

Nuclear Calcium in Cardiac (Patho)Physiology: Small Compartment, Big Impact


{kind=link}
{kind=link}
Abstract
:1. Introduction
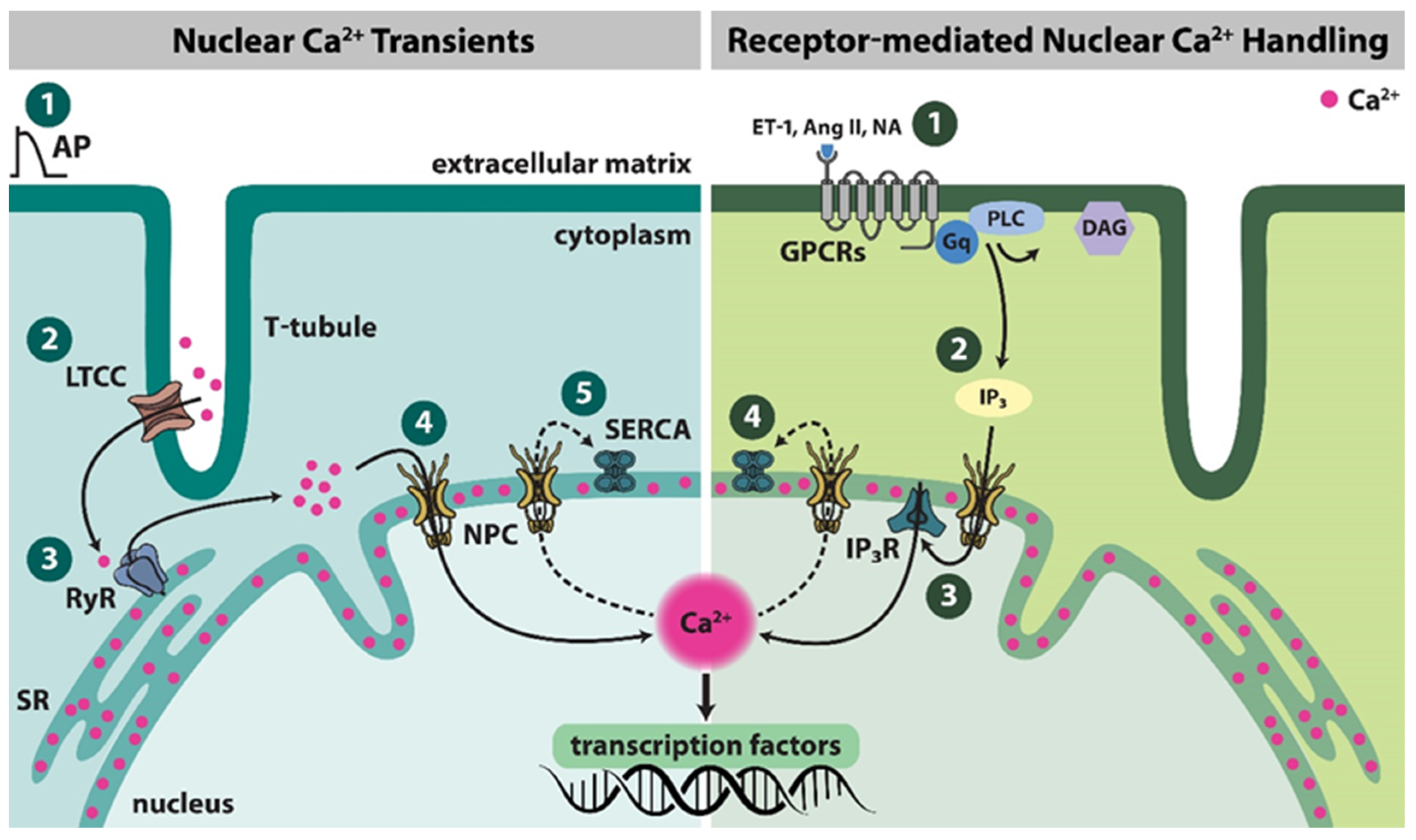
2. Nuclear Ca2+ Dynamics
2.1. Structural Basis of Nuclear Ca2+ Handling
2.2. Nuclear Ca2+ Transients
2.3. Nuclear Ca2+ Sparks and Puffs
2.4. Receptor-Mediated Nuclear Ca2+ Handling
2.4.1. IP3R
2.4.2. Adrenergic Receptors
3. Role of Nuclear Ca2+ in Cardiomyocytes
4. Nuclear Ca2+ Dysregulation
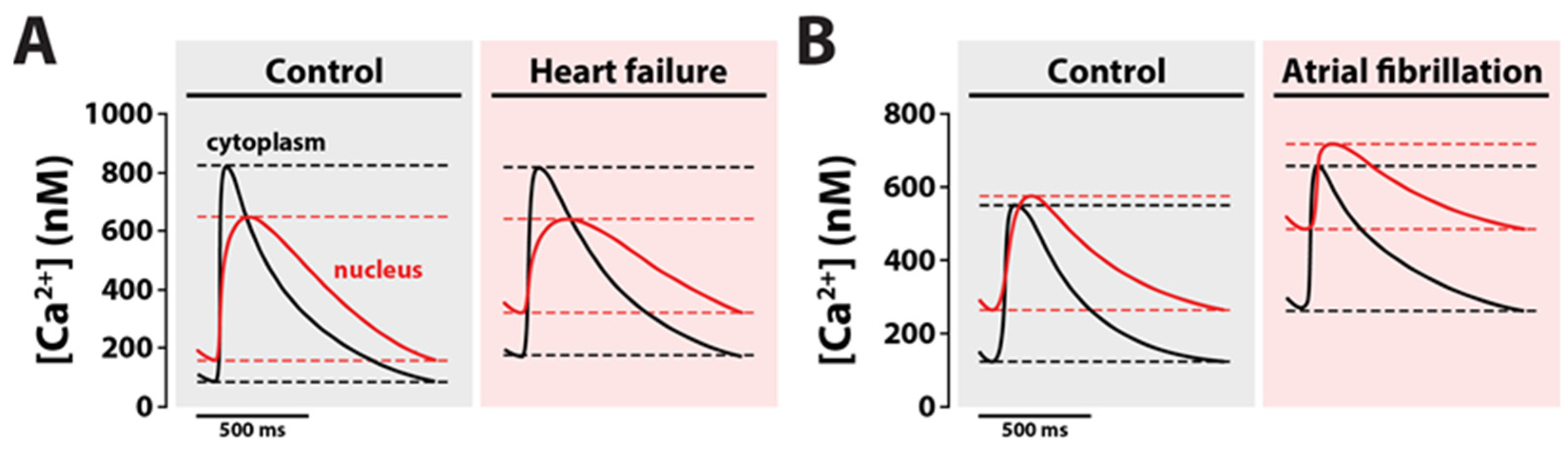
4.1. Nuclear Ca2+ in Ventricular Remodeling: Hypertrophy to Heart Failure
4.1.1. Early Cardiac Remodeling
4.1.2. Late Cardiac Remodeling

4.2. Nuclear Ca2+ in Atrial Fibrillation
4.3. Nuclear Ca2+ in Familial Cardiomyopathy
5. Nuclear Ca2+: A Practical Approach
5.1. Ca2+ Indicators
5.2. Emerging Models, Tools and Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Bers, D. Excitation-Contraction Coupling and Cardiac Contractile Force; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2001; Volume 237. [Google Scholar]
- Bers, D.M.; Guo, T. Calcium signaling in cardiac ventricular myocytes. Ann. N. Y. Acad. Sci. 2005, 1047, 86–98. [Google Scholar] [CrossRef]
- Kockskämper, J. Excitation–Contraction Coupling of Cardiomyocytes. In Cardiomyocytes–Active Players in Cardiac Disease; Schlüter, K.-D., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 67–96. [Google Scholar]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, N.; McKinsey, T.A.; Olson, E.N. Decoding calcium signals involved in cardiac growth and function. Nat. Med. 2000, 6, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Dewenter, M.; von der Lieth, A.; Katus, H.A.; Backs, J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circ. Res. 2017, 121, 1000–1020. [Google Scholar] [CrossRef]
- Shaw, R.M.; Colecraft, H.M. L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc. Res. 2013, 98, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Turcotte, M.G.; Thakur, H.; Kapiloff, M.S.; Dodge-Kafka, K.L. A perinuclear calcium compartment regulates cardiac myocyte hypertrophy. J. Mol. Cell. Cardiol. 2022, 172, 26–40. [Google Scholar] [CrossRef]
- Tanaka, H.; Kawanishi, T.; Matsuda, T.; Takahashi, M.; Shigenobu, K. Intracellular free Ca2+ movements in cultured cardiac myocytes as shown by rapid scanning confocal microscopy. J. Cardiovasc. Pharmacol. 1996, 27, 761–769. [Google Scholar] [CrossRef]
- Winogradoff, D.; Chou, H.Y.; Maffeo, C.; Aksimentiev, A. Percolation transition prescribes protein size-specific barrier to passive transport through the nuclear pore complex. Nat. Commun. 2022, 13, 5138. [Google Scholar] [CrossRef]
- Mohr, D.; Frey, S.; Fischer, T.; Güttler, T.; Görlich, D. Characterisation of the passive permeability barrier of nuclear pore complexes. EMBO J. 2009, 28, 2541–2553. [Google Scholar] [CrossRef] [Green Version]
- Panté, N.; Kann, M. Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol. Biol. Cell 2002, 13, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Bers, D.M. Sarcoplasmic reticulum and nuclear envelope are one highly interconnected Ca2+ store throughout cardiac myocyte. Circ. Res. 2006, 99, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhas, A.; Goulbourne, C.; Vaux, D.J. The nucleoplasmic reticulum: Form and function. Trends Cell Biol. 2011, 21, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Guatimosim, S.; Amaya, M.J.; Guerra, M.T.; Aguiar, C.J.; Goes, A.M.; Gómez-Viquez, N.L.; Rodrigues, M.A.; Gomes, D.A.; Martins-Cruz, J.; Lederer, W.J.; et al. Nuclear Ca2+ regulates cardiomyocyte function. Cell Calcium 2008, 44, 230–242. [Google Scholar] [CrossRef]
- Gensburger, C.; Freyermuth, S.; Klein, C.; Malviya, A.N. In vivo nuclear Ca2+-ATPase phosphorylation triggers intermediate size molecular transport to the nucleus. Biochem. Biophys. Res. Commun. 2003, 303, 1225–1228. [Google Scholar] [CrossRef]
- Wu, A.Z.; Xu, D.; Yang, N.; Lin, S.F.; Chen, P.S.; Cala, S.E.; Chen, Z. Phospholamban is concentrated in the nuclear envelope of cardiomyocytes and involved in perinuclear/nuclear calcium handling. J. Mol. Cell. Cardiol. 2016, 100, 1–8. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G. Sodium-calcium exchangers in the nucleus: An unexpected locus and an unusual regulatory mechanism. Ann. N. Y. Acad. Sci. 2007, 1099, 494–506. [Google Scholar] [CrossRef]
- Voglhuber, J.; Holzer, M.; Radulović, S.; Thai, P.N.; Djalinac, N.; Matzer, I.; Wallner, M.; Bugger, H.; Zirlik, A.; Leitinger, G.; et al. Functional remodelling of perinuclear mitochondria alters nucleoplasmic Ca2+ signalling in heart failure. Philos. Trans. R. Soc. Lond B Biol. Sci. 2022, 377, 20210320. [Google Scholar] [CrossRef]
