
Oleic Acid-Containing Phosphatidylinositol Is a Blood Biomarker Candidate for SPG28
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
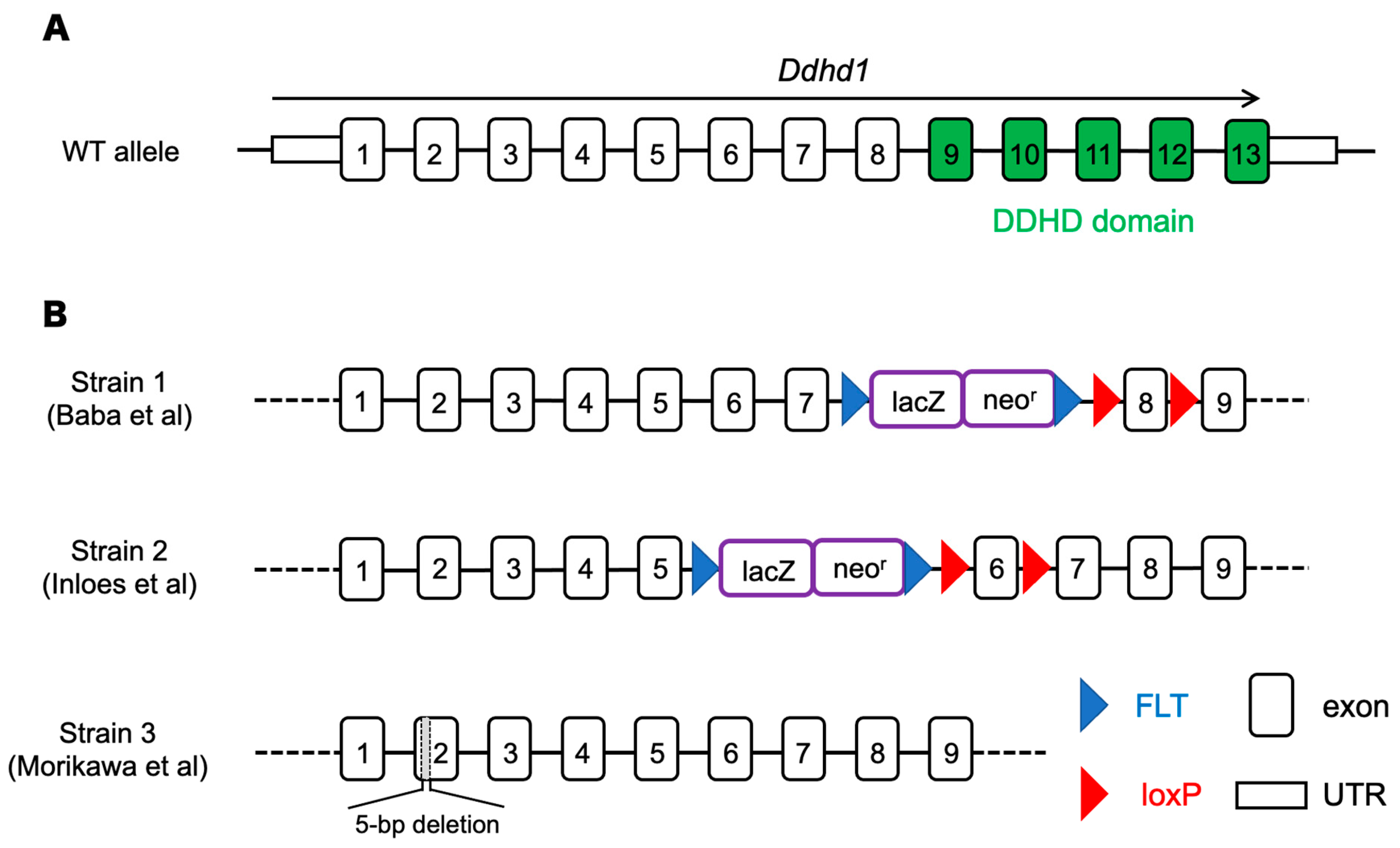
2.1. Animals
2.2. Genotyping
2.3. Sample Preparation of Mouse Plasma
2.4. Human Sample Preparation
2.5. Lipid Extraction
2.6. Lipidome Analysis for Mouse Plasma and Human Sera
3. Results
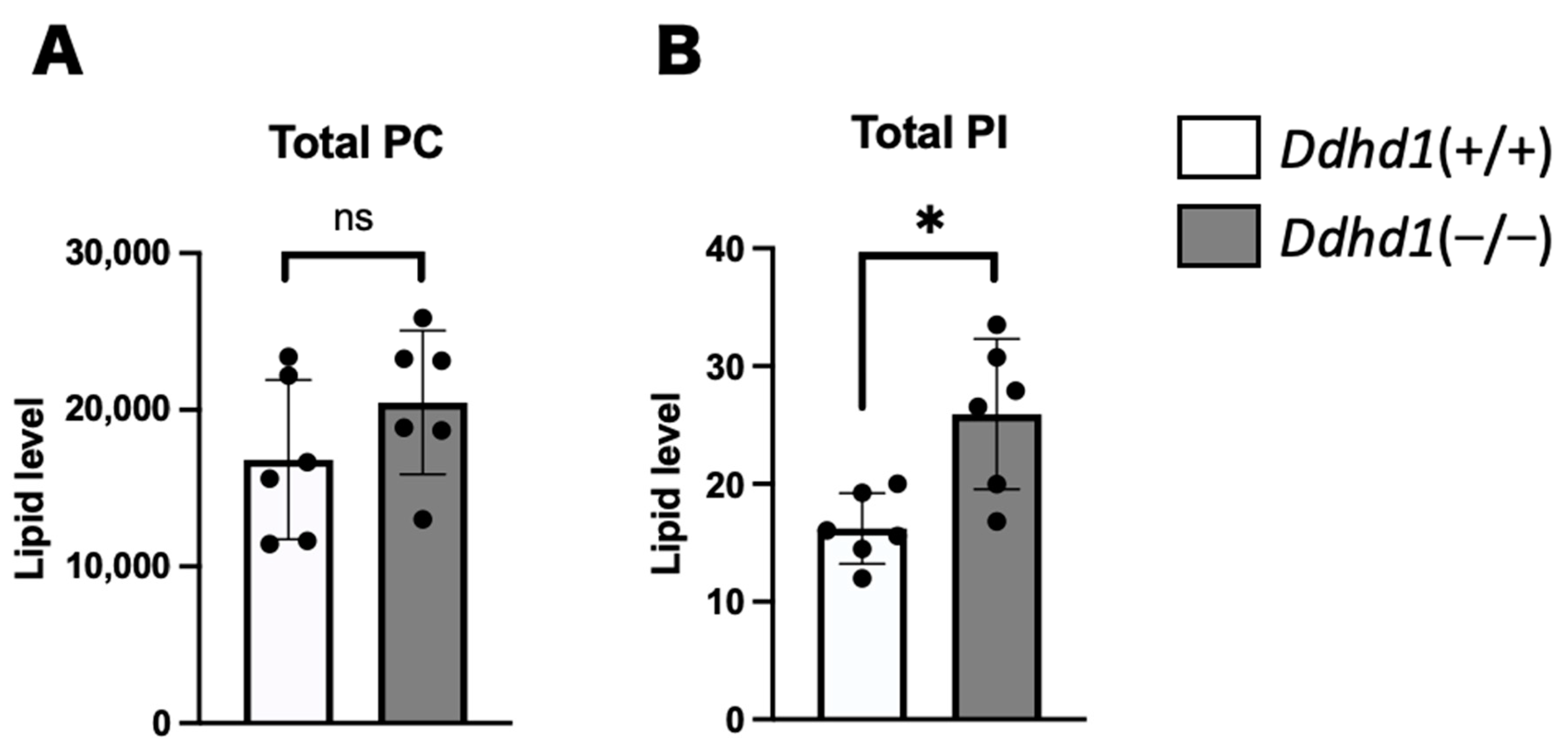
3.1. PC, PA and PI in Mouse Plasma
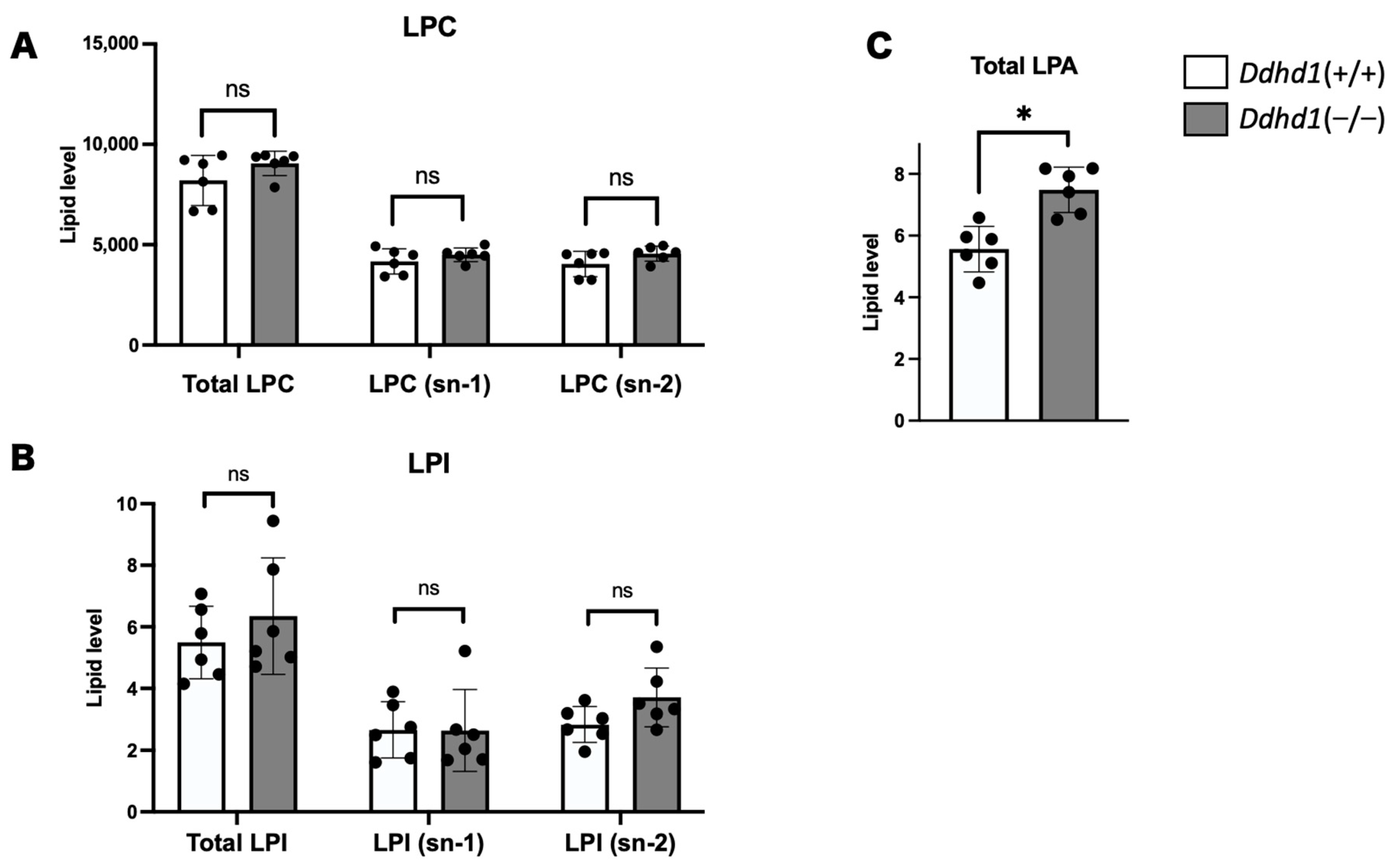
3.2. LPC, LPA and LPI in Mouse Plasma
3.3. Focused lipidome Analysis of Human Sera
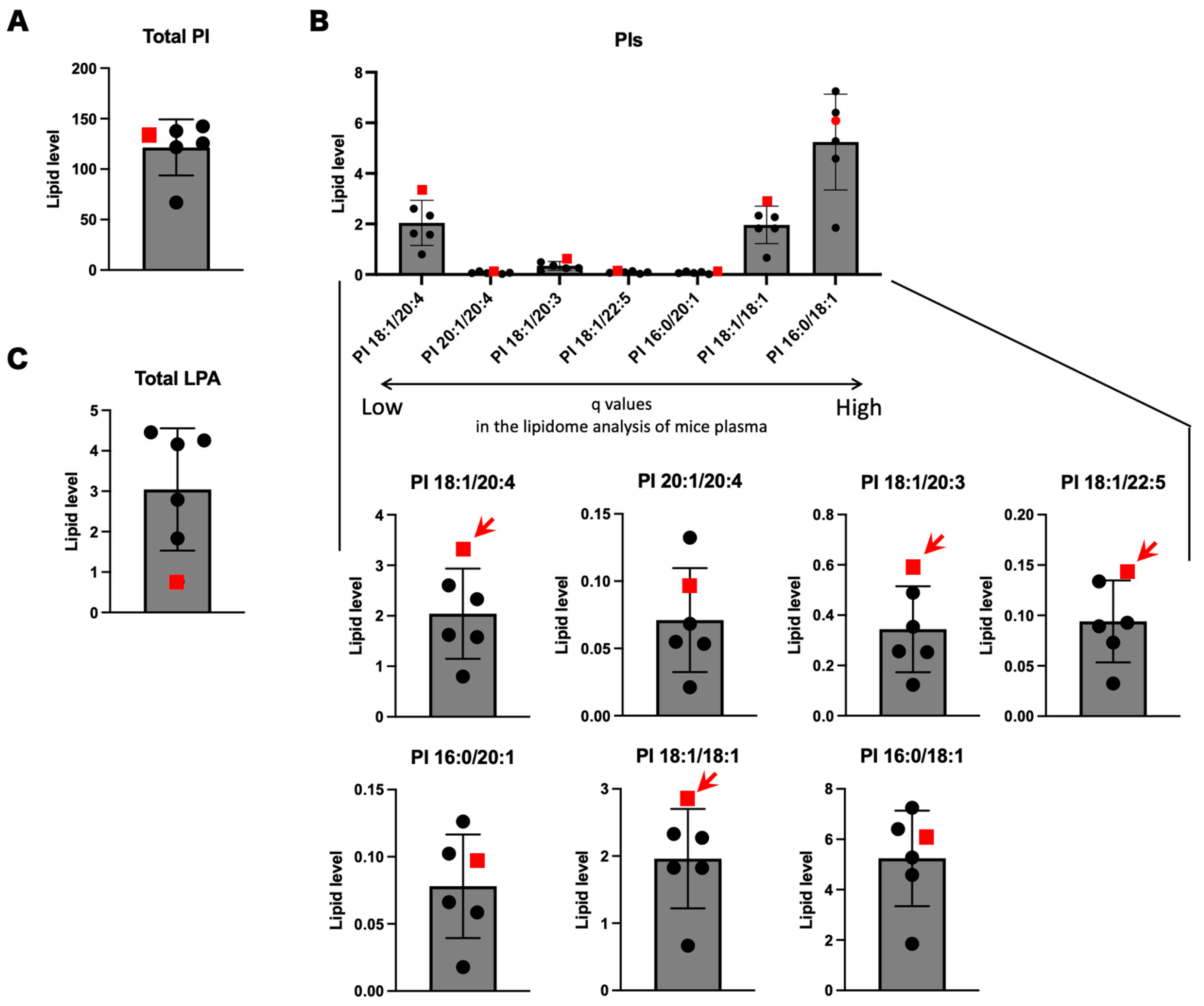
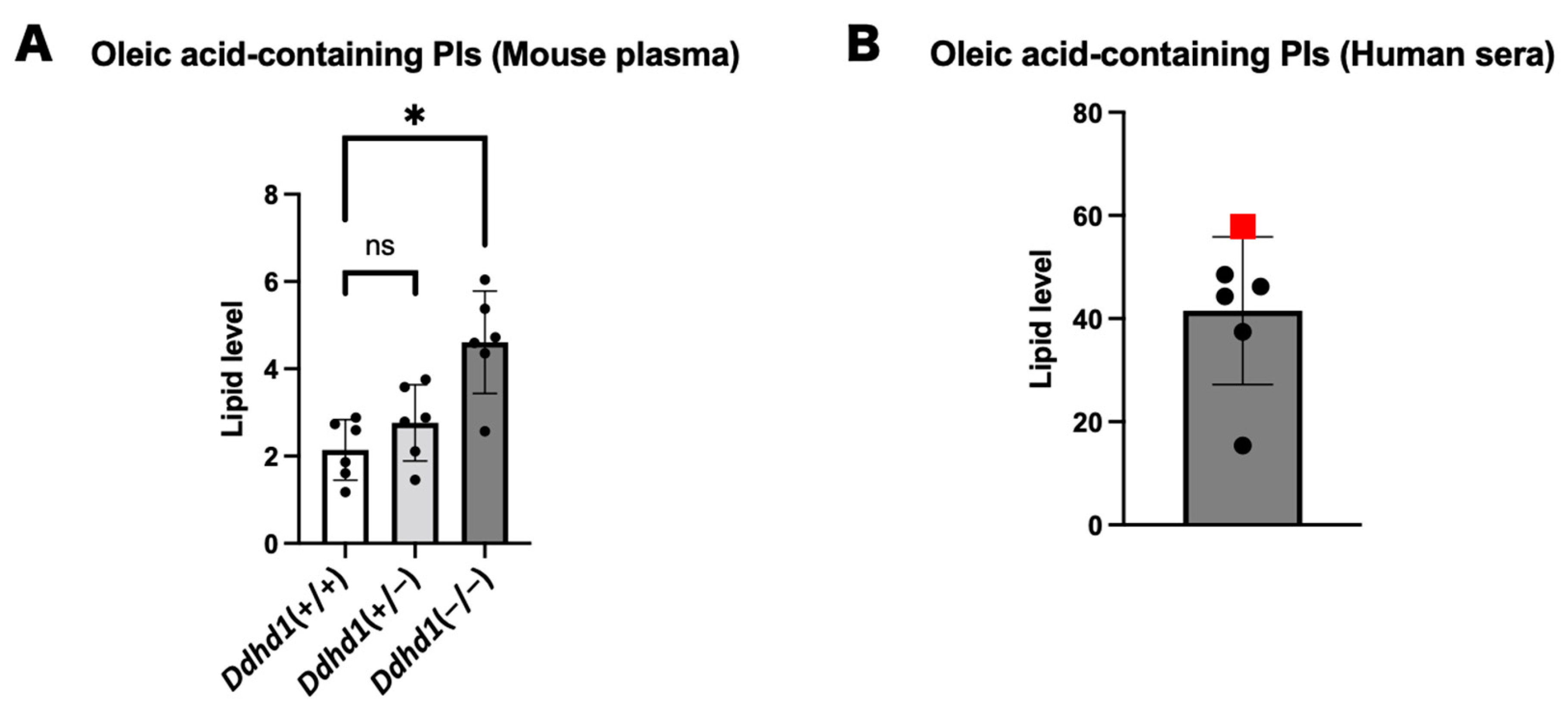
3.4. PIs and LPIs Containing Oleic Acid
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tesson, C.; Koht, J.; Stevanin, G. Delving into the complexity of hereditary spastic paraplegias: How unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum. Genet. 2015, 134, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klebe, S.; Stevanin, G.; Depienne, C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: From SPG1 to SPG72 and still counting. Rev. Neurol. 2015, 171, 505–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, J. Hereditary Spastic Paraplegia: Clinical Principles and Genetic Advances. Semin. Neurol. 2014, 34, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, C. Hereditary spastic paraplegia. Handb. Clin. Neurol. 2018, 148, 633–652. [Google Scholar] [CrossRef]
- Blackstone, C. Cellular Pathways of Hereditary Spastic Paraplegia. Annu. Rev. Neurosci. 2012, 35, 25–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef] [Green Version]
- Saputra, L.; Kumar, K.R. Challenges and Controversies in the Genetic Diagnosis of Hereditary Spastic Paraplegia. Curr. Neurol. Neurosci. Rep. 2021, 21, 15. [Google Scholar] [CrossRef]
- Elsayed, L.E.O.; Eltazi, I.Z.; Ahmed, A.E.; Stevanin, G. Insights into Clinical, Genetic, and Pathological Aspects of Hereditary Spastic Paraplegias: A Comprehensive Overview. Front. Mol. Biosci. 2021, 8, 690899. [Google Scholar] [CrossRef]
- Bouslam, N.; Benomar, A.; Azzedine, H.; Bouhouche, A.; Namekawa, M.; Klebe, S.; Durr, A.; Ruberg, M.; Brice, A.; Yahyaoui, M.; et al. Mapping of a new form of pure autosomal recessive spastic paraplegia (SPG28). Ann. Neurol. 2005, 57, 567–571. [Google Scholar] [CrossRef]
- Tesson, C.; Nawara, M.; Salih, M.A.; Rossignol, R.; Zaki, M.S.; Al Balwi, M.; Schule, R.; Mignot, C.; Obre, E.; Bouhouche, A.; et al. Alteration of Fatty-Acid-Metabolizing Enzymes Affects Mitochondrial Form and Function in Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 2012, 91, 1051–1064. [Google Scholar] [CrossRef] [Green Version]
