

Two-Pore-Domain Potassium Channel TREK–1 Mediates Pulmonary Fibrosis through Macrophage M2 Polarization and by Direct Promotion of Fibroblast Differentiation
 and
and
Abstract
:1. Introduction
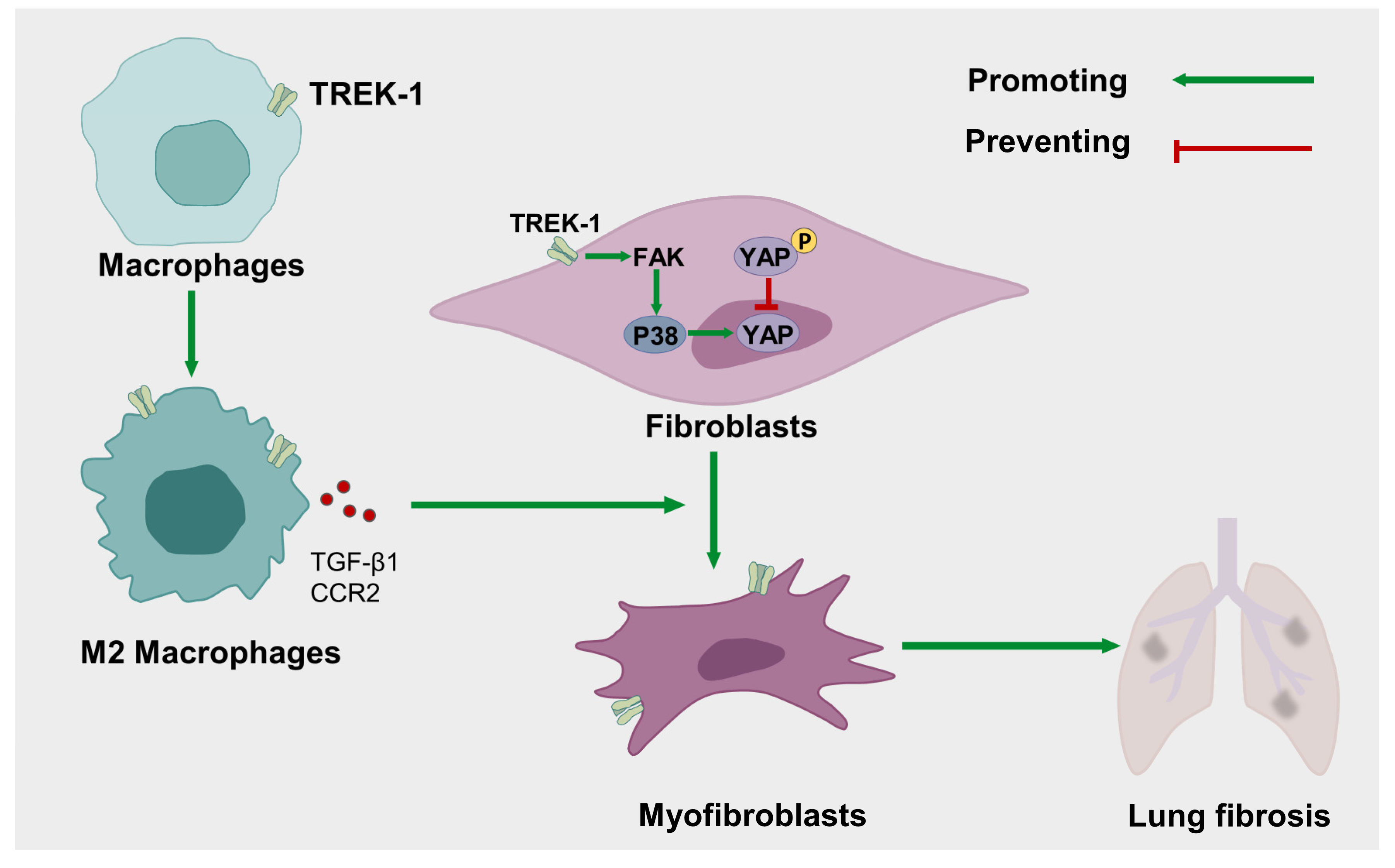
2. Results
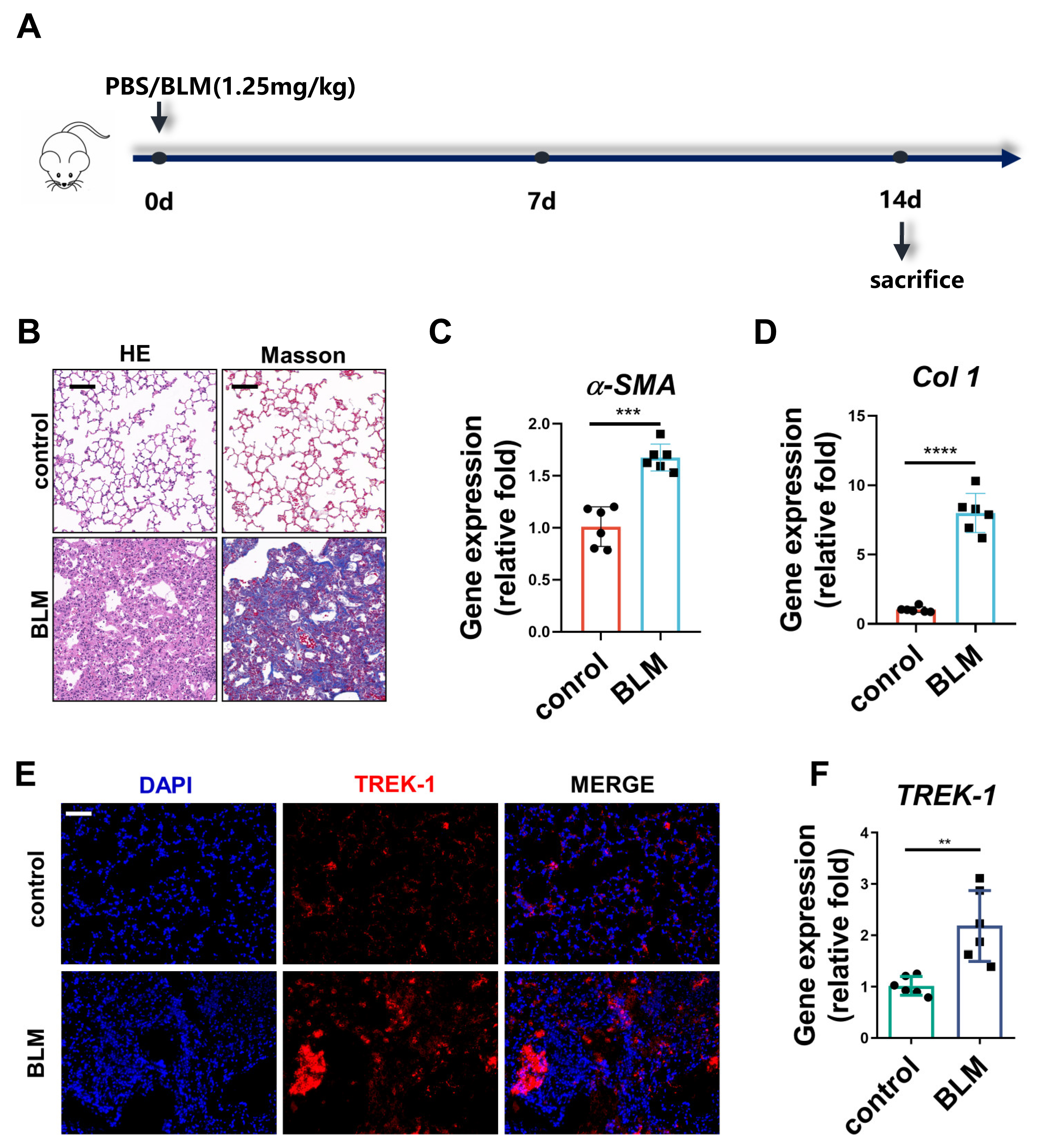
2.1. TREK–1 Expression Is Increased in the Lung Tissue of BLM-Challenged Mice
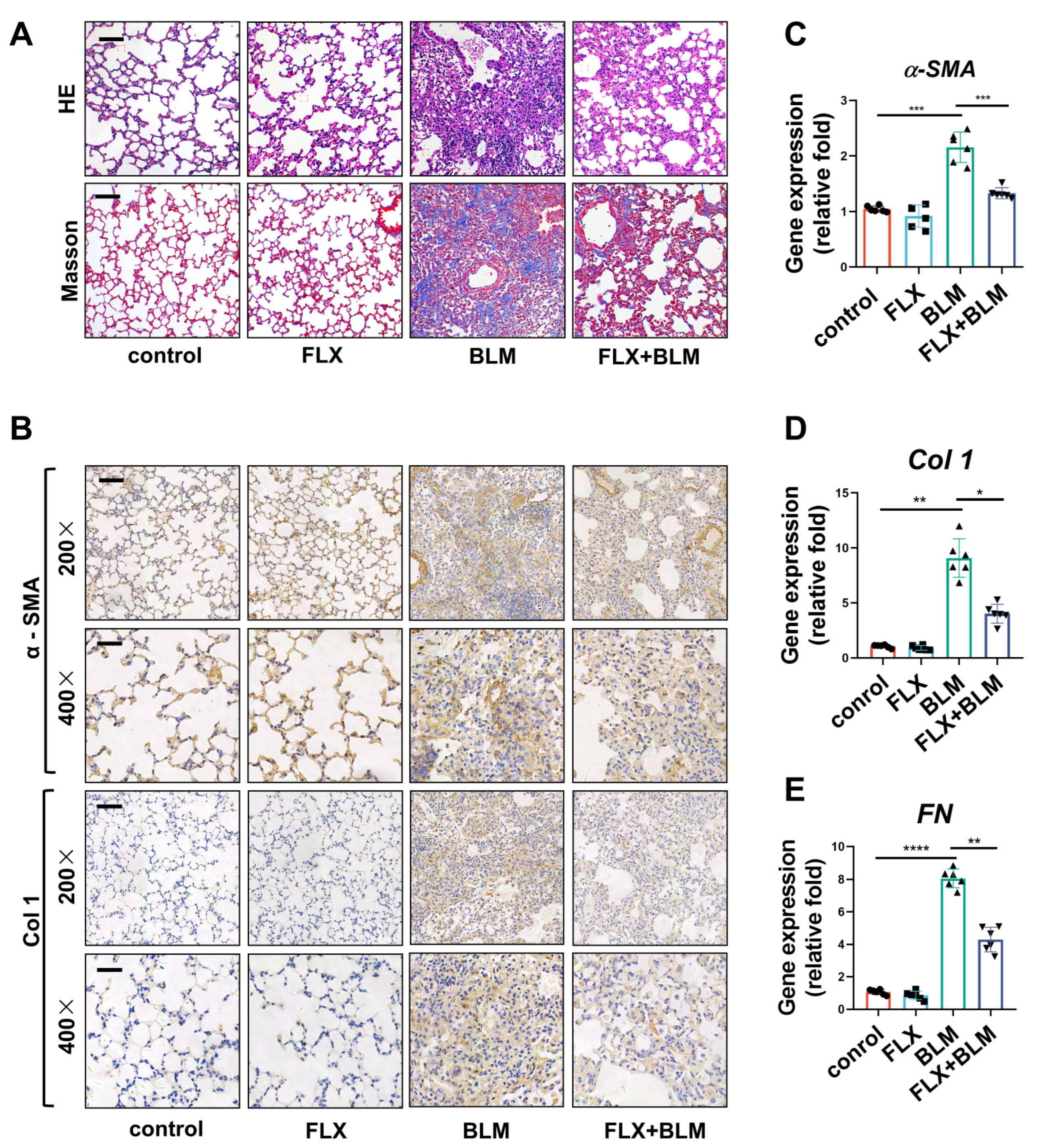
2.2. Inhibition of TREK–1 in the Lungs Attenuated BLM-Induced Fibrosis in Mice
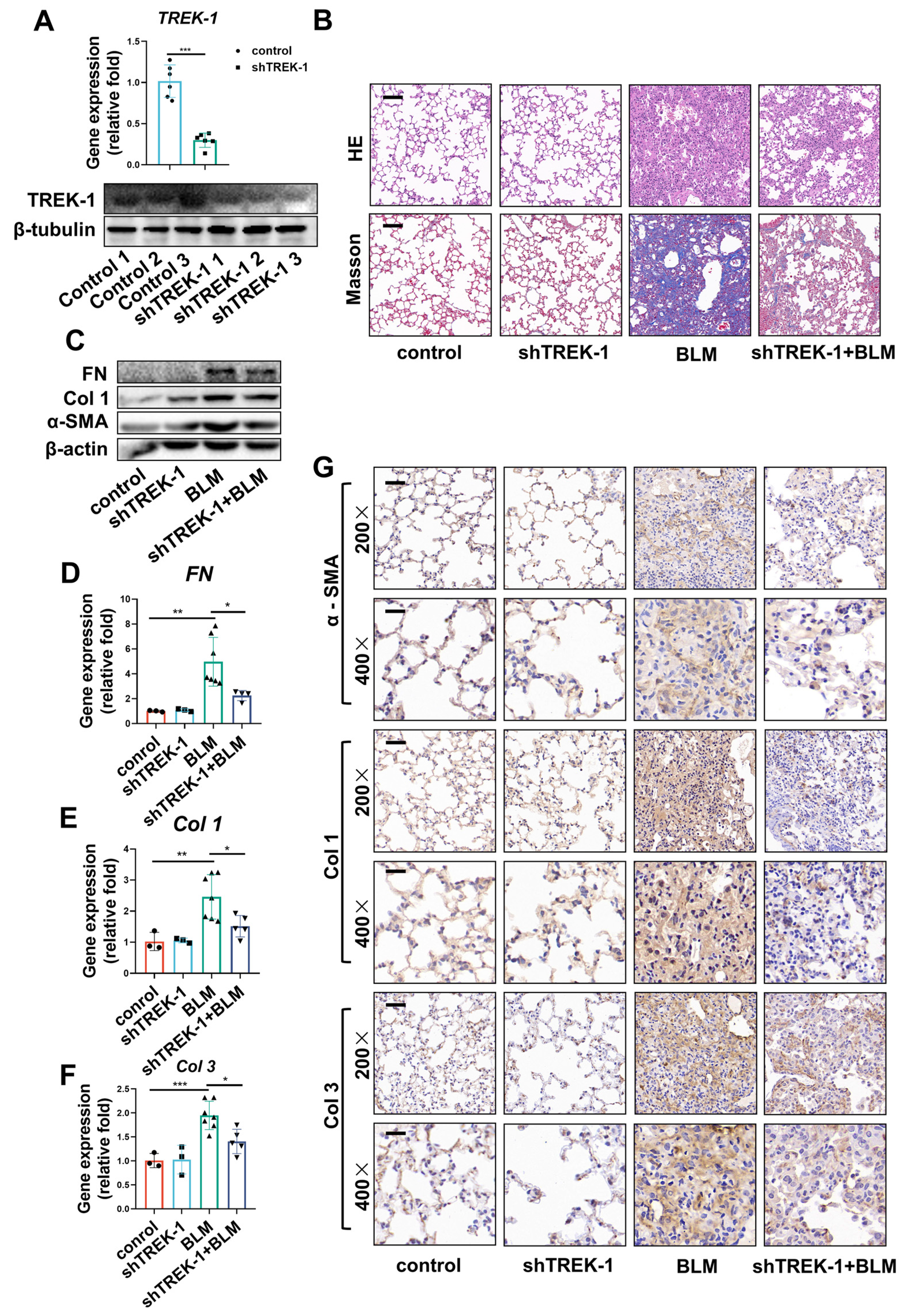
2.3. Knockdown of TREK–1 Attenuated Bleomycin-Induced Pulmonary Fibrosis
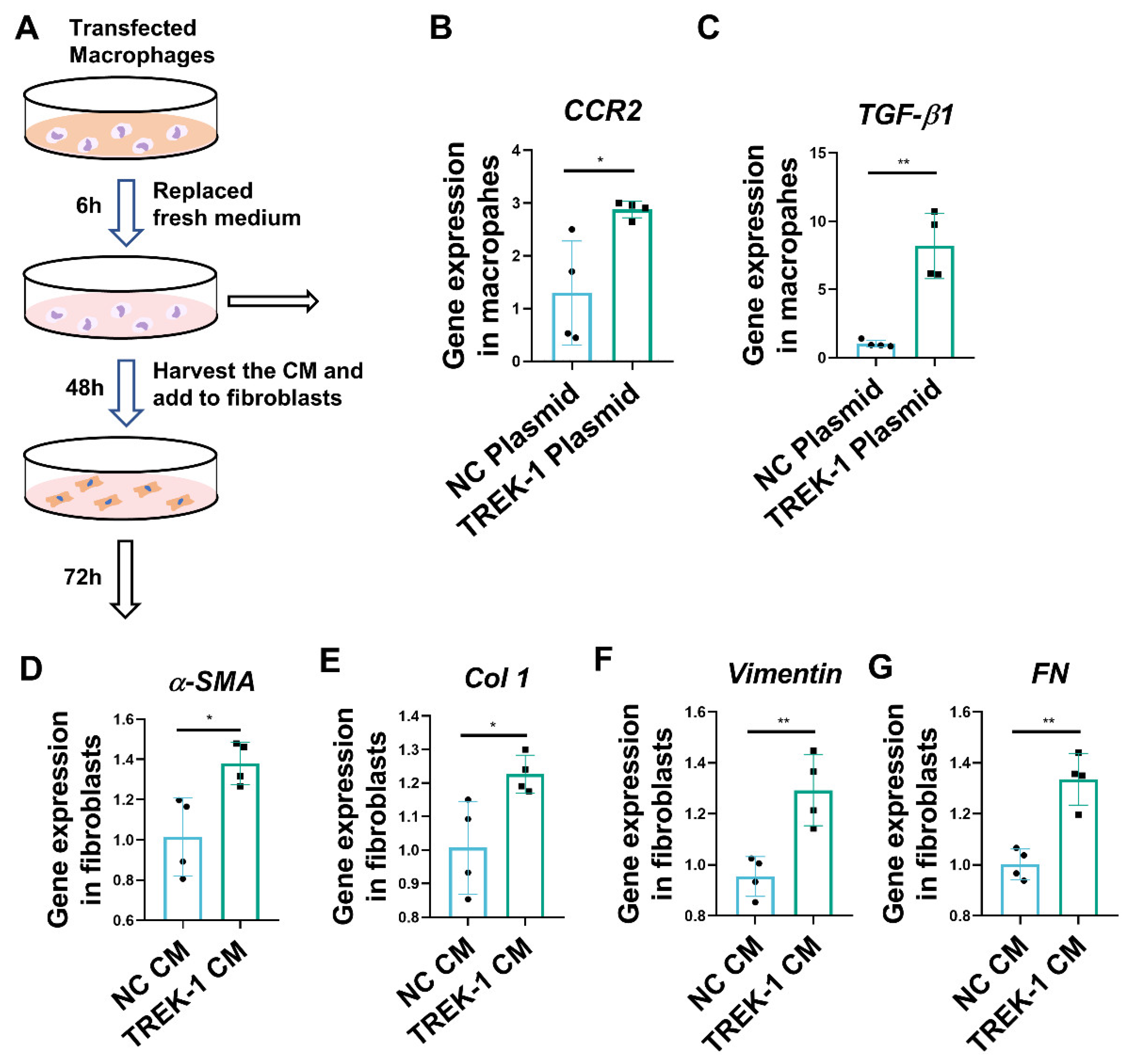
2.4. TREK–1 Overexpression Induces Macrophage Polarized to the M2 Phenotype
2.5. Macrophage Overexpression of TREK–1 Promotes Lung Fibroblast Transdifferentiation to Myofibroblasts through TGF-β1 Pathway
2.6. Knockdown and Inhibition of TREK–1 Suppress Signaling Pathways Downstream of TGF-β1
2.7. TREK–1 Is Upregulated in Lung Tissue of IPF Patients
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Histological Assessment of Pulmonary Fibrosis
4.3. Cell Culture
4.4. Plasmids and siRNA Transfections
4.5. Preparation and Collection of Macrophage-Conditioned Medium
4.6. RNA Extraction and q-PCR Experiments
4.7. Western Blotting
4.8. Immunofluorescence Staining
4.9. Immunohistochemistry
4.10. Human Microarray Data