- Lee, S.H.; Hadipour-Lakmehsari, S.; Miyake, T.; Gramolini, A.O. Three-dimensional imaging reveals endo(sarco)plasmic reticulum-containing invaginations within the nucleoplasm of muscle. Am. J. Physiol. Cell Physiol. 2018, 314, C257–C267. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Thai, P.N.; Lu, S.; Pu, J.; Bers, D.M. Intrafibrillar and perinuclear mitochondrial heterogeneity in adult cardiac myocytes. J. Mol. Cell. Cardiol. 2019, 136, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Kiess, T.O.; Kockskämper, J. SERCA Activity Controls the Systolic Calcium Increase in the Nucleus of Cardiac Myocytes. Front. Physiol. 2019, 10, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljubojevic-Holzer, S.; Herren, A.W.; Djalinac, N.; Voglhuber, J.; Morotti, S.; Holzer, M.; Wood, B.M.; Abdellatif, M.; Matzer, I.; Sacherer, M.; et al. CaMKIIδC Drives Early Adaptive Ca2+ Change and Late Eccentric Cardiac Hypertrophy. Circ. Res. 2020, 127, 1159–1178. [Google Scholar] [CrossRef] [PubMed]
- Bare, D.J.; Kettlun, C.S.; Liang, M.; Bers, D.M.; Mignery, G.A. Cardiac type 2 inositol 1,4,5-trisphosphate receptor: Interaction and modulation by calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 2005, 280, 15912–15920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, M.; Cardenas, C.; Colavita, K.; Petrenko, N.B.; Franzini-Armstrong, C. Structural evidence for perinuclear calcium microdomains in cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 50, 451–459. [Google Scholar] [CrossRef]
- Ljubojevic, S.; Radulovic, S.; Leitinger, G.; Sedej, S.; Sacherer, M.; Holzer, M.; Winkler, C.; Pritz, E.; Mittler, T.; Schmidt, A.; et al. Early remodeling of perinuclear Ca2+ stores and nucleoplasmic Ca2+ signaling during the development of hypertrophy and heart failure. Circulation 2014, 130, 244–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljubojevic, S.; Bers, D.M. Nuclear calcium in cardiac myocytes. J. Cardiovasc. Pharmacol. 2015, 65, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Shahin, V.; Danker, T.; Enss, K.; Ossig, R.; Oberleithner, H. Evidence for Ca2+- and ATP-sensitive peripheral channels in nuclear pore complexes. Faseb. J. 2001, 15, 1895–1901. [Google Scholar] [CrossRef] [Green Version]
- Martins, T.V.; Evans, M.J.; Wysham, D.B.; Morris, R.J. Nuclear pores enable sustained perinuclear calcium oscillations. BMC Syst. Biol. 2016, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Ljubojević, S.; Walther, S.; Asgarzoei, M.; Sedej, S.; Pieske, B.; Kockskämper, J. In situ calibration of nucleoplasmic versus cytoplasmic Ca2+ concentration in adult cardiomyocytes. Biophys. J. 2011, 100, 2356–2366. [Google Scholar] [CrossRef] [Green Version]
- Lipp, P.; Thomas, D.; Berridge, M.J.; Bootman, M.D. Nuclear calcium signalling by individual cytoplasmic calcium puffs. EMBO J. 1997, 16, 7166–7173. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Xu, D.; Wu, A.Z.; Kranias, E.; Lin, S.F.; Chen, P.S.; Chen, Z. Phospholamban regulates nuclear Ca2+ stores and inositol 1,4,5-trisphosphate mediated nuclear Ca2+ cycling in cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 123, 185–197. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Huang, D.; Guo, S.; Wang, D.; Guo, J.; Cala, S.E.; Chen, Z. Association with SERCA2a directs phospholamban trafficking to sarcoplasmic reticulum from a nuclear envelope pool. J. Mol. Cell. Cardiol. 2020, 143, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Amoni, M.; Gilbert, G.; Dries, E.; Doñate Puertas, R.; Tomar, A.; Nagaraju, C.K.; Pradhan, A.; Yule, D.I.; Martens, T.; et al. InsP(3)R-RyR Ca2+ channel crosstalk facilitates arrhythmias in the failing human ventricle. Basic Res. Cardiol. 2022, 117, 60. [Google Scholar] [CrossRef] [PubMed]
- Hohendanner, F.; Maxwell, J.T.; Blatter, L.A. Cytosolic and nuclear calcium signaling in atrial myocytes: IP3-mediated calcium release and the role of mitochondria. Channels 2015, 9, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.C.; Son, M.J.; Le, Q.A.; Woo, S.H. Role of inositol 1,4,5-trisphosphate receptor type 1 in ATP-induced nuclear Ca2+ signal and hypertrophy in atrial myocytes. Biochem. Biophys. Res. Commun. 2018, 503, 2998–3002. [Google Scholar] [CrossRef]
- Garcia, M.I.; Karlstaedt, A.; Chen, J.J.; Amione-Guerra, J.; Youker, K.A.; Taegtmeyer, H.; Boehning, D. Functionally redundant control of cardiac hypertrophic signaling by inositol 1,4,5-trisphosphate receptors. J. Mol. Cell. Cardiol. 2017, 112, 95–103. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, T.; Bossuyt, J.; Li, X.; McKinsey, T.A.; Dedman, J.R.; Olson, E.N.; Chen, J.; Brown, J.H.; Bers, D.M. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J. Clin. Investig. 2006, 116, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Tadevosyan, A.; Maguy, A.; Villeneuve, L.R.; Babin, J.; Bonnefoy, A.; Allen, B.G.; Nattel, S. Nuclear-delimited angiotensin receptor-mediated signaling regulates cardiomyocyte gene expression. J. Biol. Chem. 2010, 285, 22338–22349. [Google Scholar] [CrossRef] [Green Version]