- Aloulou, A.; Rahier, R.; Arhab, Y.; Noiriel, A.; Abousalham, A. Phospholipases: An Overview. Methods Mol. Biol. 2018, 1835, 69–105. [Google Scholar] [CrossRef]
- Richmond, G.S.; Smith, T.K. Phospholipases A1. Int. J. Mol. Sci. 2011, 12, 588–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tani, K.; Kogure, T.; Inoue, H. The intracellular phospholipase A1 protein family. Biomol. Concepts 2012, 3, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Higgs, H.N.; Glomset, J.A. Purification and Properties of a Phosphatidic Acid-preferring Phospholipase A1 from Bovine Testis: Examination of the Molecular Basis of Its Activation. J. Biol. Chem. 1996, 271, 10874–10883. [Google Scholar] [CrossRef] [Green Version]
- Tani, K.; Mizoguchi, T.; Iwamatsu, A.; Hatsuzawa, K.; Tagaya, M. p125 Is a Novel Mammalian Sec23p-interacting Protein with Structural Similarity to Phospholipid-modifying Proteins. J. Biol. Chem. 1999, 274, 20505–20512. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, K.-I.; Mizoguchi, T.; Nagahama, M.; Tagaya, M.; Tani, K.; Sonoda, H.; Aoki, J.; Arai, H. A Novel Phospholipase A1 with Sequence Homology to a Mammalian Sec23p-interacting Protein, p125. J. Biol. Chem. 2002, 277, 11329–11335. [Google Scholar] [CrossRef] [Green Version]
- Baba, T.; Kashiwagi, Y.; Arimitsu, N.; Kogure, T.; Edo, A.; Maruyama, T.; Nakao, K.; Nakanishi, H.; Kinoshita, M.; Frohman, M.A.; et al. Phosphatidic Acid (PA)-preferring Phospholipase A1 Regulates Mitochondrial Dynamics. J. Biol. Chem. 2014, 289, 11497. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, T.; Nakajima, K.-I.; Hatsuzawa, K.; Nagahama, M.; Hauri, H.-P.; Tagaya, M.; Tani, K. Determination of Functional Regions of p125, a Novel Mammalian Sec23p-Interacting Protein. Biochem. Biophys. Res. Commun. 2000, 279, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Shimoi, W.; Ezawa, I.; Nakamoto, K.; Uesaki, S.; Gabreski, G.; Aridor, M.; Yamamoto, A.; Nagahama, M.; Tagaya, M.; Tani, K. p125 Is Localized in Endoplasmic Reticulum Exit Sites and Involved in Their Organization. J. Biol. Chem. 2005, 280, 10141–10148. [Google Scholar] [CrossRef] [Green Version]
- Liguori, R.; Giannoccaro, M.P.; Arnoldi, A.; Citterio, A.; Tonon, C.; Lodi, R.; Bresolin, N.; Bassi, M.T. Impairment of brain and muscle energy metabolism detected by magnetic resonance spectroscopy in hereditary spastic paraparesis type 28 patients with DDHD1 mutations. J. Neurol. 2014, 261, 1789–1793. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Fan, D. A Novel Missense Mutation of the DDHD1 Gene Associated with Juvenile Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2016, 8, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, S.; Morikawa, T.; Fujioka, R.; Kosaka, K.; Yamada, K.; Hattori, G.; Motomura, M.; Taniwaki, T.; Shibata, H. A novel frameshift mutation of DDHD1 in a Japanese patient with autosomal recessive spastic paraplegia. Eur. J. Med. Genet. 