.; Fitzgerald, K.A.; Jamieson, A.M. Lung Epithelial Cell Transcriptional Regulation as a Factor in COVID-19-associated Coagulopathies. Am. J. Respir. Cell Mol. Biol. 2021, 64, 687–697. [Google Scholar] [CrossRef]
- Cañas, C.A.; Cañas, F.; Bautista-Vargas, M.; Bonilla-Abadía, F. Role of Tissue Factor in the Pathogenesis of COVID-19 and the Possible Ways to Inhibit It. Clin. Appl. Thromb. Hemost. 2021, 27, 1–9. [Google Scholar] [CrossRef]
- Stefely, J.A.; Christensen, B.B.; Gogakos, T.; Cone Sullivan, J.K.; Montgomery, G.G.; Barranco, J.P.; Van Cott, E.M. Marked factor V activity elevation in severe COVID-19 is associated with venous thromboembolism. Am. J. Hematol. 2020, 95, 1522–1530. [Google Scholar] [CrossRef]
- Wang, J.; Kotagiri, P.; Lyons, P.A.; Al-Lamki, R.S.; Mescia, F.; Bergamaschi, L.; Turner, L.; Morgan, M.D.; Calero-Nieto, F.J.; Bach, K.; et al. Coagulation factor V is a T-cell inhibitor expressed by leukocytes in COVID-19. iScience 2022, 25, 103971. [Google Scholar] [CrossRef]
- Antoniak, S. The coagulation system in host defense. Res. Pract. Thromb. Haemost. 2018, 2, 549–557. [Google Scholar] [CrossRef]
- Han, M.; Pandey, D. ZMPSTE24 Regulates SARS-CoV-2 Spike Protein-enhanced Expression of Endothelial PAI-1. Am. J. Respir. Cell Mol. Biol. 2021, 65, 300–308. [Google Scholar] [CrossRef]
- Whyte, C.S.; Simpson, M.; Morrow, G.B.; Wallace, C.A.; Mentzer, A.J.; Knight, J.C.; Shapiro, S.; Curry, N.; Bagot, C.N.; Watson, H.; et al. The suboptimal fibrinolytic response in COVID-19 is dictated by high PAI-1. J. Thromb. Haemost. 2022, 20, 2394–2406. [Google Scholar] [CrossRef]
- Bielosludtseva, K. The diagnostic and prognostic role of plasminogen activator inhibitor-1 (PAI-1) in hospitalized patients with pneumonias of different etiologies. Eur. Respir. J. 2022, 60, 2495. [Google Scholar] [CrossRef]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef]
- Morrow, G.B.; Whyte, C.S.; Mutch, N.J. A Serpin with a Finger in Many PAIs: PAI-1’s Central Function in Thromboinflammation and Cardiovascular Disease. Front. Cardiovasc. Med. 2021, 8, 653655. [Google Scholar] [CrossRef]
- Kwaan, H.C.; Lindholm, P.F. The Central Role of Fibrinolytic Response in COVID-19-A Hematologist’s Perspective. Int. J. Mol. Sci. 2021, 22, 1283. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Inoue, H.; Ono, C.; Hashimoto, S.; Kioi, Y.; Matsumoto, H.; Matsuura, H.; Matsubara, T.; Shimizu, K.; et al. IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 22351–22356. [Google Scholar] [CrossRef]
- Al-Tamimi, A.O.; Yusuf, A.M.; Jayakumar, M.N.; Ansari, A.W.; Elhassan, M.; AbdulKarim, F.; Kannan, M.; Halwani, R.; Ahmad, F. SARS-CoV-2 infection induces soluble platelet activation markers and PAI-1 in the early moderate stage of COVID-19. Int. J. Lab. Hematol. 2022, 44, 712–721. [Google Scholar] [CrossRef]