- Ibarra, C.; Vicencio, J.M.; Estrada, M.; Lin, Y.; Rocco, P.; Rebellato, P.; Munoz, J.P.; Garcia-Prieto, J.; Quest, A.F.; Chiong, M.; et al. Local control of nuclear calcium signaling in cardiac myocytes by perinuclear microdomains of sarcolemmal insulin-like growth factor 1 receptors. Circ. Res. 2013, 112, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Arantes, L.A.; Aguiar, C.J.; Amaya, M.J.; Figueiró, N.C.; Andrade, L.M.; Rocha-Resende, C.; Resende, R.R.; Franchini, K.G.; Guatimosim, S.; Leite, M.F. Nuclear inositol 1,4,5-trisphosphate is a necessary and conserved signal for the induction of both pathological and physiological cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 2012, 53, 475–486. [Google Scholar] [CrossRef]
- Olivares-Florez, S.; Czolbe, M.; Riediger, F.; Seidlmayer, L.; Williams, T.; Nordbeck, P.; Strasen, J.; Glocker, C.; Jänsch, M.; Eder-Negrin, P.; et al. Nuclear calcineurin is a sensor for detecting Ca2+ release from the nuclear envelope via IP(3)R. J. Mol. Med. 2018, 96, 1239–1249. [Google Scholar] [CrossRef]
- Ruiz-Hurtado, G.; Morel, E.; Domínguez-Rodríguez, A.; Llach, A.; Lezoualc’h, F.; Benitah, J.P.; Gomez, A.M. Epac in cardiac calcium signaling. J. Mol. Cell. Cardiol. 2013, 58, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.V.; Bare, D.J.; Mignery, G.A.; Blatter, L.A. IP3-dependent nuclear Ca2+ signalling in the mammalian heart. J. Physiol. 2007, 584, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Yang, D.; Lan, X.; Li, K.; Li, X.; Chen, J.; Zhang, Y.; Xiao, R.P.; Han, Q.; Cheng, H. Nuclear Ca2+ sparks and waves mediated by inositol 1,4,5-trisphosphate receptors in neonatal rat cardiomyocytes. Cell Calcium 2008, 43, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kockskämper, J.; Seidlmayer, L.; Walther, S.; Hellenkamp, K.; Maier, L.S.; Pieske, B. Endothelin-1 enhances nuclear Ca2+ transients in atrial myocytes through Ins(1,4,5)P3-dependent Ca2+ release from perinuclear Ca2+ stores. J. Cell Sci. 2008, 121, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L.; Ruiz-Hurtado, G.; Morel, E.; Laurent, A.C.; Métrich, M.; Domínguez-Rodríguez, A.; Lauton-Santos, S.; Lucas, A.; Benitah, J.P.; Bers, D.M.; et al. Epac enhances excitation-transcription coupling in cardiac myocytes. J. Mol. Cell. Cardiol. 2012, 52, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Nakao, S.; Wakabayashi, S.; Nakamura, T.Y. Stimulus-dependent regulation of nuclear Ca2+ signaling in cardiomyocytes: A role of neuronal calcium sensor-1. PLoS ONE 2015, 10, e0125050. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.Y.; Jeromin, A.; Mikoshiba, K.; Wakabayashi, S. Neuronal calcium sensor-1 promotes immature heart function and hypertrophy by enhancing Ca2+ signals. Circ. Res. 2011, 109, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.C.; Dahl, E.F.; Wright, C.D.; Cypher, A.L.; Healy, C.L.; O’Connell, T.D. Nuclear localization of a1A-adrenergic receptors is required for signaling in cardiac myocytes: An “inside-out” a1-AR signaling pathway. J. Am. Heart Assoc. 2014, 3, e000145. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.D.; Chen, Q.; Baye, N.L.; Huang, Y.; Healy, C.L.; Kasinathan, S.; O’Connell, T.D. Nuclear alpha1-adrenergic receptors signal activated ERK localization to caveolae in adult cardiac myocytes. Circ. Res. 2008, 103, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Wright, C.D.; Merkwan, C.L.; Baye, N.L.; Liang, Q.; Simpson, P.C.; O’Connell, T.D. An alpha1A-adrenergic-extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation 2007, 115, 763–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, C.D.; Wu, S.C.; Dahl, E.F.; Sazama, A.J.; O’Connell, T.D. Nuclear localization drives α1-adrenergic receptor oligomerization and signaling in cardiac myocytes. Cell. Signal. 2012, 24, 794–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.D.; Sun, Y.; Bourque, K.; Audet, N.; Inoue, A.; Tanny, J.C.; Hébert, T.E. Receptor- and cellular compartment-specific activation of the cAMP/PKA pathway by α(1)-adrenergic and ETA endothelin receptors. Cell. Signal. 2018, 44, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What Is the Role of β-Adrenergic Signaling in Heart Failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef] [Green Version]
- Drake, M.T.; Shenoy, S.K.; Lefkowitz, R.J. Trafficking of G protein-coupled receptors. Circ. Res. 2006, 99, 570–582. [Google Scholar] [CrossRef]