2016, 59, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, T.; Ohishi, H.; Kosaka, K.; Shimojo, T.; Nagano, A.; Taniguchi, I.; Fujioka, R.; Moriyama, K.; Unoki, M.; Takahashi, M.; et al. Ddhd1 knockout mouse as a model of locomotive and physiological abnormality in familial spastic paraplegia. Biosci. Rep. 2021, 41, BSR20204171. [Google Scholar] [CrossRef] [PubMed]
- Inloes, J.M.; Jing, H.; Cravatt, B.F. The Spastic Paraplegia-Associated Phospholipase DDHD1 Is a Primary Brain Phosphatidylinositol Lipase. Biochemistry 2018, 57, 5759–5767. [Google Scholar] [CrossRef]
- Skarnes, W.C.; Rosen, B.; West, A.P.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Mujica, A.O.; Thomas, M.; Harrow, J.; Cox, T.; et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011, 474, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Scriver, C.R. ThePAH gene, phenylketonuria, and a paradigm shift. Hum. Mutat. 2007, 28, 831–845. [Google Scholar] [CrossRef]
- Fernández-Cañón, J.M.; Granadino, B.; De Bernabé, D.B.; Renedo, N.; Fernández-Ruiz, E. The molecular basis of alkaptonuria. Nat. Genet. 1996, 14, 19–24. [Google Scholar] [CrossRef]
- Mistry, J.B.; Bukhari, M.; Taylor, A.M. Alkaptonuria. Rare Dis. 2013, 1, e27475. [Google Scholar] [CrossRef] [Green Version]
- Blau, N. Genetics of Phenylketonuria: Then and Now. Hum. Mutat. 2016, 37, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Izumi, Y.; Takahashi, M.; Paxton, T.; Tamura, S.; Koike, T.; Yu, Y.; Kato, N.; Nagase, K.; Shiomi, M.; et al. Widely-targeted quantitative lipidomics method by supercritical fluid chromatography triple quadrupole mass spectrometry. J. Lipid Res. 2018, 59, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- McDermott, M.; Wang, Y.; Wakelam, M.; Bankaitis, V. Mammalian phospholipase D: Function, and therapeutics. Prog. Lipid Res. 2020, 78, 101018. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Kumazawa, T.; Koga, H.; Suzuki, N.; Oka, S.; Sugiura, T. Generation of lysophosphatidylinositol by DDHD domain containing 1 (DDHD1): Possible involvement of phospholipase D/phosphatidic acid in the activation of DDHD1. Biochim. Biophys. Acta 2010, 1801, 711–720. [Google Scholar] [CrossRef]
- Zachowski, A. Phospholipids in animal eukaryotic membranes: Transverse asymmetry and movement. Biochem. J. 1993, 294, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, M.; Maekawa, K.; Saito, K.; Senoo, Y.; Urata, M.; Murayama, M.; Tajima, Y.; Kumagai, Y.; Saito, Y. Plasma and Serum Lipidomics of Healthy White Adults Shows Characteristic Profiles by Subjects’ Gender and Age. PLoS ONE 2014, 