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Selman, M.; King, T.E.; Pardo, A.; American Thoracic, S.; European Respiratory, S.; American College of Chest, P. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef]
- Khalil, N.; O’Connor, R. Idiopathic pulmonary fibrosis: Current understanding of the pathogenesis and the status of treatment. CMAJ 2004, 171, 153–160. [Google Scholar] [CrossRef]
- Maher, T.M.; Wells, A.U.; Laurent, G.J. Idiopathic pulmonary fibrosis: Multiple causes and multiple mechanisms? Eur. Respir. J. 2007, 30, 835–839. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 755–772. [Google Scholar] [CrossRef]
- Honore, E. The neuronal background K2P channels: Focus on TREK1. Nat. Rev. Neurosci. 2007, 8, 251–261. [Google Scholar] [CrossRef]
- Fink, M.; Duprat, F.; Lesage, F.; Reyes, R.; Romey, G.; Heurteaux, C.; Lazdunski, M. Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J. 1996, 15, 6854–6862. [Google Scholar] [CrossRef]
- Lauritzen, I.; Chemin, J.; Honore, E.; Jodar, M.; Guy, N.; Lazdunski, M.; Jane Patel, A. Cross-talk between the mechano-gated K2P channel TREK-1 and the actin cytoskeleton. EMBO Rep. 2005, 6, 642–648. [Google Scholar] [CrossRef]
- Heurteaux, C.; Guy, N.; Laigle, C.; Blondeau, N.; Duprat, F.; Mazzuca, M.; Lang-Lazdunski, L.; Widmann, C.; Zanzouri, M.; Romey, G.; et al. TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J. 2004, 23, 2684–2695. [Google Scholar] [CrossRef]
