
Genes and Microbiota Interaction in Monogenic Autoimmune Disorders

 , ,
, ,  ,
,  , , and
, , and
Abstract
:1. Introduction
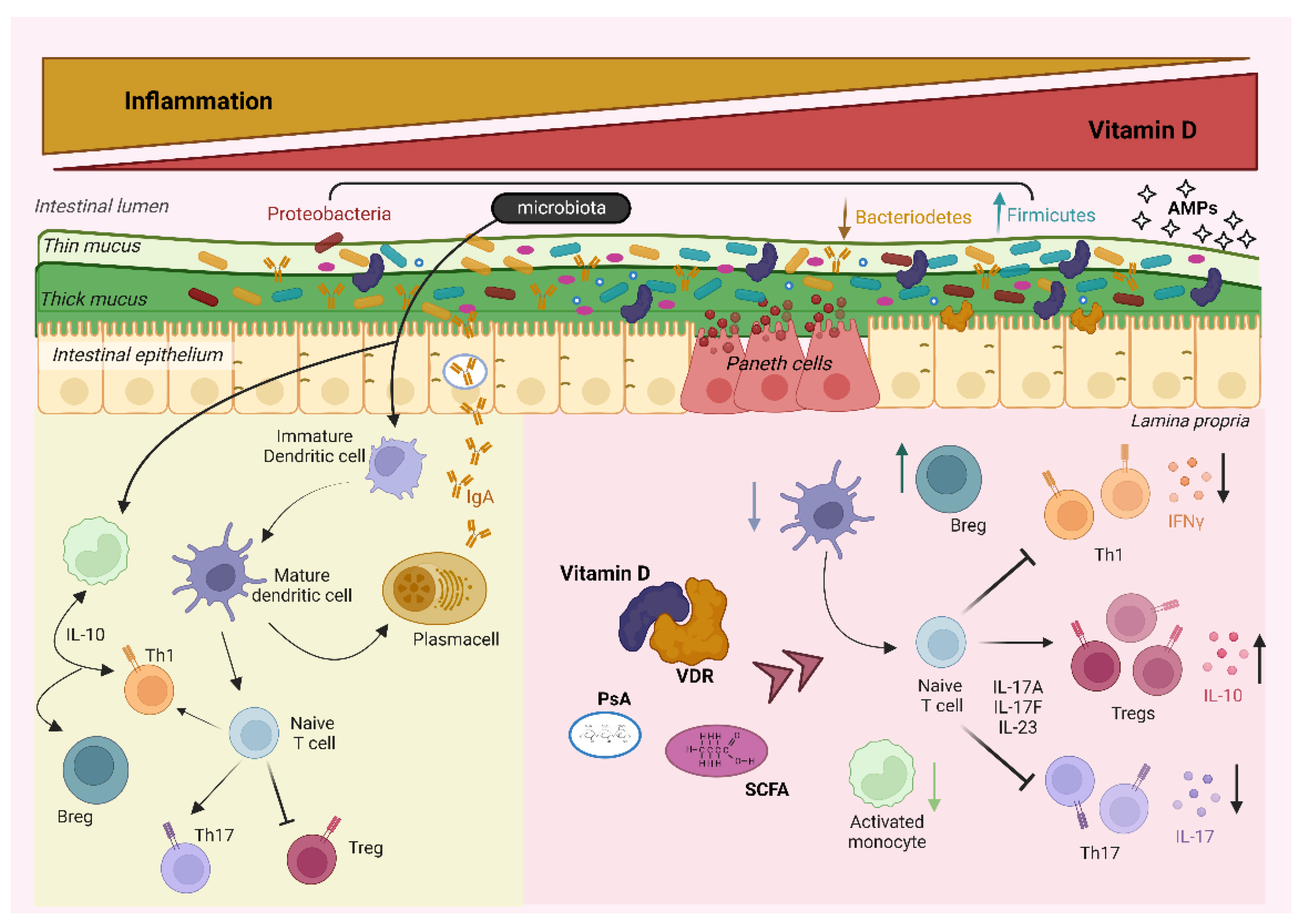
2. Microbiota and the Immune System
2.1. The Close Relationship between Microbiota and Immunity
2.2. The Role of Vitamin D in the Interplay between Microbiota and the Immune System
3. Pathophysiology of Organ-Specific Monogenic Autoimmune Disorders and Microbiota Implication
3.1. Autoimmune Polyendocrine Syndrome Type 1 (APS1)
3.2. Immunodysregulation Polyendocrinopathy Enteropathy X-Linked (IPEX) Syndrome
4. Pathophysiology of Systemic Monogenic Autoimmune Disorders and Microbiota Implication
4.1. Autoimmune Lymphoproliferative Syndrome (ALPS)
4.2. Monogenic Forms of SLE
4.2.1. Genetic Alterations
The Complement System
PRKCD
TREX1
DNASE1L3
ACP5
4.2.2. Microbiota Alterations
4.3. The Role of Microbiota in Murine Models of Systemic Autoimmune Disorders
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Wang, F.-S.; Gershwin, M.E.; Wang, A.; Wang, L.; Me, G. Human Autoimmune Diseases: A Comprehensive Update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef] [PubMed]
- Christen, U.; Herrath, M.G.V. Initiation of Autoimmunity. Curr. Opin. Immunol. 2004, 16, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Marson, A.; Housley, W.J.; Hafler, D.A. Genetic Basis of Autoimmunity. J. Clin. Investig. 2015, 125, 2234–2241. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.B.; Hsiao, E.Y. Microbiomes as Sources of Emergent Host Phenotypes. Science 2019, 365, 1405–1409. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between Microbiota and Immunity in Health and Disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Marcobal, A.; Sonnenburg, J.L. Human Milk Oligosaccharide Consumption by Intestinal Microbiota. Clin. Microbiol. Infect. 2012, 18 (Suppl. 4), 12–15. [Google Scholar] [CrossRef] [Green Version]
- An, D.; Oh, S.F.; Olszak, T.; Neves, J.F.; Avci, F.Y.; Erturk-Hasdemir, D.; Lu, X.; Zeissig, S.; Blumberg, R.S.; Kasper, D.L. Sphingolipids from a Symbiotic Microbe Regulate Homeostasis of Host Intestinal Natural Killer T Cells. Cell 2014, 156, 123. [Google Scholar] [CrossRef] [Green Version]
- McGuckin, M.A.; Lindén, S.K.; Sutton, P.; Florin, T.H. Mucin Dynamics and Enteric Pathogens. Nat. Rev. Microbiol. 2011, 9, 265–278. [Google Scholar] [CrossRef]
- MacPherson, A.J.; Slack, E.; Geuking, M.B.; McCoy, K.D. The Mucosal Firewalls against Commensal Intestinal Microbes. Semin. Immunopathol. 2009, 31, 145–149. [Google Scholar] [CrossRef]
- Sun, C.M.; Hall, J.A.; Blank, R.B.; Bouladoux, N.; Oukka, M.; Mora, J.R.; Belkaid, Y. Small Intestine Lamina Propria Dendritic Cells Promote de Novo Generation of Foxp3 T Reg Cells via Retinoic Acid. J. Exp. Med. 2007, 204, 1775. [Google Scholar] [CrossRef] [Green Version]
- Chieppa, M.; Rescigno, M.; Huang, A.Y.C.; Germain, R.N. Dynamic Imaging of Dendritic Cell Extension into the Small Bowel Lumen in Response to Epithelial Cell TLR Engagement. J. Exp. Med. 2006, 203, 2841. [Google Scholar] [CrossRef] [Green Version]
- Round, J.L.; Lee, S.M.; Li, J.; Tran, G.; Jabri, B.; Chatila, T.A.; Mazmanian, S.K. The Toll-like Receptor Pathway Establishes Commensal Gut Colonization. Science 2011, 332, 974. [Google Scholar] [CrossRef] [Green Version]
- Ramanan, D.; Tang, M.S.; Bowcutt, R.; Loke, P.; Cadwell, K. Bacterial Sensor Nod2 Prevents Inflammation of the Small Intestine by Restricting the Expansion of the Commensal Bacteroides Vulgatus. Immunity 2014, 41, 311–324. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalová, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428. [Google Scholar] [CrossRef] [Green Version]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Di, Y.; Schilter, H.C.; Rolph, M.S.; MacKay, F.; Artis, D.; et al. Regulation of Inflammatory Responses by Gut Microbiota and Chemoattractant Receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [Green Version]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; Van Der Veeken, J.; Deroos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T-Cell Generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [Green Version]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity 2019, 51, 285–297.e5. [Google Scholar] [CrossRef]
- Franchi, L.; Kamada, N.; Nakamura, Y.; Burberry, A.; Kuffa, P.; Suzuki, S.; Shaw, M.H.; Kim, Y.G.; Núñez, G. NLRC4-Driven Production of IL-1β Discriminates between Pathogenic and Commensal Bacteria and Promotes Host Intestinal Defense. Nat. Immunol. 2012, 13, 449–456. [Google Scholar] [CrossRef]
- Ivanov, I.I.; de Llanos Frutos, R.; Manel, N.; Yoshinaga, K.; Rifkin, D.B.; Sartor, R.B.; Finlay, B.B.; Littman, D.R. Specific Microbiota Direct the Differentiation of IL-17-Producing T-Helper Cells in the Mucosa of the Small Intestine. Cell Host Microbe 2008, 4, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Gaboriau-Routhiau, V.; Rakotobe, S.; Lécuyer, E.; Mulder, I.; Lan, A.; Bridonneau, C.; Rochet, V.; Pisi, A.; De Paepe, M.; Brandi, G.; et al. The Key Role of Segmented Filamentous Bacteria in the Coordinated Maturation of Gut Helper T Cell Responses. Immunity 2009, 31, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Omenetti, S.; Bussi, C.; Metidji, A.; Iseppon, A.; Lee, S.; Tolaini, M.; Li, Y.; Kelly, G.; Chakravarty, P.; Shoaie, S.; et al. The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity 2019, 51, 77–89.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbitt, N.; Kimura, S.; Isse, K.; Specht, S.; Chedwick, L.; Rosborough, B.R.; Lunz, J.G.; Murase, N.; Yokota, S.; Demetris, A.J. Gut Bacteria Drive Kupffer Cell Expansion via MAMP-Mediated ICAM-1 Induction on Sinusoidal Endothelium and Influence Preservation-Reperfusion Injury after Orthotopic Liver Transplantation. Am. J. Pathol. 2013, 182, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharon, G.; Sampson, T.R.; Geschwind, D.H.; Mazmanian, S.K. The Central Nervous System and the Gut Microbiome. Cell 2016, 167, 915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhoury, H.M.A.; Kvietys, P.R.; AlKattan, W.; Al Anouti, F.; Elahi, M.A.; Karras, S.N.; Grant, W.B. Vitamin D and Intestinal Homeostasis: Barrier, Microbiota, and Immune Modulation. J. Steroid Biochem. Mol. Biol. 2020, 200, 105663. [Google Scholar] [CrossRef]