9, e91806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Shen, Z.; Wiper, D.W.; Wu, M.; Morton, R.E.; Elson, P.; Kennedy, A.W.; Belinson, J.; Markman, M.; Casey, G. Lysophosphatidic Acid as a Potential Biomarker for Ovarian and Other Gynecologic Cancers. JAMA 1998, 280, 719–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedláková, I.; Vávrová, J.; Tošner, J.; Hanousek, L. Lysophosphatidic acid (LPA)—A perspective marker in ovarian cancer. Tumor Biol. 2010, 32, 311–316. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Zhang, W.-C.; Zhang, J.-L.; Zheng, C.-J.; Zhu, H.; Yu, H.-M.; Fan, L.-M. Plasma levels of lysophosphatidic acid in ovarian cancer versus controls: A meta-analysis. Lipids Health Dis. 2015, 14, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, J.; Taira, A.; Takanezawa, Y.; Kishi, Y.; Hama, K.; Kishimoto, T.; Mizuno, K.; Saku, K.; Taguchi, R.; Arai, H. Serum Lysophosphatidic Acid Is Produced through Diverse Phospholipase Pathways. J. Biol. Chem. 2002, 277, 48737–48744. [Google Scholar] [CrossRef] [Green Version]
- Ninou, I.; Magkrioti, C.; Aidinis, V. Autotaxin in Pathophysiology and Pulmonary Fibrosis. Front. Med. 2018, 5, 180. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, N.; Ikeda, H.; Nakamura, K.; Ohkawa, R.; Kume, Y.; Aoki, J.; Hama, K.; Okudaira, S.; Tanaka, M.; Tomiya, T.; et al. Both Plasma Lysophosphatidic Acid and Serum Autotaxin Levels are Increased in Chronic Hepatitis C. J. Clin. Gastroenterol. 2007, 41, 616–623. [Google Scholar] [CrossRef]
- Magkrioti, C.; Galaris, A.; Kanellopoulou, P.; Stylianaki, E.-A.; Kaffe, E.; Aidinis, V. Autotaxin and chronic inflammatory diseases. J. Autoimmun. 2019, 104, 102327. [Google Scholar] [CrossRef]
- Liu, S.; Murph, M.; Panupinthu, N.; Mills, G.B. ATX-LPA receptor axis in inflammation and cancer. Cell Cycle 2009, 8, 3695–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, H.; Baba, T.; Sato, S.; Ohtsuki, R.; Takemori, A.; Watanabe, T.; Tagaya, M.; Tani, K. Roles of SAM and DDHD domains in mammalian intracellular phospholipase A1 KIAA0725p. Biochim. Biophys. Acta 2012, 1823, 930–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuurs-Hoeijmakers, J.H.; Geraghty, M.T.; Kamsteeg, E.-J.; Ben-Salem, S.; de Bot, S.T.; Nijhof, B.; van de Vondervoort, I.I.; van der Graaf, M.; Nobau, A.C.; Otte-Höller, I.; et al. Mutations in DDHD2, Encoding an Intracellular Phospholipase A1, Cause a Recessive Form of Complex Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 2012, 91, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Doi, H.; Ushiyama, M.; Baba, T.; Tani, K.; Shiina, M.; Ogata, K.; Miyatake, S.; Fukuda-Yuzawa, Y.; Tsuji, S.; Nakashima, M.; et al. Late-onset spastic ataxia phenotype in a patient with a homozygous DDHD2 mutation. Sci. Rep. 2014, 4, 7132. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Phenotypes |