- Abraham, D.M.; Lee, T.E.; Watson, L.J.; Mao, L.; Chandok, G.; Wang, H.G.; Frangakis, S.; Pitt, G.S.; Shah, S.H.; Wolf, M.J.; et al. The two-pore domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction. J. Clin. Investig. 2018, 128, 4843–4855. [Google Scholar] [CrossRef]
- Di, A.; Xiong, S.; Ye, Z.; Malireddi, R.K.S.; Kometani, S.; Zhong, M.; Mittal, M.; Hong, Z.; Kanneganti, T.D.; Rehman, J.; et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity 2018, 49, 56–65.e54. [Google Scholar] [CrossRef]
- Innamaa, A.; Jackson, L.; Asher, V.; van Schalkwyk, G.; Warren, A.; Keightley, A.; Hay, D.; Bali, A.; Sowter, H.; Khan, R. Expression and effects of modulation of the K2P potassium channels TREK-1 (KCNK2) and TREK-2 (KCNK10) in the normal human ovary and epithelial ovarian cancer. Clin. Transl. Oncol. 2013, 15, 910–918. [Google Scholar] [CrossRef]
- Voloshyna, I.; Besana, A.; Castillo, M.; Matos, T.; Weinstein, I.B.; Mansukhani, M.; Robinson, R.B.; Cordon-Cardo, C.; Feinmark, S.J. TREK-1 is a novel molecular target in prostate cancer. Cancer Res. 2008, 68, 1197–1203. [Google Scholar] [CrossRef]
- Zhao, L.N.; Fu, L.; Gao, Q.P.; Xie, R.S.; Cao, J.X. Regional differential expression of TREK-1 at left ventricle in myocardial infarction. Can. J. Cardiol. 2011, 27, 826–833. [Google Scholar] [CrossRef]
- Wiedmann, F.; Schlund, D.; Faustino, F.; Kraft, M.; Ratte, A.; Thomas, D.; Katus, H.A.; Schmidt, C. N-Glycosylation of TREK-1/hK(2P)2.1 Two-Pore-Domain Potassium (K(2P)) Channels. Int. J. Mol. Sci. 2019, 20, 5193. [Google Scholar] [CrossRef]