- Hiremath, G.S.; Cettomai, D.; Baynes, M.; Ratchford, J.N.; Newsome, S.; Harrison, D.; Kerr, D.; Greenberg, B.M.; Calabresi, P.A. Vitamin D Status and Effect of Low-Dose Cholecalciferol and High-Dose Ergocalciferol Supplementation in Multiple Sclerosis. Mult. Scler. 2009, 15, 735–740. [Google Scholar] [CrossRef]
- Bener, A.; Alsaied, A.; Al-Ali, M.; Al-Kubaisi, A.; Basha, B.; Abraham, A.; Guiter, G.; Mian, M. High Prevalence of Vitamin D Deficiency in Type 1 Diabetes Mellitus and Healthy Children. Acta Diabetol. 2009, 46, 183–189. [Google Scholar] [CrossRef]
- Amital, H.; Szekanecz, Z.; Szücs, G.; Dankó, K.; Nagy, E.; Csépány, T.; Kiss, E.; Rovensky, J.; Tuchynova, A.; Kozakova, D.; et al. Serum Concentrations of 25-OH Vitamin D in Patients with Systemic Lupus Erythematosus (SLE) Are Inversely Related to Disease Activity: Is It Time to Routinely Supplement Patients with SLE with Vitamin D? Ann. Rheum. Dis. 2010, 69, 1155–1157. [Google Scholar] [CrossRef]
- Singh, P.; Kumar, M.; Al Khodor, S. Vitamin D Deficiency in the Gulf Cooperation Council: Exploring the Triad of Genetic Predisposition, the Gut Microbiome and the Immune System. Front. Immunol. 2019, 10, 1042. [Google Scholar] [CrossRef]
- Chen, S.W.; Wang, P.Y.; Zhu, J.; Chen, G.W.; Zhang, J.L.; Chen, Z.Y.; Zuo, S.; Liu, Y.C.; Pan, Y.S. Protective Effect of 1,25-Dihydroxyvitamin D3 on Lipopolysaccharide-Induced Intestinal Epithelial Tight Junction Injury in Caco-2 Cell Monolayers. Inflammation 2015, 38, 375–383. [Google Scholar] [CrossRef]
- Li, J.; Chen, N.; Wang, D.; Zhang, J.; Gong, X. Efficacy of Vitamin D in Treatment of Inflammatory Bowel Disease A Meta-Analysis. Medicine 2018, 97, e12662. [Google Scholar] [CrossRef]
- Nurminen, V.; Neme, A.; Seuter, S.; Carlberg, C. The Impact of the Vitamin D-Modulated Epigenome on VDR Target Gene Regulation. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 697–705. [Google Scholar] [CrossRef]
- Mahto, H.; Tripathy, R.; Das, B.K.; Panda, A.K. Association between Vitamin D Receptor Polymorphisms and Systemic Lupus Erythematosus in an Indian Cohort. Int. J. Rheum. Dis. 2018, 21, 468–476. [Google Scholar] [CrossRef]
- Wang, J.; Thingholm, L.B.; Skiecevičienė, J.; Rausch, P.; Kummen, M.; Hov, J.R.; Degenhardt, F.; Heinsen, F.-A.; Rühlemann, M.C.; Szymczak, S.; et al. Genome-Wide Association Analysis Identifies Variation in Vitamin D Receptor and Other Host Factors Influencing the Gut Microbiota HHS Public Access Author Manuscript. Nat. Genet. 2016, 48, 1396–1406. [Google Scholar] [CrossRef]
- Yamamoto, E.; Jørgensen, T.N. Immunological Effects of Vitamin D and Their Relations to Autoimmunity. J. Autoimmun. 2019, 100, 7–16. [Google Scholar] [CrossRef]
- Malaguarnera, L. Vitamin D and Microbiota: Two Sides of the Same Coin in the Immunomodulatory Aspects. Int. Immunopharmacol. 2020, 79, 106112. [Google Scholar] [CrossRef]
- L Bishop, E.; Ismailova, A.; Dimeloe, S.; Hewison, M.; White, J.H. Vitamin D and Immune Regulation: Antibacterial, Antiviral, Anti-Inflammatory. JBMR Plus 2020, 5. [Google Scholar] [CrossRef]
- Yamamoto, E.A.; Jørgensen, T.N. Relationships Between Vitamin D, Gut Microbiome, and Systemic Autoimmunity. Front. Immunol. 2020, 10, 3141. [Google Scholar] [CrossRef]
- Perheentupa, J. APS-I/APECED: The Clinical Disease and Therapy. Endocrinol. Metab. Clin. N. Am. 2002, 31, 295–320. [Google Scholar] [CrossRef]
- Husebye, E.S.; Perheentupa, J.; Rautemaa, R.; Kämpe, O. Clinical Manifestations and Management of Patients with Autoimmune Polyendocrine Syndrome Type I. J. Intern. Med. 2009, 265, 514–529. [Google Scholar] [CrossRef]
- Aaltonen, J.; Björses, P.; Perheentupa, J.; Horelli-Kuitunen, N.; Palotie, A.; Peltonen, L.; Lee Su, Y.; Francis, F.; Hennig, S.; Thiel, C.; et al. An Autoimmune Disease, APECED, Caused by Mutations in a Novel Gene Featuring Two PHD-Type Zinc-Finger Domains. Nat. Genet. 1997, 17, 399–403. [Google Scholar] [CrossRef]
- Nagamine, K.; Peterson, P.; Scott, H.S.; Kudoh, J.; Minoshima, S.; Heino, M.; Krohn, K.J.E.; Lalioti, M.D.; Mullis, P.E.; Antonarakis, S.E.; et al. Positional Cloning of the APECED Gene. Nat. Genet. 1997, 17, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Halonen, M.; Kangas, H.; Rüppell, T.; Ilmarinen, T.; Ollila, J.; Kolmer, M.; Vihinen, M.; Palvimo, J.; Saarela, J.; Ulmanen, I.; et al. APECED-Causing Mutations in AIRE Reveal the Functional Domains of the Protein. Hum. Mutat. 2004, 23, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Derbinski, J.; Schulte, A.; Kyewski, B.; Klein, L. Promiscuous Gene Expression in Medullary Thymic Epithelial Cells Mirrors the Peripheral Self. Nat. Immunol. 2001, 2, 2915–2922. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.S.; Venanzi, E.S.; Klein, L.; Chen, Z.; Berzins, S.P.; Turley, S.J.; Von Boehmer, H.; Bronson, R.; Dierich, A.; Benoist, C.; et al. Projection of an Immunological Self Shadow within the Thymus by the Aire Protein. Science 2002, 298, 1395–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Q.G.; She, J.X. Autoimmune Polyglandular Syndrome Type 1 and the Autoimmune Regulator. Clin. Lab. Med. 2004, 24, 305–317. [Google Scholar] [CrossRef]
- Cetani, F.; Barbesino, G.; Borsari, S.; Pardi, E.; Cianferotti, L.; Pinchera, A.; Marcocci, C. A Novel Mutation of the Autoimmune Regulator Gene in an Italian Kindred with Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy, Acting in a Dominant Fashion and Strongly Cosegregating with Hypothyroid Autoimmune Thyroiditis. J. Clin. Endocrinol. Metab. 2001, 86, 4747–4752. [Google Scholar] [CrossRef]
- Cervato, S.; Mariniello, B.; Lazzarotto, F.; Morlin, L.; Zanchetta, R.; Radetti, G.; De Luca, F.; Valenzise, M.; Giordano, R.; Rizzo, D.; et al. Evaluation of the Autoimmune Regulator (AIRE) Gene Mutations in a Cohort of Italian Patients with Autoimmune-Polyendocrinopathy-Candidiasis-Ectodermal-Dystrophy (APECED) and in Their Relatives. Clin. Endocrinol. 2009, 70, 421–428. [Google Scholar] [CrossRef]
- Kogawa, K.; Kudoh, J.; Nagafuchi, S.; Ohga, S.; Katsuta, H.; Ishibashi, H.; Harada, M.; Hara, T.; Shimizu, N. Distinct Clinical Phenotype and Immunoreactivity in Japanese Siblings with Autoimmune Polyglandular Syndrome Type 1 (APS-1) Associated with Compound Heterozygous Novel AIRE Gene Mutations. Clin. Immunol. 2002, 103, 277–283. [Google Scholar] [CrossRef]