|---|---|
| Strain 1 (Baba, 2014 [17]) | ・Shortened mitochondrial sheath in sperm ・Impaired sperm motility |
| Strain 2 (Inloes, 2018 [24]) | ・Accumulation of PI 18:1/20:4 ・Decrease of LPI 20:4 |
| Strain 3 (Morikawa, 2021 [23]) | ・Gait disturbance-like symptpms ・Decrease of LPI 20:4 (sn-2) in cerebra ・Axonal decrease in the pyramidal tract |
| PIs | Retention Time (min) | Ddhd1 (+/+) | Ddhd1 (−/−) | p-Value | q-Value | |||
|---|---|---|---|---|---|---|---|---|
| Average | ±SD | Average | ±SD | |||||
| PI 18:1/20:4 | 17.19 ± 0 | 0.645 | 0.131 | 1.495 | 0.271 | 0.0001 | 0.006 | * |
| PI 20:1/20:4 | 17.08 ± 0.01 | 0.0141 | 0.005 | 0.0410 | 0.011 | 0.0005 | 0.017 | * |
| PI 18:1/20:3 | 17.13 ± 0 | 0.153 | 0.043 | 0.435 | 0.129 | 0.0009 | 0.020 | * |
| PI 18:1/22:5 | 17.2 ± 0.01 | 0.0258 | 0.013 | 0.0782 | 0.027 | 0.0029 | 0.046 | * |
| PI 19:1/20:4 | 17.13 ± 0.01 | 0.00777 | 0.003 | 0.0239 | 0.009 | 0.0034 | 0.046 | * |
| PI 16:0/20:1 | 16.94 ± 0.01 | 0.0141 | 0.006 | 0.0367 | 0.012 | 0.0041 | 0.046 | * |
| PI 15:0/24:0 | 17.28 ± 0 | 0.0524 | 0.030 | 0.1668 | 0.062 | 0.0041 | 0.046 | * |
| PI 18:1/18:1 | 17.02 ± 0 | 0.185 | 0.078 | 0.370 | 0.085 | 0.0052 | 0.046 | * |
| PI 16:0/18:1 | 17.03 ± 0 | 0.214 | 0.065 | 0.437 | 0.126 | 0.0057 | 0.046 | * |
| PI 18:1/22:6 | 17.28 ± 0 | 0.0846 | 0.049 | 0.238 | 0.092 | 0.0079 | 0.051 | |
| PI 16:0/18:2 | 17.15 ± 0 | 0.829 | 0.219 | 1.61 | 0.493 | 0.0088 | 0.051 | |
| PI 18:2/20:1 | 17.03 ± 0.01 | 0.0131 | 0.006 | 0.0284 | 0.009 | 0.010 | 0.053 | |
| PI 16:0/18:0 | 17.01 ± 0.02 | 0.0196 | 0.006 | 0.0375 | 0.011 | 0.011 | 0.055 | |
| PI 17:0/20:3 | 17.09 ± 0.02 | 0.0205 | 0.006 | 0.0343 | 0.008 | 0.012 | 0.055 | |
| PI 18:1/20:5 | 17.3 ± 0 | 0.201 | 0.094 | 0.397 | 0.111 | 0.013 | 0.055 | |
| PI 18:1/18:2 | 17.14 ± 0 | 0.376 | 0.150 | 0.751 | 0.239 | 0.014 | 0.056 | |
| PI 16:0/16:0 | 17.08 ± 0.01 | 0.0194 | 0.007 | 0.0296 | 0.004 | 0.016 | 0.061 | |
| PI 16:0/20:4 | 17.2 ± 0 | 1.20 | 0.233 | 2.25 | 0.788 | 0.017 | 0.061 | |
| PI 20:1/22:6 | 16.23 ± 3.93 | 0.00158 | 0.001 | 0.00571 | 0.003 | 0.021 | 0.069 | |
| PI 16:0/22:6 | 17.28 ± 0 | 0.308 | 0.164 | 0.749 | 0.325 | 0.022 | 0.070 | |
| PI 15:0/22:5 | 17.24 ± 0 | 0.0243 | 0.007 | 0.0471 | 0.017 | 0.022 | 0.070 | |
| PI 18:1/18:3 | 17.23 ± 0.01 | 0.00816 | 0.006 | 0.0187 | 0.007 | 0.023 | 0.070 | |
| PI 16:0/20:3 | 17.13 ± 0 | 0.318 | 0.098 | 0.655 | 0.269 | 0.025 | 0.070 | |
| PI 17:0/18:2 | 17.1 ± 0.01 | 0.0327 | 0.010 | 0.0580 | 0.020 | 0.029 | 0.079 | |
| PI 16:1/18:2 | 17.26 ± 0.01 | 0.0228 | 0.018 | 0.0557 | 0.023 | 0.032 | 0.081 | |
| PI 16:0/22:5 | 17.2 ± 0 | 0.0888 | 0.048 | 0.227 | 0.115 | 0.032 | 0.081 | |