- Zyrianova, T.; Lopez, B.; Olcese, R.; Belperio, J.; Waters, C.M.; Wong, L.; Nguyen, V.; Talapaneni, S.; Schwingshackl, A. K(2P)2.1 (TREK-1) potassium channel activation protects against hyperoxia-induced lung injury. Sci. Rep. 2020, 10, 22011. [Google Scholar] [CrossRef]
- Zyrianova, T.; Lopez, B.; Zou, K.; Gu, C.; Pham, D.; Talapaneni, S.; Waters, C.M.; Olcese, R.; Schwingshackl, A. Activation of TREK-1 (K(2P)2.1) potassium channels protects against influenza A-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2023, 324, L64–L75. [Google Scholar] [CrossRef]
- Sari, E.; He, C.; Margaroli, C. Plasticity towards Rigidity: A Macrophage Conundrum in Pulmonary Fibrosis. Int. J. Mol. Sci. 2022, 23, 11443. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef]
- Zhang, T.; He, X.; Caldwell, L.; Goru, S.K.; Ulloa Severino, L.; Tolosa, M.F.; Misra, P.S.; McEvoy, C.M.; Christova, T.; Liu, Y.; et al. NUAK1 promotes organ fibrosis via YAP and TGF-beta/SMAD signaling. Sci. Transl. Med. 2022, 14, eaaz4028. [Google Scholar] [CrossRef]
- Djillani, A.; Mazella, J.; Heurteaux, C.; Borsotto, M. Role of TREK-1 in Health and Disease, Focus on the Central Nervous System. Front. Pharmacol. 2019, 10, 379. [Google Scholar] [CrossRef]
- Fink, M.; Lesage, F.; Duprat, F.; Heurteaux, C.; Reyes, R.; Fosset, M.; Lazdunski, M. A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J. 1998, 17, 3297–3308. [Google Scholar] [CrossRef]