- Laakso, S.M.; Laurinolli, T.T.; Rossi, L.H.; Lehtoviita, A.; Sairanen, H.; Perheentupa, J.; Kekäläinen, E.; Arstila, T.P. Regulatory T Cell Defect in APECED Patients Is Associated with Loss of Naive FOXP3+ Precursors and Impaired Activated Population. J. Autoimmun. 2010, 35, 351–357. [Google Scholar] [CrossRef]
- Pöntynen, N.; Strengell, M.; Sillanpää, N.; Saharinen, J.; Ulmanen, I.; Julkunen, I.; Peltonen, L. Critical Immunological Pathways Are Downregulated in APECED Patient Dendritic Cells. J. Mol. Med. 2008, 86, 1139–1152. [Google Scholar] [CrossRef] [Green Version]
- Gebre-Medhin, G.; Husebye, E.S.; Gustafsson, J.; Winqvist, O.; Goksøyr, A.; Rorsman, F.; Kämpe, O. Cytochrome P450IA2 and Aromatic L-Amino Acid Decarboxylase Are Hepatic Autoantigens in Autoimmune Polyendocrine Syndrome Type I. FEBS Lett. 1997, 412, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Meloni, A.; Furcas, M.; Cetani, F.; Marcocci, C.; Falorni, A.; Perniola, R.; Pura, M.; Bøe Wolff, A.S.; Husebye, E.S.; Lilic, D.; et al. Autoantibodies against Type I Interferons as an Additional Diagnostic Criterion for Autoimmune Polyendocrine Syndrome Type I. J. Clin. Endocrinol. Metab. 2008, 93, 4389–4397. [Google Scholar] [CrossRef] [Green Version]
- Alimohammadi, M.; Björklund, P.; Hallgren, Å.; Pöntynen, N.; Szinnai, G.; Shikama, N.; Keller, M.P.; Ekwall, O.; Kinkel, S.A.; Husebye, E.S.; et al. Autoimmune Polyendocrine Syndrome Type 1 and NALP5, a Parathyroid Autoantigen. N. Engl. J. Med. 2008, 358, 1018–1028. [Google Scholar] [CrossRef] [Green Version]
- Puel, A.; Döffinger, R.; Natividad, A.; Chrabieh, M.; Barcenas-Morales, G.; Picard, C.; Cobat, A.; Ouachée-Chardin, M.; Toulon, A.; Bustamante, J.; et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in Patients with Chronic Mucocutaneous Candidiasis and Autoimmune Polyendocrine Syndrome Type I. J. Exp. Med. 2010, 207, 291–297. [Google Scholar] [CrossRef] [Green Version]
- de Albuquerque, J.A.T.; Banerjee, P.P.; Castold, A.; Ma, R.; Zurro, N.B.; Ynoue, L.H.; Arslanian, C.; Barbosa-Carvalho, M.U.W.; de Menezes Correia-Deur, J.E.; Weiler, F.G.; et al. The Role of AIRE in the Immunity Against Candida Albicans in a Model of Human Macrophages. Front. Immunol. 2018, 9, 567. [Google Scholar] [CrossRef] [Green Version]
- Kluger, N.; Jokinen, M.; Krohn, K.; Ranki, A. Gastrointestinal Manifestations in APECED Syndrome. J. Clin. Gastroenterol. 2013, 47, 112–120. [Google Scholar] [CrossRef]
- Kluger, N.; Jokinen, M.; Lintulahti, A.; Krohn, K.; Ranki, A. Gastrointestinal Immunity against Tryptophan Hydroxylase-1, Aromatic L-Amino-Acid Decarboxylase, AIE-75, Villin and Paneth Cells in APECED. Clin. Immunol. 2015, 158, 212–220. [Google Scholar] [CrossRef]
- Dobeš, J.; Neuwirth, A.; Dobešová, M.; Vobořil, M.; Balounová, J.; Ballek, O.; Lebl, J.; Meloni, A.; Krohn, K.; Kluger, N.; et al. Gastrointestinal Autoimmunity Associated with Loss of Central Tolerance to Enteric α-Defensins. Gastroenterology 2015, 149, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Salzman, N.H.; Hung, K.; Haribhai, D.; Chu, H.; Karlsson-Sjöberg, J.; Amir, E.; Teggatz, P.; Barman, M.; Hayward, M.; Eastwood, D.; et al. Enteric Defensins Are Essential Regulators of Intestinal Microbial Ecology. Nat. Immunol. 2009, 11, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.; Jokinen, M.; Plichta, D.R.; Liebisch, G.; Gronwald, W.; Dettmer, K.; Oefner, P.J.; Vlamakis, H.; Chung, D.C.; Ranki, A.; et al. Cytokine-Specific Autoantibodies Shape the Gut Microbiome in Autoimmune Polyendocrine Syndrome Type 1. J. Allergy Clin. Immunol. 2021, 148, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Hetemäki, I.; Jarva, H.; Kluger, N.; Baldauf, H.-M.; Laakso, S.; Bratland, E.; Husebye, E.S.; Kisand, K.; Ranki, A.; Peterson, P.; et al. Anticommensal Responses Are Associated with Regulatory T Cell Defect in Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy Patients. J. Immunol. 2016, 196, 2955–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Li, C.; Zhao, X.; Lv, C.; He, Q.; Lei, S.; Guo, Y.; Zhi, F. Anti-Saccharomyces Cerevisiae Antibodies Associate with Phenotypes and Higher Risk for Surgery in Crohn’s Disease: A Meta-Analysis. Dig. Dis. Sci. 2012, 57, 2944–2954. [Google Scholar] [CrossRef]
- Kawamoto, S.; Maruya, M.; Kato, L.M.; Suda, W.; Atarashi, K.; Doi, Y.; Tsutsui, Y.; Qin, H.; Honda, K.; Okada, T.; et al. Foxp3(+) T Cells Regulate Immunoglobulin a Selection and Facilitate Diversification of Bacterial Species Responsible for Immune Homeostasis. Immunity 2014, 41, 152–165. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Hoang, T.K.; Wang, T.; Ferris, M.; Taylor, C.M.; Tian, X.; Luo, M.; Tran, D.Q.; Zhou, J.; Tatevian, N.; et al. Resetting Microbiota by Lactobacillus Reuteri Inhibits T Reg Deficiency-Induced Autoimmunity via Adenosine A2A Receptors. J. Exp. Med. 2017, 214, 107–123. [Google Scholar] [CrossRef]
- Winer, K.K.; Schmitt, M.M.; Ferre, E.M.N.; Fennelly, K.P.; Olivier, K.N.; Heller, T.; Lionakis, M.S. Impact of Periprocedural Subcutaneous Parathyroid Hormone on Control of Hypocalcaemia in APS-1/APECED Patients Undergoing Invasive Procedures. Clin. Endocrinol. 2021, 94, 377–383. [Google Scholar] [CrossRef]
- Halabi, I.; Barohom, M.N.; Peleg, S.; Trougouboff, P.; Elias-Assad, G.; Agbaria, R.; Tenenbaum-Rakover, Y. Case Report: Severe Hypocalcemic Episodes Due to Autoimmune Enteropathy. Front. Endocrinol. 2021, 12, 674. [Google Scholar] [CrossRef]