- O’Connell, T.D.; Jensen, B.C.; Baker, A.J.; Simpson, P.C. Cardiac alpha1-adrenergic receptors: Novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol. Rev. 2014, 66, 308–333. [Google Scholar] [CrossRef] [Green Version]
- Boivin, B.; Lavoie, C.; Vaniotis, G.; Baragli, A.; Villeneuve, L.R.; Ethier, N.; Trieu, P.; Allen, B.G.; Hébert, T.E. Functional beta-adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc. Res. 2006, 71, 69–78. [Google Scholar] [CrossRef]
- Vaniotis, G.; Allen, B.G.; Hébert, T.E. Nuclear GPCRs in cardiomyocytes: An insider’s view of β-adrenergic receptor signaling. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1754–H1764. [Google Scholar] [CrossRef] [Green Version]
- Vaniotis, G.; Del Duca, D.; Trieu, P.; Rohlicek, C.V.; Hébert, T.E.; Allen, B.G. Nuclear β-adrenergic receptors modulate gene expression in adult rat heart. Cell. Signal. 2011, 23, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shi, Q.; Li, M.; Zhao, M.; Reddy Gopireddy, R.; Teoh, J.P.; Xu, B.; Zhu, C.; Ireton, K.E.; Srinivasan, S.; et al. Intracellular β(1)-Adrenergic Receptors and Organic Cation Transporter 3 Mediate Phospholamban Phosphorylation to Enhance Cardiac Contractility. Circ. Res. 2021, 128, 246–261. [Google Scholar] [CrossRef]
- Bedioune, I.; Lefebvre, F.; Lechêne, P.; Varin, A.; Domergue, V.; Kapiloff, M.S.; Fischmeister, R.; Vandecasteele, G. PDE4 and mAKAPβ are nodal organizers of β2-ARs nuclear PKA signalling in cardiac myocytes. Cardiovasc. Res. 2018, 114, 1499–1511. [Google Scholar] [CrossRef] [Green Version]
- Bathe-Peters, M.; Gmach, P.; Boltz, H.H.; Einsiedel, J.; Gotthardt, M.; Hübner, H.; Gmeiner, P.; Lohse, M.J.; Annibale, P. Visualization of β-adrenergic receptor dynamics and differential localization in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2021, 118, 818–823. [Google Scholar] [CrossRef]
- Nash, C.A.; Wei, W.; Irannejad, R.; Smrcka, A.V. Golgi localized β1-adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCε to regulate cardiac hypertrophy. Elife 2019, 8, e48167. [Google Scholar] [CrossRef] [PubMed]
- Irannejad, R.; Pessino, V.; Mika, D.; Huang, B.; Wedegaertner, P.B.; Conti, M.; von Zastrow, M. Functional selectivity of GPCR-directed drug action through location bias. Nat. Chem. Biol. 2017, 13, 799–806. [Google Scholar] [CrossRef] [Green Version]
- Kehat, I.; Molkentin, J.D. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreusser, M.M.; Lehmann, L.H.; Keranov, S.; Hoting, M.O.; Oehl, U.; Kohlhaas, M.; Reil, J.C.; Neumann, K.; Schneider, M.D.; Hill, J.A.; et al. Cardiac CaM Kinase II genes δ and γ contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy. Circulation 2014, 130, 1262–1273. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Phan, D.; van Rooij, E.; Wang, D.Z.; McAnally, J.; Qi, X.; Richardson, J.A.; Hill, J.A.; Bassel-Duby, R.; Olson, E.N. The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J. Clin. Investig. 2008, 118, 124–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmstadter, K.G.; Ljubojevic-Holzer, S.; Wood, B.M.; Taheri, K.D.; Sedej, S.; Erickson, J.R.; Bossuyt, J.; Bers, D.M. CaMKII and PKA-dependent phosphorylation co-regulate nuclear localization of HDAC4 in adult cardiomyocytes. Basic Res. Cardiol. 2021, 116, 11. [Google Scholar] [CrossRef]
- Kreusser, M.M.; Backs, J. Integrated mechanisms of CaMKII-dependent ventricular remodeling. Front. Pharmacol. 2014, 5, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suetomi, T.; Willeford, A.; Brand, C.S.; Cho, Y.; Ross, R.S.; Miyamoto, S.; Brown, J.H. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca2+/Calmodulin-Dependent Protein Kinase II δ Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef] [Green Version]
- Subedi, K.P.; Son, M.J.; Chidipi, B.; Kim, S.W.; Wang, J.; Kim, K.H.; Woo, S.H.; Kim, J.C. Signaling Pathway for Endothelin-1- and Phenylephrine-Induced cAMP Response Element Binding Protein Activation in Rat Ventricular Myocytes: Role of Inositol 1,4,5-Trisphosphate Receptors and CaMKII. Cell. Physiol. Biochem. 2017, 41, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Ronkainen, J.J.; Hänninen, S.L.; Korhonen, T.; Koivumäki, J.T.; Skoumal, R.; Rautio, S.; Ronkainen, V.P.; Tavi, P. Ca2+-calmodulin-dependent protein kinase II represses cardiac transcription of the L-type calcium channel alpha(1C)-subunit gene (Cacna1c) by DREAM translocation. J. Physiol. 2011, 589, 2669–2686. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Aye-Han, N.N.; Ganesan, A.; Oldach, L.; Gorshkov, K.; Zhang, J. Calmodulin-controlled spatial decoding of oscillatory Ca2+ signals by calcineurin. Elife 2014, 3, e03765. [Google Scholar] [CrossRef]
- Chaklader, M.; Rothermel, B.A. Calcineurin in the heart: New horizons for an old friend. Cell. Signal. 2021, 87, 110134. [Google Scholar] [CrossRef]