- Maingret, F.; Lauritzen, I.; Patel, A.J.; Heurteaux, C.; Reyes, R.; Lesage, F.; Lazdunski, M.; Honore, E. TREK-1 is a heat-activated background K+ channel. EMBO J. 2000, 19, 2483–2491. [Google Scholar] [CrossRef]
- Mukherjee, S.; Sikdar, S.K. Intracellular activation of full-length human TREK-1 channel by hypoxia, high lactate, and low pH denotes polymodal integration by ischemic factors. Pflugers Arch. 2021, 473, 167–183. [Google Scholar] [CrossRef]
- Heurteaux, C.; Lucas, G.; Guy, N.; El Yacoubi, M.; Thummler, S.; Peng, X.D.; Noble, F.; Blondeau, N.; Widmann, C.; Borsotto, M.; et al. Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat. Neurosci. 2006, 9, 1134–1141. [Google Scholar] [CrossRef]
- Cheng, P.; Li, S.; Chen, H. Macrophages in Lung Injury, Repair, and Fibrosis. Cells 2021, 10, 436. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Wu, G.R.; Zhou, Q.; Yue, H.; Rao, L.Z.; Yuan, T.; Mo, B.; Wang, F.X.; Chen, L.M.; et al. MBD2 serves as a viable target against pulmonary fibrosis by inhibiting macrophage M2 program. Sci. Adv. 2021, 7, eabb6075. [Google Scholar] [CrossRef]
- Locatelli, L.; Cadamuro, M.; Spirli, C.; Fiorotto, R.; Lecchi, S.; Morell, C.M.; Popov, Y.; Scirpo, R.; De Matteis, M.; Amenduni, M.; et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology 2016, 63, 965–982. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xiong, M.; Chen, C.; Du, L.; Liu, Z.; Shi, Y.; Zhang, M.; Gong, J.; Song, X.; Xiang, R.; et al. Legumain, an asparaginyl endopeptidase, mediates the effect of M2 macrophages on attenuating renal interstitial fibrosis in obstructive nephropathy. Kidney Int. 2018, 94, 91–101. [Google Scholar] [CrossRef]
- Zaiss, D.M.W.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015, 42, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Scotton, C.J.; Chambers, R.C. Molecular targets in pulmonary fibrosis: The myofibroblast in focus. Chest 2007, 132, 1311–1321. [Google Scholar] [CrossRef]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38(MAPK): Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef]
- Chopra, P.; Kanoje, V.; Semwal, A.; Ray, A. Therapeutic potential of inhaled p38 mitogen-activated protein kinase inhibitors for inflammatory pulmonary diseases. Expert. Opin. Investig. Drugs 2008, 17, 1411–1425. [Google Scholar] [CrossRef]
- Ding, Q.; Gladson, C.L.; Wu, H.; Hayasaka, H.; Olman, M.A. Focal adhesion kinase (FAK)-related non-kinase inhibits myofibroblast differentiation through differential MAPK activation in a FAK-dependent manner. J. Biol. Chem. 2008, 283, 26839–26849. [Google Scholar] [CrossRef]
- Bugg, D.; Bretherton, R.; Kim, P.; Olszewski, E.; Nagle, A.; Schumacher, A.E.; Chu, N.; Gunaje, J.; DeForest, C.A.; Stevens, K.; et al. Infarct Collagen Topography Regulates Fibroblast Fate via p38-Yes-Associated Protein Transcriptional Enhanced Associate Domain Signals. Circ. Res. 2020, 127, 1306–1322. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef]