- Powell, B.R.; Buist, N.R.M.; Stenzel, P. An X-Linked Syndrome of Diarrhea, Polyendocrinopathy, and Fatal Infection in Infancy. J. Pediatr. 1982, 100, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Wildin, R.S.; Smyk-Pearson, S.; Filipovich, A.H. Clinical and Molecular Features of the Immunodysregulation, Polyendocrinopathy, Enteropathy, X Linked (IPEX) Syndrome. J. Med. Genet. 2002, 39, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.L.; Yoshioka, R.; Kiyosawa, H.; Barker, D.F.; Fain, P.R.; Shigeoka, A.O.; Chance, P.F. X-Linked Syndrome of Polyendocrinopathy, Immune Dysfunction, and Diarrhea Maps to Xp11.23-Xq13.3. Am. J. Hum. Genet. 2000, 66, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Passerini, L.; Santoni De Sio, F.R.; Roncarolo, M.G.; Bacchetta, R. Forkhead Box P3: The Peacekeeper of the Immune System. Int. Rev. Immunol. 2014, 33, 129–145. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome (IPEX) Is Caused by Mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Bacchetta, R.; Passerini, L.; Gambineri, E.; Dai, M.; Allan, S.E.; Perroni, L.; Dagna-Bricarelli, F.; Sartirana, C.; Matthes-Martin, S.; Lawitschka, A.; et al. Defective Regulatory and Effector T Cell Functions in Patients with FOXP3 Mutations. J. Clin. Investig. 2006, 116, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Gambineri, E.; Perroni, L.; Passerini, L.; Bianchi, L.; Doglioni, C.; Meschi, F.; Bonfanti, R.; Sznajer, Y.; Tommasini, A.; Lawitschka, A.; et al. Clinical and Molecular Profile of a New Series of Patients with Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome: Inconsistent Correlation between Forkhead Box Protein 3 Expression and Disease Severity. J. Allergy Clin. Immunol. 2008, 122. [Google Scholar] [CrossRef] [PubMed]
- Passerini, L.; Olek, S.; Di Nunzio, S.; Barzaghi, F.; Hambleton, S.; Abinun, M.; Tommasini, A.; Vignola, S.; Cipolli, M.; Amendola, M.; et al. Forkhead Box Protein 3 (FOXP3) Mutations Lead to Increased TH17 Cell Numbers and Regulatory T-Cell Instability. J. Allergy Clin. Immunol. 2011, 128, 1376–1379. [Google Scholar] [CrossRef]
- Nieves, D.S.; Phipps, R.P.; Pollock, S.J.; Ochs, H.D.; Zhu, Q.; Scott, G.A.; Ryan, C.K.; Kobayashi, I.; Rossi, T.M.; Goldsmith, L.A. Dermatologic and Immunologic Findings in the Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome. Arch. Derm. 2004, 140, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Chatila, T.A.; Blaeser, F.; Ho, N.; Lederman, H.M.; Voulgaropoulos, C.; Helms, C.; Bowcock, A.M. JM2, Encoding a Fork Head-Related Protein, Is Mutated in X-Linked Autoimmunity-Allergic Disregulation Syndrome. J. Clin. Investig. 2000, 106, R75–R81. [Google Scholar] [CrossRef] [Green Version]
- Ben-skowronek, I. IPEX Syndrome: Genetics and Treatment Options. Genes 2021, 12, 323. [Google Scholar] [CrossRef]
- Goodwin, M.; Lee, E.; Lakshmanan, U.; Shipp, S.; Froessl, L.; Barzaghi, F.; Passerini, L.; Narula, M.; Sheikali, A.; Lee, C.M.; et al. CRISPR-Based Gene Editing Enables FOXP3 Gene Repair in IPEX Patient Cells. Sci. Adv. 2020, 6, eaaz0571. [Google Scholar] [CrossRef]
- Wu, W.; Shen, N.; Luo, L.; Deng, Z.; Chen, J.; Tao, Y.; Mo, X.; Cao, Q. Fecal Microbiota Transplantation before Hematopoietic Stem Cell Transplantation in a Pediatric Case of Chronic Diarrhea with a FOXP3 Mutation. Pediatr. Neonatol. 2021, 62, 172–180. [Google Scholar] [CrossRef]
- Chinen, T.; Volchkov, P.Y.; Chervonsky, A.V.; Rudensky, A.Y. A Critical Role for Regulatory T Cell-Mediated Control of Inflammation in the Absence of Commensal Microbiota. J. Exp. Med. 2010, 207, 2323–2330. [Google Scholar] [CrossRef] [Green Version]
- Wildin, R.S.; Ramsdell, F.; Peake, J.; Faravelli, F.; Casanova, J.L.; Buist, N.; Levy-Lahad, E.; Mazzella, M.; Goulet, O.; Perroni, L.; et al. X-Linked Neonatal Diabetes Mellitus, Enteropathy and Endocrinopathy Syndrome Is the Human Equivalent of Mouse Scurfy. Nat. Genet. 2001, 27, 18–20. [Google Scholar] [CrossRef]
- Bleesing, J.J.H. Autoimmune Lymphoproliferative Syndrome: A Genetic Disorder of Abnormal Lymphocyte Apoptosis. Immunol. Allergy Clin. N. Am. 2002, 22, 339–355. [Google Scholar] [CrossRef]
- Straus, S.E.; Jaffe, E.S.; Puck, J.M.; Dale, J.K.; Elkon, K.B.; Rösen-Wolff, A.; Peters, A.M.J.; Sneller, M.C.; Hallahan, C.W.; Wang, J.; et al. The Development of Lymphomas in Families with Autoimmune Lymphoproliferative Syndrome with Germline Fas Mutations and Defective Lymphocyte Apoptosis. Blood 2001, 98, 194–200. [Google Scholar] [CrossRef]
- Oliveira, J.B.; Bleesing, J.J.; Dianzani, U.; Fleisher, T.A.; Jaffe, E.S.; Lenardo, M.J.; Rieux-Laucat, F.; Siegel, R.M.; Su, H.C.; Teachey, D.T.; et al. Revised Diagnostic Criteria and Classification for the Autoimmune Lymphoproliferative Syndrome (ALPS): Report from the 2009 NIH International Workshop. Blood 2010, 116, e35. [Google Scholar] [CrossRef] [Green Version]
- Sneller, M.C.; Straus, S.E.; Jaffe, E.S.; Jaffe, J.S.; Fleisher, T.A.; Stetler- Stevenson, M.; Strober, W. A Novel Lymphoproliferative/Autoimmune Syndrome Resembling Murine Lpr/Gld Disease. J. Clin. Investig. 1992, 90, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Dianzani, U.; Bragardo, M.; DiFranco, D.; Alliaudi, C.; Scagni, P.; Buonfiglio, D.; Redoglia, V.; Bonissoni, S.; Correra, A.; Dianzani, I.; et al. Deficiency of the Fas Apoptosis Pathway Without Fas Gene Mutations in Pediatric Patients With Autoimmunity/Lymphoproliferation. Blood 1997, 89, 2871–2879. [Google Scholar] [CrossRef]
- Dean, G.S.; Anand, A.; Blofeld, A.; Isenberg, D.A.; Lydyard, P.M. Characterization of CD3+ CD4- CD8- (Double Negative) T Cells in Patients with Systemic Lupus Erythematosus: Production of IL-4. Lupus 2002, 11, 501–507. [Google Scholar] [CrossRef]
- Rensing-Ehl, A.; Völkl, S.; Speckmann, C.; Lorenz, M.R.; Ritter, J.; Janda, A.; Abinun, M.; Pircher, H.; Bengsch, B.; Thimme, R.; et al. Abnormally Differentiated CD4+ or CD8+ T Cells with Phenotypic and Genetic Features of Double Negative T Cells in Human Fas Deficiency. Blood 2014, 124, 851–860. [Google Scholar] [CrossRef] [Green Version]
- Bristeau-Leprince, A.; Mateo, V.; Lim, A.; Magerus-Chatinet, A.; Solary, E.; Fischer, A.; Rieux-Laucat, F.; Gougeon, M.-L. Human TCR Alpha/Beta+ CD4-CD8- Double-Negative T Cells in Patients with Autoimmune Lymphoproliferative Syndrome Express Restricted Vbeta TCR Diversity and Are Clonally Related to CD8+ T Cells. J. Immunol. 2008, 181, 440–448. [Google Scholar] [CrossRef] [Green Version]
- Bowen, R.A.R.; Dowdell, K.C.; Dale, J.K.; Drake, S.K.; Fleisher, T.A.; Hortin, G.L.; Remaley, A.T.; Nexo, E.; Rao, V.K. Elevated Vitamin B₁₂ Levels in Autoimmune Lymphoproliferative Syndrome Attributable to Elevated Haptocorrin in Lymphocytes. Clin. Biochem. 2012, 45, 490–492. [Google Scholar] [CrossRef] [Green Version]