- Poole, L.G.; Massey, V.L.; Siow, D.L.; Torres-Gonzáles, E.; Warner, N.L.; Luyendyk, J.P.; Ritzenthaler, J.D.; Roman, J.; Arteel, G.E. Plasminogen Activator Inhibitor-1 Is Critical in Alcohol-Enhanced Acute Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2017, 57, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Warnock, M.; Harbaugh, A.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Knight, J.S.; Kanthi, Y.; Lawrence, D.A. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci. Rep. 2021, 11, 1580. [Google Scholar] [CrossRef] [PubMed]
- Bouchla, A.; Kriebardis, A.G.; Georgatzakou, H.T.; Fortis, S.P.; Thomopoulos, T.P.; Lekkakou, L.; Markakis, K.; Gkotzias, D.; Panagiotou, A.; Papageorgiou, E.G.; et al. Red Blood Cell Abnormalities as the Mirror of SARS-CoV-2 Disease Severity: A Pilot Study. Front. Physiol. 2021, 12, 825055. [Google Scholar] [CrossRef] [PubMed]
- Espallargas, I.; Rodriguez Sevilla, J.J.; Rodriguez Chiaradia, D.A.; Salar, A.; Casamayor, G.; Villar-Garcia, J.; Rodo-Pin, A.; Marsico, S.; Carbullanca, S.; Ramal, D.; et al. CT imaging of pulmonary embolism in patients with COVID-19 pneumonia: A retrospective analysis. Eur. Radiol. 2021, 31, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Longchamp, G.; Manzocchi-Besson, S.; Longchamp, A.; Righini, M.; Robert-Ebadi, H.; Blondon, M. Proximal deep vein thrombosis and pulmonary embolism in COVID-19 patients: A systematic review and meta-analysis. Thromb. J. 2021, 19, 15. [Google Scholar] [CrossRef]
- Valle, C.; Bonaffini, P.A.; Dal Corso, M.; Mercanzin, E.; Franco, P.N.; Sonzogni, A.; Vacca, G.; Gianatti, A.; Sironi, S. Association between pulmonary embolism and COVID-19 severe pneumonia: Experience from two centers in the core of the infection Italian peak. Eur. J. Radiol. 2021, 137, 109613. [Google Scholar] [CrossRef]
- Niculae, C.M.; Anghel, A.M.; Militaru, E.D.; Tirlescu, L.G.; Lazar, M.; Hristea, A. Acute Pulmonary Artery Thrombosis despite Anticoagulation in Patients with COVID-19 Pneumonia: A Single-Center Retrospective Cohort Study. J. Clin. Med. 2022, 11, 2633. [Google Scholar] [CrossRef]
- Trunz, L.M.; Lee, P.; Lange, S.M.; Pomeranz, C.L.; Needleman, L.; Ford, R.W.; Karambelkar, A.; Sundaram, B. Imaging approach to COVID-19 associated pulmonary embolism. Int. J. Clin. Pract. 2021, 75, e14340. [Google Scholar] [CrossRef]
- Barnett, N.; Leith, D.; Govind, D.; Ramnani, V.; Williamson, H.; Chung, J.; Drebes, A. Prevalence of pulmonary embolism and deep venous thrombosis during the COVID-19 pandemic in an intensive care unit cohort: A service evaluation. Br. J. Anaesth. 2022, 129, e124–e126. [Google Scholar] [CrossRef]
- Cau, R.; Pacielli, A.; Fatemeh, H.; Vaudano, P.; Arru, C.; Crivelli, P.; Stranieri, G.; Suri, J.S.; Mannelli, L.; Conti, M.; et al. Complications in COVID-19 patients: Characteristics of pulmonary embolism. Clin. Imaging 2021, 77, 244–249. [Google Scholar] [CrossRef]
- Scialpi, M.; Sielaszuk, E.B.; Vitale, M.E.; Scalera, G.B.; Nicola, R.; Mancioli, F.A. Pulmonary embolism in COVID-19: Ancillary findings on chest CT angiography. Lung India 2021, 38, S123–S125. [Google Scholar] [CrossRef]