- Parra, V.; Rothermel, B.A. Calcineurin signaling in the heart: The importance of time and place. J. Mol. Cell. Cardiol. 2017, 103, 121–136. [Google Scholar] [CrossRef] [Green Version]
- Plačkić, J.; Preissl, S.; Nikonova, Y.; Pluteanu, F.; Hein, L.; Kockskämper, J. Enhanced nucleoplasmic Ca2+ signaling in ventricular myocytes from young hypertensive rats. J. Mol. Cell. Cardiol. 2016, 101, 58–68. [Google Scholar] [CrossRef]
- Pérez-Hernández, M.; van Opbergen, C.J.M.; Bagwan, N.; Vissing, C.R.; Marrón-Liñares, G.M.; Zhang, M.; Torres Vega, E.; Sorrentino, A.; Drici, L.; Sulek, K.; et al. Loss of Nuclear Envelope Integrity and Increased Oxidant Production Cause DNA Damage in Adult Hearts Deficient in PKP2: A Molecular Substrate of ARVC. Circulation 2022, 146, 851–867. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling--concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.; Zornoff, L.A. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Zhang, T.; Pereira, L.; Means, C.K.; Cheng, H.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Chen, J.; Bers, D.; et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J. Clin. Investig. 2009, 119, 1230–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakata, Y.; Masuyama, T.; Yamamoto, K.; Nishikawa, N.; Yamamoto, H.; Kondo, H.; Ono, K.; Otsu, K.; Kuzuya, T.; Miwa, T.; et al. Calcineurin inhibitor attenuates left ventricular hypertrophy, leading to prevention of heart failure in hypertensive rats. Circulation 2000, 102, 2269–2275. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.H.; Eiringhaus, J.; Dybkova, N.; Förster, A.; Herting, J.; Kleinwächter, A.; Ljubojevic, S.; Schmitto, J.D.; Streckfuß-Bömeke, K.; Renner, A.; et al. Ca2+ /calmodulin-dependent protein kinase II equally induces sarcoplasmic reticulum Ca2+ leak in human ischaemic and dilated cardiomyopathy. Eur. J. Heart Fail. 2014, 16, 1292–1300. [Google Scholar] [CrossRef]
- Lou, Q.; Janardhan, A.; Efimov, I.R. Remodeling of calcium handling in human heart failure. Adv. Exp. Med. Biol. 2012, 740, 1145–1174. [Google Scholar] [CrossRef] [Green Version]
- Sossalla, S.; Fluschnik, N.; Schotola, H.; Ort, K.R.; Neef, S.; Schulte, T.; Wittköpper, K.; Renner, A.; Schmitto, J.D.; Gummert, J.; et al. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ. Res. 2010, 107, 1150–1161. [Google Scholar] [CrossRef] [Green Version]
- Westenbrink, B.D.; Edwards, A.G.; McCulloch, A.D.; Brown, J.H. The promise of CaMKII inhibition for heart disease: Preventing heart failure and arrhythmias. Expert Opin. Ther. Targets 2013, 17, 889–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panther, F.; Williams, T.; Ritter, O. Inhibition of the calcineurin-NFAT signalling cascade in the treatment of heart failure. Recent Pat. Cardiovasc. Drug Discov. 2009, 4, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Lin, J.W.; Huang, C.Y.; Yeh, Y.L.; Shen, C.Y.; Badrealam, K.F.; Ho, T.J.; Padma, V.V.; Kuo, W.W.; Huang, C.Y. The combined inhibition of the CaMKIIδ and calcineurin signaling cascade attenuates IGF-IIR-induced cardiac hypertrophy. J. Cell. Physiol. 2020, 235, 3539–3547. [Google Scholar] [CrossRef]
- Pellicena, P.; Schulman, H. CaMKII inhibitors: From research tools to therapeutic agents. Front. Pharmacol. 2014, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Kawase, Y.; Ly, H.Q.; Prunier, F.; Lebeche, D.; Shi, Y.; Jin, H.; Hadri, L.; Yoneyama, R.; Hoshino, K.; Takewa, Y.; et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 2008, 51, 1112–1119. [Google Scholar] [CrossRef] [Green Version]
- Hulot, J.S.; Ishikawa, K.; Hajjar, R.J. Gene therapy for the treatment of heart failure: Promise postponed. Eur. Heart J. 2016, 37, 1651–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, B.; Butler, J.; Felker, G.M.; Ponikowski, P.; Voors, A.A.; Desai, A.S.; Barnard, D.; Bouchard, A.; Jaski, B.; Lyon, A.R.; et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): A randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 2016, 387, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Lebek, S.; Chemello, F.; Caravia, X.M.; Tan, W.; Li, H.; Chen, K.; Xu, L.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Ablation of CaMKIIδ oxidation by CRISPR-Cas9 base editing as a therapy for cardiac disease. Science 2023, 379, 179–185. [Google Scholar] [CrossRef]
- Shimojima, M.; Yuasa, S.; Motoda, C.; Yozu, G.; Nagai, T.; Ito, S.; Lachmann, M.; Kashimura, S.; Takei, M.; Kusumoto, D.; et al. Emerin plays a crucial role in nuclear invagination and in the nuclear calcium transient. Sci. Rep. 2017, 7, 44312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzer, I.; Voglhuber, J.; Kiessling, M.; Djalinac, N.; Trummer-Herbst, V.; Mabotuwana, N.; Rech, L.; Holzer, M.; Sossalla, S.; Rainer, P.P.; et al. β-Adrenergic Receptor Stimulation Maintains NCX-CaMKII Axis and Prevents Overactivation of IL6R-Signaling in Cardiomyocytes upon Increased Workload. Biomedicines 2022, 10, 1648. [Google Scholar] [CrossRef]