- Magerus-Chatinet, A.; Stolzenberg, M.C.; Loffredo, M.S.; Neven, B.; Schaffner, C.; Ducrot, N.; Arkwright, P.D.; Bader-Meunier, B.; Barbot, J.; Blanche, S.; et al. FAS-L, IL-10, and Double-Negative CD4- CD8- TCR Alpha/Beta+ T Cells Are Reliable Markers of Autoimmune Lymphoproliferative Syndrome (ALPS) Associated with FAS Loss of Function. Blood 2009, 113, 3027–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe-Fukunaga, R.; Brannan, C.I.; Copeland, N.G.; Jenkins, N.A.; Nagata, S. Lymphoproliferation Disorder in Mice Explained by Defects in Fas Antigen That Mediates Apoptosis. Nature 1992, 356, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tanaka, M.; Brannan, C.I.; Jenkins, N.A.; Copeland, N.G.; Suda, T.; Nagata, S. Generalized Lymphoproliferative Disease in Mice, Caused by a Point Mutation in the Fas Ligand. Cell 1994, 76, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Consonni, F.; Gambineri, E.; Favre, C. ALPS, FAS, and beyond: From Inborn Errors of Immunity to Acquired Immunodeficiencies. Ann. Hematol. 2022, 101, 469–484. [Google Scholar] [CrossRef]
- Strasser, A.; Jost, P.J.; Nagata, S. The Many Roles of FAS Receptor Signaling in the Immune System. Immunity 2009, 30, 180–192. [Google Scholar] [CrossRef] [Green Version]
- Clementi, R.; Dagna, L.; Dianzani, U.; Dupré, L.; Dianzani, I.; Ponzoni, M.; Cometa, A.; Chiocchetti, A.; Sabbadini, M.G.; Rugarli, C.; et al. Inherited Perforin and Fas Mutations in a Patient with Autoimmune Lymphoproliferative Syndrome and Lymphoma. N. Engl. J. Med. 2004, 351, 1419–1424. [Google Scholar] [CrossRef]
- Cerutti, E.; Campagnoli, M.F.; Ferretti, M.; Garelli, E.; Crescenzio, N.; Rosolen, A.; Chiocchetti, A.; Lenardo, M.J.; Ramenghi, U.; Dianzani, U. Co-Inherited Mutations of Fas and Caspase-10 in Development of the Autoimmune Lymphoproliferative Syndrome. BMC Immunol. 2007, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Holzelova, E.; Vonarbourg, C.; Stolzenberg, M.-C.; Arkwright, P.D.; Selz, F.; Prieur, A.-M.; Blanche, S.; Bartunkova, J.; Vilmer, E.; Fischer, A.; et al. Autoimmune Lymphoproliferative Syndrome with Somatic Fas Mutations. N. Engl. J. Med. 2004, 351, 1409–1418. [Google Scholar] [CrossRef]
- Dowdell, K.C.; Niemela, J.E.; Price, S.; Davis, J.; Hornung, R.L.; Oliveira, J.B.; Puck, J.M.; Jaffe, E.S.; Pittaluga, S.; Cohen, J.I.; et al. Somatic FAS Mutations Are Common in Patients with Genetically Undefined Autoimmune Lymphoproliferative Syndrome. Blood 2010, 115, 5164–5169. [Google Scholar] [CrossRef] [Green Version]
- Campagnoli, M.F.; Garbarini, L.; Quarello, P.; Garelli, E.; Carando, A.; Baravalle, V.; Doria, A.; Biava, A.; Chiocchetti, A.; Rosolen, A.; et al. The Broad Spectrum of Autoimmune Lymphoproliferative Disease: Molecular Bases, Clinical Features and Long-Term Follow-up in 31 Patients. Haematologica 2006, 91, 538–541. [Google Scholar]
- Aricò, M.; Boggio, E.; Cetica, V.; Melensi, M.; Orilieri, E.; Clemente, N.; Cappellano, G.; Buttini, S.; Soluri, M.F.; Comi, C.; et al. Variations of the UNC13D Gene in Patients with Autoimmune Lymphoproliferative Syndrome. PLoS ONE 2013, 8, e68045. [Google Scholar] [CrossRef]
- Clementi, R.; Chiocchetti, A.; Cappellano, G.; Cerutti, E.; Ferretti, M.; Orilieri, E.; Dianzani, I.; Ferrarini, M.; Bregni, M.; Danesino, C.; et al. Variations of the Perforin Gene in Patients with Autoimmunity/Lymphoproliferation and Defective Fas Function. Blood 2006, 108, 3079–3084. [Google Scholar] [CrossRef]
- Booth, C.; Gilmour, K.C.; Veys, P.; Gennery, A.R.; Slatter, M.A.; Chapel, H.; Heath, P.T.; Steward, C.G.; Smith, O.; O’Meara, A.; et al. X-Linked Lymphoproliferative Disease Due to SAP/SH2D1A Deficiency: A Multicenter Study on the Manifestations, Management and Outcome of the Disease. Blood 2011, 117, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Comi, C.; Leone, M.; Bonissoni, S.; DeFranco, S.; Bottarel, F.; Mezzatesta, C.; Chiocchetti, A.; Perla, F.; Monaco, F.; Dianzani, U. Defective T Cell Fas Function in Patients with Multiple Sclerosis. Neurology 2000, 55, 921–927. [Google Scholar] [CrossRef]
- DeFranco, S.; Bonissoni, S.; Cerutti, F.; Bona, G.; Bottarel, F.; Cadario, F.; Brusco, A.; Loffredo, G.; Rabbone, I.; Corrias, A.; et al. Defective Function of Fas in Patients with Type 1 Diabetes Associated with Other Autoimmune Diseases. Diabetes 2001, 50, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Zhou, T.; Liu, C.; Shapiro, J.P.; Brauer, M.J.; Kiefer, M.C.; Barr, P.J.; Mountz, J.D. Protection from Fas-Mediated Apoptosis by a Soluble Form of the Fas Molecule. Science 1994, 263, 1759–1762. [Google Scholar] [CrossRef]
- Teachey, D.T.; Seif, A.E.; Grupp, S.A. Advances in the Management and Understanding of Autoimmune Lymphoproliferative Syndrome (ALPS). Br. J. Haematol. 2010, 148, 205–216. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, L.; Lobito, A.; Chan, F.K.M.; Dale, J.; Sneller, M.; Yao, X.; Puck, J.M.; Straus, S.E.; Lenardo, M.J. Inherited Human Caspase 10 Mutations Underlie Defective Lymphocyte and Dendritic Cell Apoptosis in Autoimmune Lymphoproliferative Syndrome Type II. Cell 1999, 98, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Stranges, P.B.; Watson, J.; Cooper, C.J.; Choisy-Rossi, C.M.; Stonebraker, A.C.C.; Beighton, R.A.; Hartig, H.; Sundberg, J.P.; Servick, S.; Kaufmann, G.; et al. Elimination of Antigen-Presenting Cells and Autoreactive T Cells by Fas Contributes to Prevention of Autoimmunity. Immunity 2007, 26, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Gershwin, M.E.; Chang, C. Diagnostic Criteria for Systemic Lupus Erythematosus: A Critical Review. J. Autoimmun. 2014, 48–49, 10–13. [Google Scholar] [CrossRef]
- Namjou, B.; Kilpatrick, J.; Harley, J.B. Genetics of Clinical Expression in SLE. Autoimmunity 2007, 40, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Belot, A.; Cimaz, R. Monogenic Forms of Systemic Lupus Erythematosus: New Insights into SLE Pathogenesis. Pediatr. Rheumatol. Online J. 2012, 10, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leffler, J.; Bengtsson, A.A.; Blom, A.M. The Complement System in Systemic Lupus Erythematosus: An Update. Ann. Rheum. Dis. 2014, 73, 1601–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuveson, D.A.; Ahearn, J.M.; Matsumoto, A.K.; Fearon, D.T. Molecular Interactions of Complement Receptors on B Lymphocytes: A CR1/CR2 Complex Distinct from the CR2/CD19 Complex. J. Exp. Med. 1991, 173, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Kemper, C.; Chan, A.C.; Green, J.M.; Brett, K.A.; Murphy, K.M.; Atkinson, J.P. Activation of Human CD4+ Cells with CD3 and CD46 Induces a T-Regulatory Cell 1 Phenotype. Nature 2003, 421, 388–392. [Google Scholar] [CrossRef]