| PI 16:1/20:4 | 17.31 ± 0.01 | 0.0105 | 0.006 | 0.0338 | 0.020 | 0.035 | 0.082 | |
| PI 18:0/18:0 | 16.92 ± 0.01 | 0.0272 | 0.007 | 0.0419 | 0.012 | 0.037 | 0.084 | |
| PI 18:0/20:4 | 17.07 ± 0 | 5.60 | 0.756 | 7.11 | 1.174 | 0.037 | 0.084 | |
| PI 18:0/20:2 | 16.99 ± 0.01 | 0.0975 | 0.028 | 0.1571 | 0.048 | 0.037 | 0.084 | |
| PI 18:0/18:1 | 16.93 ± 0 | 0.226 | 0.061 | 0.337 | 0.084 | 0.037 | 0.084 | |
| PI 18:0/20:3 | 17.01 ± 0.01 | 1.32 | 0.350 | 1.91 | 0.434 | 0.041 | 0.084 | |
| PI 15:0/22:2 | 16.98 ± 0 | 0.0265 | 0.010 | 0.0469 | 0.017 | 0.042 | 0.084 | |
| PI 18:2/23:0 | 17.16 ± 0.02 | 0.00120 | 0.001 | 0.00268 | 0.001 | 0.042 | 0.084 | |
| PI 18:0/20:1 | 16.84 ± 0.02 | 0.0156 | 0.006 | 0.0279 | 0.011 | 0.046 | 0.084 | |
| PI 18:2/20:4 | 17.29 ± 0.02 | 0.0274 | 0.014 | 0.0543 | 0.023 | 0.048 | 0.086 | |
| PI 18:0/18:2 | 17.02 ± 0 | 1.09 | 0.265 | 1.55 | 0.383 | 0.050 | 0.086 | |
| PI 16:0/18:3 | 17.24 ± 0.01 | 0.0168 | 0.007 | 0.0341 | 0.017 | 0.058 | 0.097 | |
| PI 18:0/18:3 | 17.12 ± 0.01 | 0.0167 | 0.007 | 0.0265 | 0.007 | 0.059 | 0.097 | |
| PI 18:2/25:0 | 17.06 ± 0.05 | 0.00225 | 0.001 | 0.00328 | 0.001 | 0.059 | 0.097 | |
| PI 19:0/20:4 | 17 ± 0 | 0.0809 | 0.029 | 0.131 | 0.046 | 0.064 | 0.10 | |
| PI 16:1/18:1 | 17.15 ± 0 | 0.0182 | 0.012 | 0.0393 | 0.019 | 0.066 | 0.10 | |
| PI 16:0/20:5 | 17.31 ± 0 | 0.389 | 0.170 | 0.764 | 0.370 | 0.067 | 0.10 | |
| PI 16:0/16:1 | 17.16 ± 0.01 | 0.0198 | 0.011 | 0.0389 | 0.018 | 0.075 | 0.11 | |
| PI 22:1/22:6 | 16.32 ± 3.95 | 0.000925 | 0.001 | 0.00280 | 0.002 | 0.078 | 0.11 | |
| PI 19:0/22:6 | 17.12 ± 0.02 | 0.00492 | 0.003 | 0.0095 | 0.005 | 0.080 | 0.11 | |
| PI 18:0/22:6 | 17.16 ± 0 | 0.712 | 0.286 | 1.122 | 0.377 | 0.081 | 0.11 | |
| PI 18:2/19:0 | 16.97 ± 0 | 0.0180 | 0.009 | 0.0323 | 0.014 | 0.090 | 0.12 | |
| PI 18:2/18:2 | 17.25 ± 0 | 0.0799 | 0.051 | 0.1649 | 0.088 | 0.091 | 0.12 | |
| PI 15:0/22:0 | 17.41 ± 0.02 | 0.0116 | 0.010 | 0.0232 | 0.011 | 0.11 | 0.14 | |
| PI 17:0/20:4 | 17.13 ± 0 | 0.0969 | 0.014 | 0.1305 | 0.041 | 0.12 | 0.14 | |
| PI 17:0/18:1 | 16.98 ± 0.01 | 0.0092 | 0.003 | 0.0149 | 0.007 | 0.12 | 0.15 | |
| PI 20:0/20:4 | 16.96 ± 0.01 | 0.0302 | 0.023 | 0.0568 | 0.027 | 0.12 | 0.15 | |
| PI 15:0/22:1 | 17 ± 0.22 | 0.00741 | 0.004 | 0.0126 | 0.006 | 0.13 | 0.15 | |
| PI 18:0/22:4 | 16.97 ± 0 | 0.0325 | 0.011 | 0.0478 | 0.017 | 0.13 | 0.15 | |
| PI 19:0/20:3 | 16.96 ± 0.01 | 0.0154 | 0.009 | 0.0268 | 0.013 | 0.13 | 0.15 | |
| PI 18:0/22:5 | 17.09 ± 0.01 | 0.181 | 0.083 | 0.278 | 0.103 | 0.13 | 0.15 | |
| PI 15:0/24:1 | 17.37 ± 0.02 | 0.0067 | 0.006 | 0.0140 | 0.009 | 0.16 | 0.18 | |
| PI 16:1/18:0 | 17.02 ± 0.01 | 0.0175 | 0.007 | 0.0251 | 0.008 | 0.16 | 0.18 | |
| PI 18:0/20:5 | 17.18 ± 0 | 1.10 | 0.229 | 1.33 | 0.324 | 0.22 | 0.23 | |