- Hwang, E.M.; Kim, E.; Yarishkin, O.; Woo, D.H.; Han, K.S.; Park, N.; Bae, Y.; Woo, J.; Kim, D.; Park, M.; et al. A disulphide-linked heterodimer of TWIK-1 and TREK-1 mediates passive conductance in astrocytes. Nat. Commun. 2014, 5, 3227. [Google Scholar] [CrossRef]
- Boutanquoi, P.M.; Burgy, O.; Beltramo, G.; Bellaye, P.S.; Dondaine, L.; Marcion, G.; Pommerolle, L.; Vadel, A.; Spanjaard, M.; Demidov, O.; et al. TRIM33 prevents pulmonary fibrosis by impairing TGF-beta1 signalling. Eur. Respir. J. 2020, 55, 1901346. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | |
|---|---|
| β-actin | F: TTCCAGCCTTCCTTCTTG, R: GGAGCCAGAGCAGTAATC |
| α-SMA | F: CTTCGCTGGTGATGATGCTC, R: GTTGGTGATGATGCCGTGTT |
| Col 1 | F: GAGCGGAGAGTACTGGATCG, R: GCTTCTTTTCCTTGGGGTTC |
| Col 3 | F: GCACAGCAGTCCAACGTAGA, R: TCTCCAAATGGGATCTCTGG |
| FN | F: CCGACCAGAAGTTTGGGTTCT, R: CAATGCGGTACATGACCCCT |
| TREK–1 | F: TTTCCTGGTGGTCGTCCTCTA, R: CTCGGTGGAGTTGACGCAG |
| TGF-β1 | F: TTGCTTCAGCTCCACAGAGA, R: TGGTTGTAGAGGGCAAGGAC |
| IL-1β | F: GCCCATCCTCTGTGACTCAT, R: AGGCCACAGGTATTTTGTCG |
| IL-10 | F: CCCATTCCTCGTCACGATCTC, R: TCAGACTGGTTTGGGATAGGTTT |
| Arg1 | F: CTCCAAGCCAAAGTCCTTAGAG, R: AGGAGCTGTCATTAGGGACATC |
| Ym1 | F: CAGCTCCTCTCAAAAGGATGTG, R: CTTGGGCAAACTGCTATCAGTAT |
| CD206 | F: CTCTGTTCAGCTATTGGACGC, R: CGGAATTTCTGGGATTCAGCTTC |