- Harzheim, D.; Talasila, A.; Movassagh, M.; Foo, R.S.; Figg, N.; Bootman, M.D.; Roderick, H.L. Elevated InsP3R expression underlies enhanced calcium fluxes and spontaneous extra-systolic calcium release events in hypertrophic cardiac myocytes. Channels 2010, 4, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M. Cardiac ryanodine receptor phosphorylation: Target sites and functional consequences. Biochem. J. 2006, 396, e1–e3. [Google Scholar] [CrossRef] [Green Version]
- Wood, B.M.; Simon, M.; Galice, S.; Alim, C.C.; Ferrero, M.; Pinna, N.N.; Bers, D.M.; Bossuyt, J. Cardiac CaMKII activation promotes rapid translocation to its extra-dyadic targets. J. Mol. Cell. Cardiol. 2018, 125, 18–28. [Google Scholar] [CrossRef]
- Gómez, A.M.; Ruiz-Hurtado, G.; Benitah, J.P.; Domínguez-Rodríguez, A. Ca2+ fluxes involvement in gene expression during cardiac hypertrophy. Curr. Vasc. Pharmacol. 2013, 11, 497–506. [Google Scholar] [CrossRef]
- Wang, P.; Xu, S.; Xu, J.; Xin, Y.; Lu, Y.; Zhang, H.; Zhou, B.; Xu, H.; Sheu, S.S.; Tian, R.; et al. Elevated MCU Expression by CaMKIIδB Limits Pathological Cardiac Remodeling. Circulation 2022, 145, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yang, N.; Sidor, A.; O’Rourke, B. MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca2+ Leak. Circ. Res. 2021, 128, 1191–1204. [Google Scholar] [CrossRef]
- Chahine, M.N.; Mioulane, M.; Sikkel, M.B.; O’Gara, P.; Dos Remedios, C.G.; Pierce, G.N.; Lyon, A.R.; Földes, G.; Harding, S.E. Nuclear pore rearrangements and nuclear trafficking in cardiomyocytes from rat and human failing hearts. Cardiovasc. Res. 2015, 105, 31–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarazón, E.; Rivera, M.; Roselló-Lletí, E.; Molina-Navarro, M.M.; Sánchez-Lázaro, I.J.; España, F.; Montero, J.A.; Lago, F.; González-Juanatey, J.R.; Portolés, M. Heart failure induces significant changes in nuclear pore complex of human cardiomyocytes. PLoS ONE 2012, 7, e48957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.Y.; Vahdati Hassani, F.; Hoffmann, D.; Xiao, J.; Xiong, F.; Villeneuve, L.R.; Ljubojevic-Holzer, S.; Kamler, M.; Abu-Taha, I.; Heijman, J.; et al. Inositol Trisphosphate Receptors and Nuclear Calcium in Atrial Fibrillation. Circ. Res. 2021, 128, 619–635. [Google Scholar] [CrossRef]
- Staerk, L.; Wang, B.; Preis, S.R.; Larson, M.G.; Lubitz, S.A.; Ellinor, P.T.; McManus, D.D.; Ko, D.; Weng, L.C.; Lunetta, K.L.; et al. Lifetime risk of atrial fibrillation according to optimal, borderline, or elevated levels of risk factors: Cohort study based on longitudinal data from the Framingham Heart Study. BMJ 2018, 361, k1453. [Google Scholar] [CrossRef] [Green Version]
- Andrade, J.; Khairy, P.; Dobrev, D.; Nattel, S. The clinical profile and pathophysiology of atrial fibrillation: Relationships among clinical features, epidemiology, and mechanisms. Circ. Res. 2014, 114, 1453–1468. [Google Scholar] [CrossRef]
- Kornej, J.; Börschel, C.S.; Benjamin, E.J.; Schnabel, R.B. Epidemiology of Atrial Fibrillation in the 21st Century: Novel Methods and New Insights. Circ. Res. 2020, 127, 4–20. [Google Scholar] [CrossRef]
- Nattel, S.; Dobrev, D. The multidimensional role of calcium in atrial fibrillation pathophysiology: Mechanistic insights and therapeutic opportunities. Eur. Heart J. 2012, 33, 1870–1877. [Google Scholar] [CrossRef] [Green Version]
- Pabel, S.; Knierim, M.; Stehle, T.; Alebrand, F.; Paulus, M.; Sieme, M.; Herwig, M.; Barsch, F.; Körtl, T.; Pöppl, A.; et al. Effects of Atrial Fibrillation on the Human Ventricle. Circ. Res. 2022, 130, 994–1010. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Siegfried, J.D. Update 2011: Clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2011, 57, 1641–1649. [Google Scholar] [CrossRef] [Green Version]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J., Jr.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A. Molecular basis of hereditary cardiomyopathy: Abnormalities in calcium sensitivity, stretch response, stress response and beyond. J. Hum. Genet. 2010, 55, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holaska, J.M. Emerin and the nuclear lamina in muscle and cardiac disease. Circ. Res. 2008, 103, 16–23. [Google Scholar] [CrossRef]