| PI 17:0/20:5 | 17.25 ± 0 | 0.0259 | 0.011 | 0.0365 | 0.015 | 0.23 | 0.24 | |
| PI 20:4/22:1 | 17.21 ± 0.01 | 0.00481 | 0.002 | 0.0065 | 0.003 | 0.28 | 0.29 | |
| PI 20:0/22:6 | 14.22 ± 6.35 | 0.00153 | 0.002 | 0.00233 | 0.001 | 0.46 | 0.47 | |
| PI 20:5/24:1 | 17.19 ± 0.03 | 0.00325 | 0.001 | 0.00305 | 0.002 | 0.84 | 0.84 | |
| LPAs | Retention Time | Ddhd1 (+/+) | Ddhd1 (−/−) | p-Value | q-Value | |||
|---|---|---|---|---|---|---|---|---|
| (min) | Average | SD | Average | SD | ||||
| LPA 24:0 | 22.89 ± 0.04 | 1.17 | 0.47 | 3.12 | 0.45 | 0.0001 | 0.0004 | * |
| LPA 18:0 | 22.89 ± 0.03 | 0.329 | 0.04 | 0.437 | 0.07 | 0.015 | 0.061 | |
| LPA 22:6 | 22.89 ± 0.13 | 1.48 | 0.18 | 1.38 | 0.10 | 0.29 | 0.78 | |
| LPA 22:5 | 22.84 ± 0.06 | 0.905 | 0.10 | 0.879 | 0.04 | 0.61 | >0.99 | |
| LPA 20:2 | 22.89 ± 0.02 | 0.275 | 0.07 | 0.267 | 0.04 | 0.83 | >0.99 | |
| LPA 20:4 | 22.32 ± 0.06 | 0.0825 | 0.02 | 0.0786 | 0.03 | 0.84 | >0.99 | |
| LPA 20:3 | 22.89 ± 0.03 | 0.778 | 0.18 | 0.796 | 0.11 | 0.86 | >0.99 | |
| LPA 18:2 | 22.89 ± 0.03 | 0.546 | 0.24 | 0.531 | 0.10 | 0.90 | >0.99 | |
| PIs | Retention Time (min) | SPG28 | Control 1 | Control 2 | Control 3 | Control 4 | Control 5 | Rank of SPG28 among All Samples | Average of Conrols | SD of Conrols | 95% CI | SPG28 out of 95% CI? | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PI 18:1/20:4 * | 17.20 ± 0.01 | 3.33 | 2.60 | 1.58 | 1.62 | 2.33 | 0.797 | 1 | 1.79 | 0.634 | 0.556 | (1.23–2.34) | Yes |
| PI 20:1/20:4 | 17.10 ± 0.02 | 0.0963 | 0.0534 | 0.0549 | 0.0684 | 0.1323 | 0.0212 | 2 | 0.0660 | 0.037 | 0.0321 | (0.0340–0.0981) | No |
| PI 18:1/20:3 * | 17.17 ± 0.01 | 0.589 | 0.489 | 0.253 | 0.257 | 0.353 | 0.123 | 1 | 0.295 | 0.121 | 0.106 | (0.189–0.401) | Yes |
| PI 18:1/22:5 * | 17.21 ± 0.01 | 0.143 | 0.134 | 0.0928 | 0.0730 | 0.0892 | 0.0325 | 1 | 0.0843 | 0.0327 | 0.0287 | (0.0556–0.113) | Yes |
| PI 16:0/20:1 | 16.97 ± 0.01 | 0.0971 | 0.0661 | 0.0586 | 0.1023 | 0.1261 | 0.0178 | 3 | 0.0742 | 0.0374 | 0.0328 | (0.0414–0.107) | No |
| PI 18:1/18:1 * | 17.04 ± 0.01 | 2.85 | 2.27 | 1.83 | 2.33 | 1.82 | 0.665 | 1 | 1.78 | 0.598 | 0.524 | (1.26–2.31) | Yes |
| PI 16:0/18:1 | 17.06 ± 0.01 | 6.08 | 6.40 | 5.28 | 7.25 | 4.59 | 1.85 | 3 | 5.07 | 1.85 | 1.62 | (3.45–6.70) | No |
| PIs | Retention Time (min) | SPG28 | Control 1 | Control 2 | Control 3 | Control 4 | Control 5 | Rank of SPG28 among All Samples | Average of Conrols | ±SD of Conrols | 95% CI | SPG28 out of 95% CI? | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PI 16:1/18:1 | 17.17 ± 0.01 | 0.374 | 0.362 | 0.306 | 0.252 | 0.415 | 0.0819 | 2 | 0.283 | 0.114 | 0.100 | (0.183–0.384) | No |
| PI 16:0/18:1 | 17.06 ± 0.01 | 6.08 | 6.40 | 5.28 | 7.25 | 4.59 | 1.85 | 3 | 5.07 | 1.85 | 1.62 | (3.45–6.70) | No |