- Mandal, A.K.J.; Kho, J.; Ioannou, A.; Van den Abbeele, K.; Missouris, C.G. Covid-19 and in situ pulmonary artery thrombosis. Respir. Med. 2021, 176, 106176. [Google Scholar] [CrossRef]
- De Cobelli, F.; Palumbo, D.; Ciceri, F.; Landoni, G.; Ruggeri, A.; Rovere-Querini, P.; D’Angelo, A.; Steidler, S.; Galli, L.; Poli, A.; et al. Pulmonary Vascular Thrombosis in COVID-19 Pneumonia. J. Cardiothorac. Vasc. Anesth. 2021, 35, 3631–3641. [Google Scholar] [CrossRef]
- Bompard, F.; Monnier, H.; Saab, I.; Tordjman, M.; Abdoul, H.; Fournier, L.; Sanchez, O.; Lorut, C.; Chassagnon, G.; Revel, M.P. Pulmonary embolism in patients with COVID-19 pneumonia. Eur. Respir. J. 2020, 56, 2001365. [Google Scholar] [CrossRef]
- Lazar, M.; Barbu, E.C.; Chitu, C.E.; Anghel, A.M.; Niculae, C.M.; Manea, E.D.; Damalan, A.C.; Bel, A.A.; Patrascu, R.E.; Hristea, A.; et al. Mortality Predictors in Severe SARS-CoV-2 Infection. Medicina 2022, 58, 945. [Google Scholar] [CrossRef]
- Birocchi, S.; Manzoni, M.; Podda, G.M.; Casazza, G.; Cattaneo, M. High rates of pulmonary artery occlusions in COVID-19. A meta-analysis. Eur. J. Clin. Investig. 2021, 51, e13433. [Google Scholar] [CrossRef]
- Fauvel, C.; Weizman, O.; Trimaille, A.; Mika, D.; Pommier, T.; Pace, N.; Douair, A.; Barbin, E.; Fraix, A.; Bouchot, O.; et al. Pulmonary embolism in COVID-19 patients: A French multicentre cohort study. Eur. Heart J. 2020, 41, 3058–3068. [Google Scholar] [CrossRef]
- Farouk, N.; Ashry, W.M.O.; El-Hagrasy, H.A.; Mohamed, E.F.; Eltrawy, H.H.; El-Nasser, A.M.; Shipl, W.; Attar, S.E.; Kh Sakr, L.; Abdel Wahab, M.A.; et al. Admission Levels of Serum P-Selectin and IL-6 Can Predict Development of Deep Venous Thrombosis in Hospitalized Covid-19 Patients. Int. J. Gen. Med. 2022, 15, 5599–5607. [Google Scholar] [CrossRef]
- Marone, E.M.; Bonalumi, G.; Curci, R.; Arzini, A.; Chierico, S.; Marazzi, G.; Diaco, D.A.; Rossini, R.; Boschini, S.; Rinaldi, L.F. Characteristics of Venous Thromboembolism in COVID-19 Patients: A Multicenter Experience from Northern Italy. Ann. Vasc. Surg. 2020, 68, 83–87. [Google Scholar] [CrossRef]
- Alonso Martinez, J.L.; Anniccherico Sánchez, F.J.; Urbieta Echezarreta, M.A.; García, I.V.; Álvaro, J.R. Central Versus Peripheral Pulmonary Embolism: Analysis of the Impact on the Physiological Parameters and Long-term Survival. N. Am. J. Med. Sci. 2016, 8, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Cha, S.-I.; Shin, K.-M.; Lim, J.-K.; Yoo, S.-S.; Lee, S.-Y.; Lee, J.; Kim, C.-H.; Park, J.-Y.; Lee, W.-K.; Jung, C.-Y. Pulmonary embolism concurrent with lung cancer and central emboli predict mortality in patients with lung cancer and pulmonary embolism. J. Thorac. Dis. 2017, 10, 262–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loffredo, L.; Vidili, G.; Sciacqua, A.; Cogliati, C.; Di Giulio, R.; Bernardini, S.; Ciacci, P.; Pietrangelo, A.; Orlando, F.; Paraninfi, A.; et al. Asymptomatic and symptomatic deep venous thrombosis in hospitalized acutely ill medical patients: Risk factors and therapeutic implications. Thromb. J. 2022, 20, 72. [Google Scholar] [CrossRef] [PubMed]