- Nattel, S.; Heijman, J.; Zhou, L.; Dobrev, D. Molecular Basis of Atrial Fibrillation Pathophysiology and Therapy: A Translational Perspective. Circ. Res. 2020, 127, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, B.; Pölönen, R.P.; Hellgren, K.T.; Ko, C.Y.; Ginsburg, K.S.; Bossuyt, J.; Mercola, M.; Bers, D.M. Cardiomyocyte Na+ and Ca2+ mishandling drives vicious cycle involving CaMKII, ROS, and ryanodine receptors. Basic Res. Cardiol. 2021, 116, 58. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Higo, S.; Shiba, M.; Kohama, Y.; Kameda, S.; Tabata, T.; Inoue, H.; Okuno, S.; Ogawa, S.; Nakamura, S.; et al. Human-Induced Pluripotent Stem Cell-Derived Cardiomyocyte Model for TNNT2 Δ160E-Induced Cardiomyopathy. Circ. Genom. Precis. Med. 2022, 15, e003522. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.; Middleton, D.A. Comparison of the structure and function of phospholamban and the arginine-14 deficient mutant associated with dilated cardiomyopathy. PLoS ONE 2014, 9, e106746. [Google Scholar] [CrossRef] [Green Version]
- van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 Mutations Induce an Arrhythmogenic Dilated Cardiomyopathy Related to Disturbed Calcium Handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Dibb, K.M.; Graham, H.K.; Venetucci, L.A.; Eisner, D.A.; Trafford, A.W. Analysis of cellular calcium fluxes in cardiac muscle to understand calcium homeostasis in the heart. Cell Calcium 2007, 42, 503–512. [Google Scholar] [CrossRef]
- Bootman, M.D.; Rietdorf, K.; Collins, T.; Walker, S.; Sanderson, M. Ca2+-sensitive fluorescent dyes and intracellular Ca2+ imaging. Cold Spring Harb. Protoc. 2013, 2013, 83–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Camacho, P.; Lechleiter, J.D.; Herman, B. Measurement of Intracellular Calcium. Physiol. Rev. 1999, 79, 1089–1125. [Google Scholar] [CrossRef] [PubMed]
- Ljubojević, S.; Bers, D.M. Measuring intranuclear and nuclear envelope [Ca2+] vs. cytosolic [Ca2+]. Methods Mol. Biol. 2015, 1234, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.M.; Taylor, C.W. Reliable measurement of free Ca2+ concentrations in the ER lumen using Mag-Fluo-4. Cell Calcium 2020, 87, 102188. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shen, L.; Zhao, F.; Zou, X.; He, Y.; Zhang, F.; Zhang, C.; Yu, B.; Cao, Z. Modification of distinct ion channels differentially modulates Ca2+ dynamics in primary cultured rat ventricular cardiomyocytes. Sci. Rep. 2017, 7, 40952. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Patel, D.; Zhao, Q.; Peng, R.; Guo, H.; Diwu, Z. A novel Ca2+ indicator for long-term tracking of intracellular calcium flux. BioTechniques 2021, 70, 271–277. [Google Scholar] [CrossRef]
- Daily, N.J.; Santos, R.; Vecchi, J.; Kemanli, P.; Wakatsuki, T. Calcium Transient Assays for Compound Screening with Human iPSC-derived Cardiomyocytes: Evaluating New Tools. J. Evol. Stem Cell Res. 2017, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.; Tovey, S.C.; Collins, T.J.; Bootman, M.D.; Berridge, M.J.; Lipp, P. A comparison of fluorescent Ca2+ indicator properties and their use in measuring elementary and global Ca2+ signals. Cell Calcium 2000, 28, 213–223. [Google Scholar] [CrossRef]
- Smith, N.A.; Kress, B.T.; Lu, Y.; Chandler-Militello, D.; Benraiss, A.; Nedergaard, M. Fluorescent Ca2+ indicators directly inhibit the Na,K-ATPase and disrupt cellular functions. Sci. Signal. 2018, 11, eaal2039. [Google Scholar] [CrossRef] [Green Version]
- Nakai, J.; Ohkura, M.; Imoto, K. A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat. Biotechnol. 2001, 19, 137–141. [Google Scholar] [CrossRef]
- Chen, T.W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Liu, N.; He, Y.; Liu, Y.; Ge, L.; Zou, L.; Song, S.; Xiong, W.; Liu, X. Improved calcium sensor GCaMP-X overcomes the calcium channel perturbations induced by the calmodulin in GCaMP. Nat. Commun. 2018, 9, 1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Zhou, Y.; Bao, X.; Chen, C.; Randolph, L.N.; Du, J.; Lian, X.L. An Ultrasensitive Calcium Reporter System via CRISPR-Cas9-Mediated Genome Editing in Human Pluripotent Stem Cells. iScience 2018, 9, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Safaralizade, M.; Fuderer, R.; Grosse, R.; Zhao, B. Measuring nuclear calcium and actin assembly in living cells. J. Biochem. 2021, 169, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Mertes, N.; Busch, M.; Huppertz, M.C.; Hacker, C.N.; Wilhelm, J.; Gürth, C.M.; Kühn, S.; Hiblot, J.; Koch, B.; Johnsson, K. Fluorescent and Bioluminescent Calcium Indicators with Tuneable Colors and Affinities. J. Am. Chem. Soc. 2022, 144, 6928–6935. [Google Scholar] [CrossRef] [PubMed]
- Voigt, N.; Heijman, J.; Wang, Q.; Chiang, D.Y.; Li, N.; Karck, M.; Wehrens, X.H.T.; Nattel, S.; Dobrev, D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 2014, 129, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljubojević-Holzer, S.; Kraler, S.; Djalinac, N.; Abdellatif, M.; Voglhuber, J.; Schipke, J.; Schmidt, M.; Kling, K.M.; Franke, G.T.; Herbst, V.; et al. Loss of autophagy protein ATG5 impairs cardiac capacity in mice and humans through diminishing mitochondrial abundance and disrupting Ca2+ cycling. Cardiovasc. Res. 2022, 118, 1492–1505. [Google Scholar] [CrossRef]