| CCR2 | F: ATCCACGGCATACTATCAACATC, R: CAAGGCTCACCATCATCGTAG |
| Vimentin | F: CGGCTGCGAGAGAAATTGC, R: CCACTTTCCGTTCAAGGTCAAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Fu, J.; Han, Y.; Feng, D.; Yue, S.; Zhou, Y.; Luo, Z. Two-Pore-Domain Potassium Channel TREK–1 Mediates Pulmonary Fibrosis through Macrophage M2 Polarization and by Direct Promotion of Fibroblast Differentiation. Biomedicines 2023, 11, 1279. https://doi.org/10.3390/biomedicines11051279
Zhang Y, Fu J, Han Y, Feng D, Yue S, Zhou Y, Luo Z. Two-Pore-Domain Potassium Channel TREK–1 Mediates Pulmonary Fibrosis through Macrophage M2 Polarization and by Direct Promotion of Fibroblast Differentiation. Biomedicines. 2023; 11(5):1279. https://doi.org/10.3390/biomedicines11051279
Chicago/Turabian StyleZhang, Yunna, Jiafeng Fu, Yang Han, Dandan Feng, Shaojie Yue, Yan Zhou, and Ziqiang Luo. 2023. "Two-Pore-Domain Potassium Channel TREK–1 Mediates Pulmonary Fibrosis through Macrophage M2 Polarization and by Direct Promotion of Fibroblast Differentiation" Biomedicines 11, no. 5: 1279. https://doi.org/10.3390/biomedicines11051279
APA StyleZhang, Y., Fu, J., Han, Y., Feng, D., Yue, S., Zhou, Y., & Luo, Z. (2023). Two-Pore-Domain Potassium Channel TREK–1 Mediates Pulmonary Fibrosis through Macrophage M2 Polarization and by Direct Promotion of Fibroblast Differentiation. Biomedicines, 11(5), 1279. https://doi.org/10.3390/biomedicines11051279