- Lood, C.; Gullstrand, B.; Truedsson, L.; Olin, A.I.; Alm, G.V.; Rönnblom, L.; Sturfelt, G.; Eloranta, M.L.; Bengtsson, A.A. C1q Inhibits Immune Complex-Induced Interferon-α Production in Plasmacytoid Dendritic Cells. Arthritis Rheum. 2009, 60, 3081–3090. [Google Scholar] [CrossRef]
- Ling, G.S.; Crawford, G.; Buang, N.; Bartok, I.; Tian, K.; Thielens, N.M.; Bally, I.; Harker, J.A.; Ashton-Rickardt, P.G.; Rutschmann, S.; et al. C1q Restrains Autoimmunity and Viral Infection by Regulating CD8+ T Cell Metabolism. Science (1979) 2018, 360, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Stegert, M.; Bock, M.; Trendelenburg, M. Clinical Presentation of Human C1q Deficiency: How Much of a Lupus? Mol. Immunol. 2015, 67, 3–11. [Google Scholar] [CrossRef]
- Macedo, A.C.L.; Isaac, L. Systemic Lupus Erythematosus and Deficiencies of Early Components of the Complement Classical Pathway. Front. Immunol. 2016, 7, 55. [Google Scholar] [CrossRef] [Green Version]
- Elkon, K.B. Cell Death, Nucleic Acids, and Immunity: Inflammation Beyond the Grave. Arthritis Rheumatol. 2018, 70, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wilson, J.; He, J.; Xiang, L.; Schur, P.H.; Mountz, J.D. Fas Ligand Mutation in a Patient with Systemic Lupus Erythematosus and Lymphoproliferative Disease. J. Clin. Investig. 1996, 98, 1107–1113. [Google Scholar] [CrossRef] [Green Version]
- Vaishnaw, A.K.; Toubi, E.; Ohsako, S.; Drappa, J.; Buys, S.; Estrada, J.; Sitarz, A.; Zemel, L.; Chu, J.-L.; Elkon, K.B. THE SPECTRUM OF APOPTOTIC DEFECTS AND CLINICAL MANIFESTATIONS, INCLUDING SYSTEMIC LUPUS ERYTHEMATOSUS, IN HUMANS WITH CD95 (Fas/APO-1) MUTATIONS. Arthritis Rheum. 1999, 42, 1833–1842. [Google Scholar] [CrossRef]
- Belot, A.; Kasher, P.R.; Trotter, E.W.; Foray, A.P.; Debaud, A.L.; Rice, G.I.; Szynkiewicz, M.; Zabot, M.T.; Rouvet, I.; Bhaskar, S.S.; et al. Protein Kinase Cδ Deficiency Causes Mendelian Systemic Lupus Erythematosus with B Cell-Defective Apoptosis and Hyperproliferation. Arthritis Rheum. 2013, 65, 2161–2171. [Google Scholar] [CrossRef] [Green Version]
- Limnander, A.; Zikherman, J.; Lau, T.; Leitges, M.; Weiss, A.; Roose, J.P. Protein Kinase Cδ Promotes Transitional B Cell-Negative Selection and Limits Proximal B Cell Receptor Signaling to Enforce Tolerance. Mol. Cell Biol. 2014, 34, 1474–1485. [Google Scholar] [CrossRef] [Green Version]
- Kiykim, A.; Ogulur, I.; Baris, S.; Salzer, E.; Karakoc-Aydiner, E.; Ozen, A.O.; Garncarz, W.; Hirschmugl, T.; Krolo, A.; Yucelten, A.D.; et al. Potentially Beneficial Effect of Hydroxychloroquine in a Patient with a Novel Mutation in Protein Kinase Cδ Deficiency. J. Clin. Immunol. 2015, 35, 523–526. [Google Scholar] [CrossRef]
- Pullabhatla, V.; Roberts, A.L.; Lewis, M.J.; Mauro, D.; Morris, D.L.; Odhams, C.A.; Tombleson, P.; Liljedahl, U.; Vyse, S.; Simpson, M.A.; et al. De Novo Mutations Implicate Novel Genes in Systemic Lupus Erythematosus. Hum. Mol. Genet. 2018, 27, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Walter, J.E.; Lo, M.S.; Kis-Toth, K.; Tirosh, I.; Frugoni, F.; Lee, Y.N.; Csomos, K.; Chen, K.; Pillai, S.; Dunham, J.; et al. Impaired Receptor Editing and Heterozygous RAG2 Mutation in a Patient with Systemic Lupus Erythematosus and Erosive Arthritis. J. Allergy Clin. Immunol. 2015, 135, 272–273. [Google Scholar] [CrossRef] [Green Version]
- Ragotte, R.J.; Dhanrajani, A.; Pleydell-Pearce, J.; Del Bel, K.L.; Tarailo-Graovac, M.; van Karnebeek, C.; Terry, J.; Senger, C.; McKinnon, M.L.; Seear, M.; et al. The Importance of Considering Monogenic Causes of Autoimmunity: A Somatic Mutation in KRAS Causing Pediatric Rosai-Dorfman Syndrome and Systemic Lupus Erythematosus. Clin. Immunol. 2017, 175, 143–146. [Google Scholar] [CrossRef]
- Hebron, K.E.; Hernandez, E.R.; Yohe, M.E. The RASopathies: From Pathogenetics to Therapeutics. Dis. Model. Mech. 2022, 15. [Google Scholar] [CrossRef]
- Leventopoulos, G.; Denayer, E.; Makrythanasis, P.; Papapolychroniou, C.; Fryssira, H. Noonan Syndrome and Systemic Lupus Erythematosus in a Patient with a Novel KRAS Mutation. Clin. Exp. Rheumatol. 2010, 28, 556–557. [Google Scholar]
- Hiraki, L.T.; Silverman, E.D. Genomics of Systemic Lupus Erythematosus: Insights Gained by Studying Monogenic Young-Onset Systemic Lupus Erythematosus. Rheum. Dis. Clin. N. Am. 2017, 43, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 Prevents Cell-Intrinsic Initiation of Autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, J.L.; Chon, H.; Cerritelli, S.M.; Kunkel, T.A.; Johansson, E.; Crouch, R.J.; Burgers, P.M. RNase H2-Initiated Ribonucleotide Excision Repair. Mol. Cell 2012, 47, 980–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, G.I.; Rodero, M.P.; Crow, Y.J. Human Disease Phenotypes Associated with Mutations in TREX1. J. Clin. Immunol. 2015, 35, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.A.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of Human Disease Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 2015, 167A, 296–312. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, K.J.; Carroll, P.; Lettice, L.; Tarnauskait, Z.; Reddy, K.; Dix, F.; Revuelta, A.; Abbondati, E.; Rigby, R.E.; Rabe, B.; et al. Ribonuclease H2 Mutations Induce a CGAS/STING-Dependent Innate Immune Response. EMBO J. 2016, 35, 831–844. [Google Scholar] [CrossRef]
- Ramantani, G.; Kohlhase, J.; Hertzberg, C.; Micheil Innes, A.; Engel, K.; Hunger, S.; Borozdin, W.; Mah, J.K.; Ungerath, K.; Walkenhorst, H.; et al. Expanding the Phenotypic Spectrum of Lupus Erythematosus in Aicardi-Goutières Syndrome. Arthritis Rheum. 2010, 62, 1469–1477. [Google Scholar] [CrossRef]
- Ellyard, J.I.; Jerjen, R.; Martin, J.L.; Lee, A.Y.S.; Field, M.A.; Jiang, S.H.; Cappello, J.; Naumann, S.K.; Andrews, T.D.; Scott, H.S.; et al. Brief Report: Identification of a Pathogenic Variant in Trex1 in Early-Onset Cerebral Systemic Lupus Erythematosus by Whole-Exome Sequencing. Arthritis Rheumatol.. 2014, 66, 3382–3386. [Google Scholar] [CrossRef]
- Lebon, P.; Badoual, J.; Ponsot, G.; Goutières, F.; Hémeury-Cukier, F.; Aicardi, J. Intrathecal Synthesis of Interferon-Alpha in Infants with Progressive Familial Encephalopathy. J. Neurol. Sci. 1988, 84, 201–208. [Google Scholar] [CrossRef]