- Liang, P.; Lan, F.; Lee, A.S.; Gong, T.; Sanchez-Freire, V.; Wang, Y.; Diecke, S.; Sallam, K.; Knowles, J.W.; Wang, P.J.; et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 2013, 127, 1677–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itzhaki, I.; Rapoport, S.; Huber, I.; Mizrahi, I.; Zwi-Dantsis, L.; Arbel, G.; Schiller, J.; Gepstein, L. Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PLoS ONE 2011, 6, e18037. [Google Scholar] [CrossRef]
- Karakikes, I.; Ameen, M.; Termglinchan, V.; Wu, J.C. Human induced pluripotent stem cell-derived cardiomyocytes: Insights into molecular, cellular, and functional phenotypes. Circ. Res. 2015, 117, 80–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.S.; Kryshtal, D.O.; Feaster, T.K.; Sánchez-Freire, V.; Zhang, J.; Kamp, T.J.; Hong, C.C.; Wu, J.C.; Knollmann, B.C. Comparable calcium handling of human iPSC-derived cardiomyocytes generated by multiple laboratories. J. Mol. Cell. Cardiol. 2015, 85, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Klos, M.; Wilson, G.F.; Herman, A.M.; Lian, X.; Raval, K.K.; Barron, M.R.; Hou, L.; Soerens, A.G.; Yu, J.; et al. Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: The matrix sandwich method. Circ. Res. 2012, 111, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Lundy, S.D.; Zhu, W.Z.; Regnier, M.; Laflamme, M.A. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 2013, 22, 1991–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Guo, L.; Fiene, S.J.; Anson, B.D.; Thomson, J.A.; Kamp, T.J.; Kolaja, K.L.; Swanson, B.J.; January, C.T. High purity human-induced pluripotent stem cell-derived cardiomyocytes: Electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2006–H2017. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Keung, W.; Cheng, H.; Li, R.A. Structural and Mechanistic Bases of Nuclear Calcium Signaling in Human Pluripotent Stem Cell-Derived Ventricular Cardiomyocytes. Stem Cells Int. 2019, 2019, 8765752. [Google Scholar] [CrossRef]
- Lee, J.; Termglinchan, V.; Diecke, S.; Itzhaki, I.; Lam, C.K.; Garg, P.; Lau, E.; Greenhaw, M.; Seeger, T.; Wu, H.; et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 2019, 572, 335–340. [Google Scholar] [CrossRef]
- Funakoshi, M.; Tsuchiya, Y.; Arahata, K. Emerin and cardiomyopathy in Emery-Dreifuss muscular dystrophy. Neuromuscul. Disord. 1999, 9, 108–114. [Google Scholar] [CrossRef]
- Thomas, D.; Choi, S.; Alamana, C.; Parker, K.K.; Wu, J.C. Cellular and Engineered Organoids for Cardiovascular Models. Circ. Res. 2022, 130, 1780–1802. [Google Scholar] [CrossRef]
- Campostrini, G.; Windt, L.M.; van Meer, B.J.; Bellin, M.; Mummery, C.L. Cardiac Tissues From Stem Cells: New Routes to Maturation and Cardiac Regeneration. Circ. Res. 2021, 128, 775–801. [Google Scholar] [CrossRef]
- Richards, D.J.; Li, Y.; Kerr, C.M.; Yao, J.; Beeson, G.C.; Coyle, R.C.; Chen, X.; Jia, J.; Damon, B.; Wilson, R.; et al. Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat. Biomed. Eng. 2020, 4, 446–462. [Google Scholar] [CrossRef]
- Higazi, D.R.; Fearnley, C.J.; Drawnel, F.M.; Talasila, A.; Corps, E.M.; Ritter, O.; McDonald, F.; Mikoshiba, K.; Bootman, M.D.; Roderick, H.L. Endothelin-1-stimulated InsP3-induced Ca2+ release is a nexus for hypertrophic signaling in cardiac myocytes. Mol. Cell 2009, 33, 472–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfenniger, A.; Yoo, S.; Arora, R. Nucleoplasmic Ca2+: The ‘Mastermind’ Behind Pathological Atrial Remodeling? Circ. Res. 2021, 128, 636–638. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiessling, M.; Djalinac, N.; Voglhuber, J.; Ljubojevic-Holzer, S. Nuclear Calcium in Cardiac (Patho)Physiology: Small Compartment, Big Impact. Biomedicines 2023, 11, 960. https://doi.org/10.3390/biomedicines11030960
Kiessling M, Djalinac N, Voglhuber J, Ljubojevic-Holzer S. Nuclear Calcium in Cardiac (Patho)Physiology: Small Compartment, Big Impact. Biomedicines. 2023; 11(3):960. https://doi.org/10.3390/biomedicines11030960
Chicago/Turabian StyleKiessling, Mara, Nataša Djalinac, Julia Voglhuber, and Senka Ljubojevic-Holzer. 2023. "Nuclear Calcium in Cardiac (Patho)Physiology: Small Compartment, Big Impact" Biomedicines 11, no. 3: 960. https://doi.org/10.3390/biomedicines11030960
APA StyleKiessling, M., Djalinac, N., Voglhuber, J., & Ljubojevic-Holzer, S. (2023). Nuclear Calcium in Cardiac (Patho)Physiology: Small Compartment, Big Impact. Biomedicines, 11(3), 960. https://doi.org/10.3390/biomedicines11030960