- Katsoularis, I.; Fonseca-Rodríguez, O.; Farrington, P.; Jerndal, H.; Lundevaller, E.H.; Sund, M.; Lindmark, K.; Fors Connolly, A.M. Risks of deep vein thrombosis, pulmonary embolism, and bleeding after covid-19: Nationwide self-controlled cases series and matched cohort study. BMJ 2022, 377, e069590. [Google Scholar] [CrossRef] [PubMed]
- Poor, H.D. Pulmonary Thrombosis and Thromboembolism in COVID-19. Chest 2021, 160, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 1421–1424. [Google Scholar] [CrossRef] [Green Version]
- Beretta, S.; Da Re, F.; Francioni, V.; Remida, P.; Storti, B.; Fumagalli, L.; Piatti, M.L.; Santoro, P.; Cereda, D.; Cutellè, C.; et al. Case Report: Concomitant Massive Cerebral Venous Thrombosis and Internal Iliac Vein Thrombosis Related to Paucisymptomatic COVID-19 Infection. Front. Neurol. 2021, 12, 622130. [Google Scholar] [CrossRef]
- Malentacchi, M.; Gned, D.; Angelino, V.; Demichelis, S.; Perboni, A.; Veltri, A.; Bertolotto, A.; Capobianco, M. Concomitant brain arterial and venous thrombosis in a COVID-19 patient. Eur. J. Neurol. 2020, 27, e38–e39. [Google Scholar] [CrossRef]
- Lucijanic, M.; Piskac Zivkovic, N.; Ivic, M.; Sedinic, M.; Brkljacic, B.; Mutvar, A.; Atic, A.; Rudan, D.; Barsic, B.; Luksic, I.; et al. Asymptomatic deep vein thromboses in prolonged hospitalized COVID-19 patients. Wien. Klin. Wochenschr. 2021, 133, 1281–1288. [Google Scholar] [CrossRef]
- Horne, M.K., III. The dark side of deep venous thrombosis: The failure of anticoagulation. Am. J. Med. 2001, 110, 589–590. [Google Scholar] [CrossRef]
- Rodger, M.A.; Miranda, S.; Delluc, A.; Carrier, M. Management of suspected and confirmed recurrent venous thrombosis while on anticoagulant therapy. What next? Thromb. Res. 2019, 180, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Mosarla, R.C.; Vaduganathan, M.; Qamar, A.; Moslehi, J.; Piazza, G.; Giugliano, R.P. Anticoagulation Strategies in Patients with Cancer: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 1336–1349. [Google Scholar] [CrossRef]
- Paez Vargas, J.; Vidal GonzáLez, Á.; Robaglia, D.; Piris, M.; AléN, J.F.; Gorgolas, M.; Llamas, P.; Perez Calvo, C.; Flandes, J.; Prieto-PéRez, L. ANTICOAGULATION, BLEEDING, AND IMMUNOTHROMBOSIS IN CRITICALLY ILL PATIENTS WITH COVID-19. CHEST 2021, 160, A994–A995. [Google Scholar] [CrossRef]
- Tan, B.K.; Mainbourg, S.; Friggeri, A.; Bertoletti, L.; Douplat, M.; Dargaud, Y.; Grange, C.; Lobbes, H.; Provencher, S.; Lega, J.C. Arterial and venous thromboembolism in COVID-19: A study-level meta-analysis. Thorax 2021, 76, 970–979. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Llitjos, J.F.; Leclerc, M.; Chochois, C.; Monsallier, J.M.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef]