- Crow, M.K. Pathogenesis of Systemic Lupus Erythematosus: Risks, Mechanisms and Therapeutic Targets. Ann. Rheum. Dis. 2023, ard-2022-223741. [Google Scholar] [CrossRef]
- Bengtsson, A.A.; Rönnblom, L. Role of Interferons in SLE. Best. Pract. Res. Clin. Rheumatol. 2017, 31, 415–428. [Google Scholar] [CrossRef]
- Al-Mayouf, S.M.; Sunker, A.; Abdwani, R.; Al Abrawi, S.; Almurshedi, F.; Alhashmi, N.; Al Sonbul, A.; Sewairi, W.; Qari, A.; Abdallah, E.; et al. Loss-of-Function Variant in DNASE1L3 Causes a Familial Form of Systemic Lupus Erythematosus. Nat. Genet. 2011, 43, 1186–1188. [Google Scholar] [CrossRef]
- Hartl, J.; Serpas, L.; Wang, Y.; Rashidfarrokhi, A.; Perez, O.A.; Sally, B.; Sisirak, V.; Soni, C.; Khodadadi-Jamayran, A.; Tsirigos, A.; et al. Autoantibody-Mediated Impairment of DNASE1L3 Activity in Sporadic Systemic Lupus Erythematosus. J. Exp. Med. 2021, 218, e20201138. [Google Scholar] [CrossRef]
- Pisetsky, D.S. The Expression of HMGB1 on Microparticles Released during Cell Activation and Cell Death in Vitro and in Vivo. Mol. Med. 2014, 20, 158–163. [Google Scholar] [CrossRef]
- Bodaño, A.; Amarelo, J.; González, A.; Gómez-Reino, J.J.; Conde, C. Novel DNASE I Mutations Related to Systemic Lupus Erythematosus. Arthritis Rheum. 2004, 50, 4070–4071. [Google Scholar] [CrossRef]
- Kara, B.; Ekinci, Z.; Sahin, S.; Gungor, M.; Gunes, A.S.; Ozturk, K.; Adrovic, A.; Cefle, A.; Inanç, M.; Gul, A.; et al. Monogenic Lupus Due to Spondyloenchondrodysplasia with Spastic Paraparesis and Intracranial Calcification: Case-Based Review. Rheumatol. Int. 2020, 40, 1903–1910. [Google Scholar] [CrossRef]
- Bilginer, Y.; Düzova, A.; Topaloaylu, R.; Batu, E.D.; Boduroaylu, K.; Güçer, A.; Bodur, A.; Alanay, Y. Three Cases of Spondyloenchondrodysplasia (SPENCD) with Systemic Lupus Erythematosus: A Case Series and Review of the Literature. Lupus 2016, 25, 760–765. [Google Scholar] [CrossRef]
- Weber, G.F.; Zawaideh, S.; Hikita, S.; Kumar, V.A.; Cantor, H.; Ashkar, S. Phosphorylation-Dependent Interaction of Osteopontin with Its Receptors Regulates Macrophage Migration and Activation. J. Leukoc. Biol. 2002, 72, 752–761. [Google Scholar] [CrossRef]
- Shinohara, M.L.; Lu, L.; Bu, J.; Werneck, M.B.F.; Kobayashi, K.S.; Glimcher, L.H.; Cantor, H. Osteopontin Expression Is Essential for Interferon-α Production by Plasmacytoid Dendritic Cells. Nat. Immunol. 2006, 7, 498–506. [Google Scholar] [CrossRef]
- Ouyang, S.; Gong, B.; Li, J.Z.; Zhao, L.X.; Wu, W.; Zhang, F.S.; Sun, L.; Wang, S.J.; Pan, M.; Li, C.; et al. Structural Insights into a Human Anti-IFN Antibody Exerting Therapeutic Potential for Systemic Lupus Erythematosus. J. Mol. Med. 2012, 90, 837–846. [Google Scholar] [CrossRef]
- Rodríguez-Carrio, J.; Burska, A.; Conaghan, P.G.; Dik, W.A.; Biesen, R.; Eloranta, M.-L.; Cavalli, G.; Visser, M.; Boumpas, D.T.; Bertsias, G.; et al. Association between Type I Interferon Pathway Activation and Clinical Outcomes in Rheumatic and Musculoskeletal Diseases: A Systematic Literature Review Informing EULAR Points to Consider. RMD Open 2023, 9, e002864. [Google Scholar] [CrossRef] [PubMed]
- Wahadat, M.J.; Bodewes, I.L.A.; Maria, N.I.; van Helden-Meeuwsen, C.G.; van Dijk-Hummelman, A.; Steenwijk, E.C.; Kamphuis, S.; Versnel, M.A. Type I IFN Signature in Childhood-Onset Systemic Lupus Erythematosus: A Conspiracy of DNA- and RNA-Sensing Receptors? Arthritis Res. 2018, 20, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, Y.J.; Vanderver, A.; Orcesi, S.; Kuijpers, T.W.; Rice, G.I. Therapies in Aicardi–Goutières Syndrome. Clin. Exp. Immunol. 2013, 175, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dasdemir, S.; Yildiz, M.; Celebi, D.; Sahin, S.; Aliyeva, N.; Haslak, F.; Gunalp, A.; Adrovic, A.; Barut, K.; Artim Esen, B.; et al. Genetic Screening of Early-Onset Patients with Systemic Lupus Erythematosus by a Targeted next-Generation Sequencing Gene Panel. Lupus 2022, 31, 330–337. [Google Scholar] [CrossRef]
- Eloi, M.; Horvath, D.V.; Ortega, C.; Prado, S.; Andrade, L.E.C.; Lú Cia Szejnfeld, V.; Heldan, C.; Castro, M. 25-Hydroxivitamin D Serum Concentration, Not Free and Bioavailable Vitamin D, Is Associated with Disease Activity in Systemic Lupus Erythematosus Patients. PLoS ONE 2017, 12, e0170323. [Google Scholar] [CrossRef] [Green Version]
- Meza-Meza, M.R.; Francisco Muñoz-Valle, J.; Ruiz-Ballesteros, A.I.; Vizmanos-Lamotte, B.; Parra-Rojas, I.; Martínez-López, E.; Oregon-Romero, E.; Fabiola Márquez-Sandoval, Y.; Cerpa-Cruz, S.; De La Cruz-Mosso, U. Association of High Calcitriol Serum Levels and Its Hydroxylation Efficiency Ratio with Disease Risk in SLE Patients with Vitamin D Deficiency. Hindawi J. Immunol. Res. 2021, 2021, 16. [Google Scholar] [CrossRef]
- Swee Gaik, O.; Hui Jen, D. Vitamin D Status in a Monocentric Cohort of Systemic Lupus Erythematosus (SLE) Patients and Correlations with Clinical and Immunological Profile. Med. J. Malays. 2019, 74, 493. [Google Scholar]
- di Chen, B.; Jia, X.; Xu, J.; Zhao, L.; Ji, J.; Wu, B.; Ma, Y.; Li, H.; Zuo, X.; Pan, W.; et al. An Autoimmunogenic and Proinflammatory Profile Defined by the Gut Microbiota of Patients with Untreated Systemic Lupus Erythematosus. Arthritis Rheumatol. 2021, 73, 232–243. [Google Scholar] [CrossRef]
- Zhang, L.; Qing, P.; Yang, H.; Wu, Y.; Liu, Y.; Luo, Y. Gut Microbiome and Metabolites in Systemic Lupus Erythematosus: Link, Mechanisms and Intervention. Front. Immunol. 2021, 12, 686501. [Google Scholar] [CrossRef]
- López, P.; De Paz, B.; Rodríguez-Carrio, J.; Hevia, A.; Sánchez, B.; Margolles, A.; Suárez, A. Th17 Responses and Natural IgM Antibodies Are Related to Gut Microbiota Composition in Systemic Lupus Erythematosus Patients. Sci. Rep. 2016, 6, 24072. [Google Scholar] [CrossRef] [Green Version]
- Azzouz, D.; Omarbekova, A.; Heguy, A.; Schwudke, D.; Gisch, N.; Rovin, B.H.; Caricchio, R.; Buyon, J.P.; Alekseyenko, A.V.; Silverman, G.J. Lupus Nephritis Is Linked to Disease-Activity Associated Expansions and Immunity to a Gut Commensal. Ann. Rheum. Dis. 2019, 78, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, H.F.; Li, X.; Li, H.X.; Zhang, Q.; Zhou, H.W.; He, Y.; Li, P.; Fu, C.; Zhang, X.H.; et al. Disordered Intestinal Microbes Are Associated with the Activity of Systemic Lupus Erythematosus. Clin. Sci. 2019, 133, 821–838. [Google Scholar] [CrossRef]