| PI 17:0/18:1 | 17.00 ± 0.01 | 0.288 | 0.316 | 0.179 | 0.212 | 0.211 | 0.0951 | 2 | 0.203 | 0.0710 | 0.0622 | (0.141–0.265) | No |
| PI 18:3/18:1 | 17.28 ± 0.02 | 0.125 | 0.0620 | 0.0826 | 0.0281 | 0.0316 | 0.0172 | 1 | 0.0443 | 0.0243 | 0.0213 | (0.0230–0.0656) | Yes |
| PI 18:1/18:2 | 17.17 ± 0.01 | 3.25 | 1.93 | 2.33 | 1.59 | 2.19 | 0.803 | 1 | 1.77 | 0.544 | 0.477 | (1.29–2.24) | Yes |
| PI 18:1/18:1 | 17.04 ± 0.01 | 2.85 | 2.27 | 1.83 | 2.33 | 1.82 | 0.665 | 1 | 1.78 | 0.598 | 0.524 | (1.26–2.31) | Yes |
| PI 18:0/18:1 | 16.95 ± 0.01 | 11.2 | 9.47 | 10.0 | 9.28 | 6.50 | 3.17 | 1 | 7.68 | 2.57 | 2.25 | (5.44–9.93) | Yes |
| PI 18:1/20:5 | 17.32 ± 0.02 | 0.107 | 0.0714 | 0.0916 | 0.0444 | 0.0362 | 0.0200 | 1 | 0.0527 | 0.0256 | 0.0224 | (0.0303–0.0751) | Yes |
| PI 18:1/20:4 | 17.20 ± 0.01 | 3.33 | 2.60 | 1.58 | 1.62 | 2.33 | 0.797 | 1 | 1.79 | 0.634 | 0.556 | (1.23–2.34) | Yes |
| PI 18:1/20:3 | 17.17 ± 0.01 | 0.589 | 0.489 | 0.253 | 0.257 | 0.353 | 0.123 | 1 | 0.295 | 0.121 | 0.106 | (0.189–0.401) | Yes |
| PI 18:1/22:6 | 17.30 ± 0.01 | 0.250 | 0.138 | 0.132 | 0.161 | 0.156 | 0.0502 | 1 | 0.127 | 0.0400 | 0.0351 | (0.0922–0.162) | Yes |
| PI 18:1/22:5 | 17.21 ± 0.01 | 0.143 | 0.134 | 0.0928 | 0.0730 | 0.0892 | 0.0325 | 1 | 0.0843 | 0.0327 | 0.0287 | (0.0556–0.113) | Yes |
| Total Oleic acid-containing PI | 28.6 | 24.3 | 22.2 | 23.1 | 18.7 | 7.70 | 1 | 19.2 | 6.03 | 5.29 | (13.9–24.5) | Yes | |
| Total PI | 134 | 138 | 142 | 122 | 126 | 67.0 | 3 | 119 | 27.1 | 23.7 | (95.2–143) | No | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morikawa, T.; Takahashi, M.; Izumi, Y.; Bamba, T.; Moriyama, K.; Hattori, G.; Fujioka, R.; Miura, S.; Shibata, H. Oleic Acid-Containing Phosphatidylinositol Is a Blood Biomarker Candidate for SPG28. Biomedicines 2023, 11, 1092. https://doi.org/10.3390/biomedicines11041092
Morikawa T, Takahashi M, Izumi Y, Bamba T, Moriyama K, Hattori G, Fujioka R, Miura S, Shibata H. Oleic Acid-Containing Phosphatidylinositol Is a Blood Biomarker Candidate for SPG28. Biomedicines. 2023; 11(4):1092. https://doi.org/10.3390/biomedicines11041092
Chicago/Turabian StyleMorikawa, Takuya, Masatomo Takahashi, Yoshihiro Izumi, Takeshi Bamba, Kosei Moriyama, Gohsuke Hattori, Ryuta Fujioka, Shiroh Miura, and Hiroki Shibata. 2023. "Oleic Acid-Containing Phosphatidylinositol Is a Blood Biomarker Candidate for SPG28" Biomedicines 11, no. 4: 1092. https://doi.org/10.3390/biomedicines11041092
APA StyleMorikawa, T., Takahashi, M., Izumi, Y., Bamba, T., Moriyama, K., Hattori, G., Fujioka, R., Miura, S., & Shibata, H. (2023). Oleic Acid-Containing Phosphatidylinositol Is a Blood Biomarker Candidate for SPG28. Biomedicines, 11(4), 1092. https://doi.org/10.3390/biomedicines11041092