- Porembskaya, O.; Lobastov, K.; Pashovkina, O.; Tsaplin, S.; Schastlivtsev, I.; Zhuravlev, S.; Laberko, L.; Rodoman, G.; Kravchuk, V.; Skvortsov, A.; et al. Thrombosis of pulmonary vasculature despite anticoagulation and thrombolysis: The findings from seven autopsies. Thromb. Update 2020, 1, 100017. [Google Scholar] [CrossRef]
- Sadeghipour, P.; Talasaz, A.H.; Rashidi, F.; Sharif-Kashani, B.; Beigmohammadi, M.T.; Farrokhpour, M.; Sezavar, S.H.; Payandemehr, P.; Dabbagh, A.; Moghadam, K.G.; et al. Effect of Intermediate-Dose vs Standard-Dose Prophylactic Anticoagulation on Thrombotic Events, Extracorporeal Membrane Oxygenation Treatment, or Mortality among Patients with COVID-19 Admitted to the Intensive Care Unit: The INSPIRATION Randomized Clinical Trial. JAMA 2021, 325, 1620–1630. [Google Scholar] [CrossRef]
- Akácsos-Szász, O.-Z.; Pál, S.; Nyulas, K.-I.; Nemes-Nagy, E.; Fárr, A.-M.; Dénes, L.; Szilveszter, M.; Bán, E.-G.; Tilinca, M.C.; Simon-Szabó, Z. Pathways of Coagulopathy and Inflammatory Response in SARS-CoV-2 Infection among Type 2 Diabetic Patients. Int. J. Mol. Sci. 2023, 24, 4319. [Google Scholar] [CrossRef]
- Kaiafa, G.; Savopoulos, C.; Karlafti, E.; Pantazi, K.; Paramythiotis, D.; Thomaidou, E.; Daios, S.; Ztriva, E.; Gionis, M.; Fyntanidou, V.; et al. Coagulation Profile of COVID-19 Patients. Life 2022, 12, 1658. [Google Scholar] [CrossRef]
- Słomka, A.; Kowalewski, M.; Żekanowska, E. Hemostasis in Coronavirus Disease 2019-Lesson from Viscoelastic Methods: A Systematic Review. Thromb. Haemost. 2021, 121, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Lawler, P.R.; Goligher, E.C.; Berger, J.S.; Neal, M.D.; McVerry, B.J.; Nicolau, J.C.; Gong, M.N.; Carrier, M.; Rosenson, R.S.; Reynolds, H.R.; et al. Therapeutic Anticoagulation with Heparin in Noncritically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 385, 790–802. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Inflammation | Endotheliopathy | Coagulation |
|---|---|---|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niculae, C.-M.; Hristea, A.; Moroti, R. Mechanisms of COVID-19 Associated Pulmonary Thrombosis: A Narrative Review. Biomedicines 2023, 11, 929. https://doi.org/10.3390/biomedicines11030929
Niculae C-M, Hristea A, Moroti R. Mechanisms of COVID-19 Associated Pulmonary Thrombosis: A Narrative Review. Biomedicines. 2023; 11(3):929. https://doi.org/10.3390/biomedicines11030929
Chicago/Turabian StyleNiculae, Cristian-Mihail, Adriana Hristea, and Ruxandra Moroti. 2023. "Mechanisms of COVID-19 Associated Pulmonary Thrombosis: A Narrative Review" Biomedicines 11, no. 3: 929. https://doi.org/10.3390/biomedicines11030929
APA StyleNiculae, C. -M., Hristea, A., & Moroti, R. (2023). Mechanisms of COVID-19 Associated Pulmonary Thrombosis: A Narrative Review. Biomedicines, 11(3), 929. https://doi.org/10.3390/biomedicines11030929