- Wang, H.; Wang, G.; Banerjee, N.; Liang, Y.; Du, X.; Boor, P.J.; Hoffman, K.L.; Khan, M.F. Aberrant Gut Microbiome Contributes to Intestinal Oxidative Stress, Barrier Dysfunction, Inflammation and Systemic Autoimmune Responses in MRL/Lpr Mice. Front. Immunol. 2021, 12, 1. [Google Scholar] [CrossRef]
- Mu, Q.; Zhang, H.; Liao, X.; Lin, K.; Liu, H.; Edwards, M.R.; Ahmed, S.A.; Yuan, R.; Li, L.; Cecere, T.E.; et al. Control of Lupus Nephritis by Changes of Gut Microbiota. Microbiome 2017, 5, 73. [Google Scholar] [CrossRef] [Green Version]
- Cabana-Puig, X.; Mu, Q.; Lu, R.; Swartwout, B.; Abdelhamid, L.; Zhu, J.; Prakash, M.; Cecere, T.E.; Wang, Z.; Callaway, S.; et al. Lactobacillus Spp. Act in Synergy to Attenuate Splenomegaly and Lymphadenopathy in Lupus-Prone MRL/Lpr Mice. Front. Immunol. 2022, 13, 4103. [Google Scholar] [CrossRef]
- Niess, J.H. What Are CX3CR1+ Mononuclear Cells in the Intestinal Mucosa? Gut. Microbes 2010, 1, 396–400. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.S.; Park, Y.; Choi, J.W.; Park, S.H.; La Cho, M.; Kwok, S.K. Lactobacillus Acidophilus Supplementation Exerts a Synergistic Effect on Tacrolimus Efficacy by Modulating Th17/Treg Balance in Lupus-Prone Mice via the SIGNR3 Pathway. Front. Immunol. 2021, 12, 5246. [Google Scholar] [CrossRef]
- Toral, M.; Robles-Vera, I.; Romero, M.; de la Visitación, N.; Sánchez, M.; O’Valle, F.; Rodriguez-Nogales, A.; Gálvez, J.; Duarte, J.; Jiménez, R. Lactobacillus Fermentum CECT5716: A Novel Alternative for the Prevention of Vascular Disorders in a Mouse Model of Systemic Lupus Erythematosus. FASEB J. 2019, 33, 10005–10018. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Xu, H.; Xu, J.; Zhao, H.; Lin, Q.; Zhou, Y.; Nie, Y. Sodium Butyrate Ameliorates Gut Microbiota Dysbiosis in Lupus-Like Mice. Front. Nutr. 2020, 7, 604283. [Google Scholar] [CrossRef]
- Li, Z.; Yi, C.X.; Katiraei, S.; Kooijman, S.; Zhou, E.; Chung, C.K.; Gao, Y.; Van Den Heuvel, J.K.; Meijer, O.C.; Berbée, J.F.P.; et al. Butyrate Reduces Appetite and Activates Brown Adipose Tissue via the Gut-Brain Neural Circuit. Gut 2018, 67, 1269–1279. [Google Scholar] [CrossRef] [Green Version]
- Frostegård, J.; Svenungsson, E.; Wu, R.; Gunnarsson, I.; Lundberg, I.E.; Klareskog, L.; Hörkkö, S.; Witztum, J.L. Lipid Peroxidation Is Enhanced in Patients with Systemic Lupus Erythematosus and Is Associated with Arterial and Renal Disease Manifestations. Arthritis Rheum. 2005, 52, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhu, Z.; Lin, X.; Li, H.; Wen, C.; Bao, J.; He, Z. Gut Microbiota Mediated the Therapeutic Efficacies and the Side Effects of Prednisone in the Treatment of MRL/Lpr Mice. Arthritis Res. 2021, 23, 240. [Google Scholar] [CrossRef] [PubMed]
- Trzonkowski, P.; Bieniaszewska, M.; Juścińska, J.; Dobyszuk, A.; Krzystyniak, A.; Marek, N.; Myśliwska, J.; Hellmann, A. First-in-Man Clinical Results of the Treatment of Patients with Graft versus Host Disease with Human Ex Vivo Expanded CD4+CD25+CD127- T Regulatory Cells. Clin. Immunol. 2009, 133, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Shum, A.K.; DeVoss, J.; Tan, C.L.; Hou, Y.; Johannes, K.; O’Gorman, C.S.; Jones, K.D.; Sochett, E.B.; Fong, L.; Anderson, M.S. Identification of an Autoantigen Demonstrates a Link between Interstitial Lung Disease and a Defect in Central Tolerance. Sci. Transl. Med. 2009, 1, 9ra20. [Google Scholar] [CrossRef] [Green Version]
- Van Belleghem, J.D.; Dąbrowska, K.; Vaneechoutte, M.; Barr, J.J.; Bollyky, P.L. Interactions between Bacteriophage, Bacteria, and the Mammalian Immune System. Viruses 2019, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Hahn, J.; Cook, N.R.; Alexander, E.K.; Friedman, S.; Walter, J.; Bubes, V.; Kotler, G.; Lee, I.M.; Manson, J.A.E.; Costenbader, K.H. Vitamin D and Marine Omega 3 Fatty Acid Supplementation and Incident Autoimmune Disease: VITAL Randomized Controlled Trial. BMJ 2022, 376, e066452. [Google Scholar] [CrossRef]


{kind=link}
| Disease | Defective Gene(s) | Inheritance | Autoimmunity | Mechanism of Autoimmunity | Major Symptoms | Microbiota Changes |
|---|---|---|---|---|---|---|
| APS1 [42,49,50,61] | AIRE | Autosomal recessive | Organ specific | Defective Tregs Abnormal DCs Lymphocyte Infiltration AutoAbs | Hypoparathyroidism Mucocutaneous candidiasis Adrenal insufficiency | ↓Firmicutes ↑Proteobacteria ↑Bacteroidetes |
| IPEX [69,72,80] | FOXP3 | X-linked | Organ specific | Dysfunctional Tregs AutoAbs | Diarrhea Type I diabetes Thyroiditis Eczema | ↓Firmicutes ↑Bacteroidetes |
| ALPS [92,97,98,110] | FAS FASL CASP10 | Autosomal dominant Autosomal recessive (variable penetrance) | Systemic, organ specific | Defective lymphocyte apoptosis | Lymphadenopathy Splenomegaly Autoimmune Cytopenias Malignancy | N.A. |
| SLE [120,124,132,143,161] | Clq, Cls, Clr, C2, C4 TREX1 RNase H2 DNASE1L3 ACP5 | Autosomal recessive | Systemic | Complement deficiencies Impaired apoptosis Nucleic acid degradation Nucleic acid sensing self-tolerance type I interferonopathies | Arthritis Cutaneous vasculitis Nephritis Infections | ↓Firmicutes * ↓Lactobacillus * ↑Bacteroidetes * ↑Lachnospiraceae * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, F.; Beltrami, E.; Mellone, S.; Sacchetti, S.; Boggio, E.; Gigliotti, C.L.; Stoppa, I.; Dianzani, U.; Rolla, R.; Giordano, M. Genes and Microbiota Interaction in Monogenic Autoimmune Disorders. Biomedicines 2023, 11, 1127. https://doi.org/10.3390/biomedicines11041127
Costa F, Beltrami E, Mellone S, Sacchetti S, Boggio E, Gigliotti CL, Stoppa I, Dianzani U, Rolla R, Giordano M. Genes and Microbiota Interaction in Monogenic Autoimmune Disorders. Biomedicines. 2023; 11(4):1127. https://doi.org/10.3390/biomedicines11041127
Chicago/Turabian StyleCosta, Federica, Eleonora Beltrami, Simona Mellone, Sara Sacchetti, Elena Boggio, Casimiro Luca Gigliotti, Ian Stoppa, Umberto Dianzani, Roberta Rolla, and Mara Giordano. 2023. "Genes and Microbiota Interaction in Monogenic Autoimmune Disorders" Biomedicines 11, no. 4: 1127. https://doi.org/10.3390/biomedicines11041127
APA StyleCosta, F., Beltrami, E., Mellone, S., Sacchetti, S., Boggio, E., Gigliotti, C. L., Stoppa, I., Dianzani, U., Rolla, R., & Giordano, M. (2023). Genes and Microbiota Interaction in Monogenic Autoimmune Disorders. Biomedicines, 11(4), 1127. https://doi.org/10.3390/biomedicines11041127