
Compromised Myelin and Axonal Molecular Organization Following Adult-Onset Sulfatide Depletion
 , , and
, , and 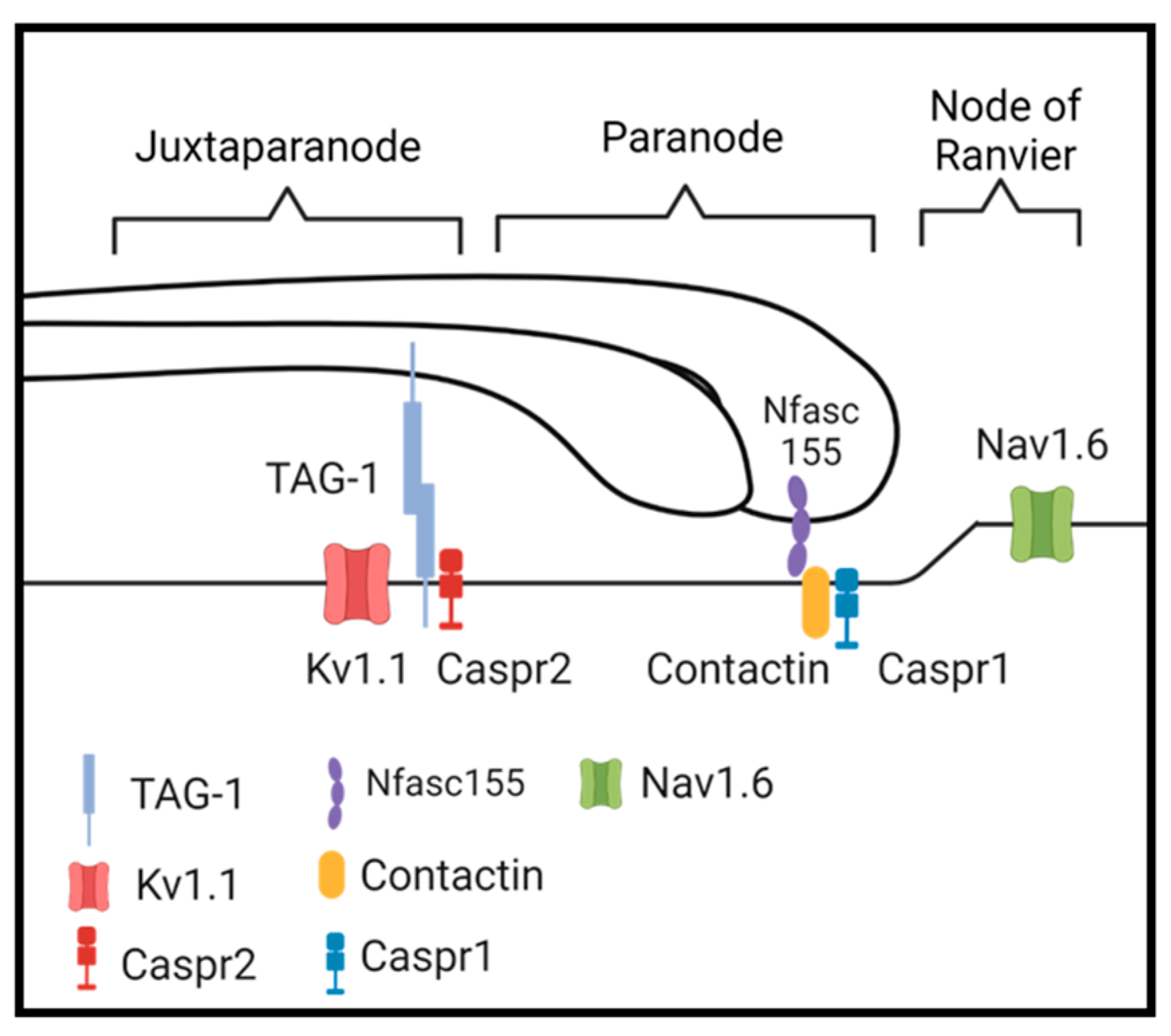{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Polymerase Chain Reaction
- Gal3st1 Forward (5′-GATTGTAGCCTTCCGTATGAACCG-3′)
- Gal3st1 Reverse 1 (5′-CGAACTCAACTCAAAGAGAGCAGG-3′) and
- Gal3st1 Reverse 2 (5′-TAATCTCTGCTCTAACCTGGTCGC-3′).
2.3. Mass Spectrometry
2.4. Rotarod Assessment
2.5. Electron Microscopy
2.6. Measurements/Quantification of Myelin and Axonal Endpoints
2.7. Immunohistochemistry
2.8. In Situ Detergent Extraction
2.9. Western Blot Analysis
2.10. Antibodies Used for Western Blot Analysis
2.11. Statistical Analysis
3. Results
3.1. Confirmation of Gene Ablation and Sulfatide Depletion
3.2. Deficits in Motor Function
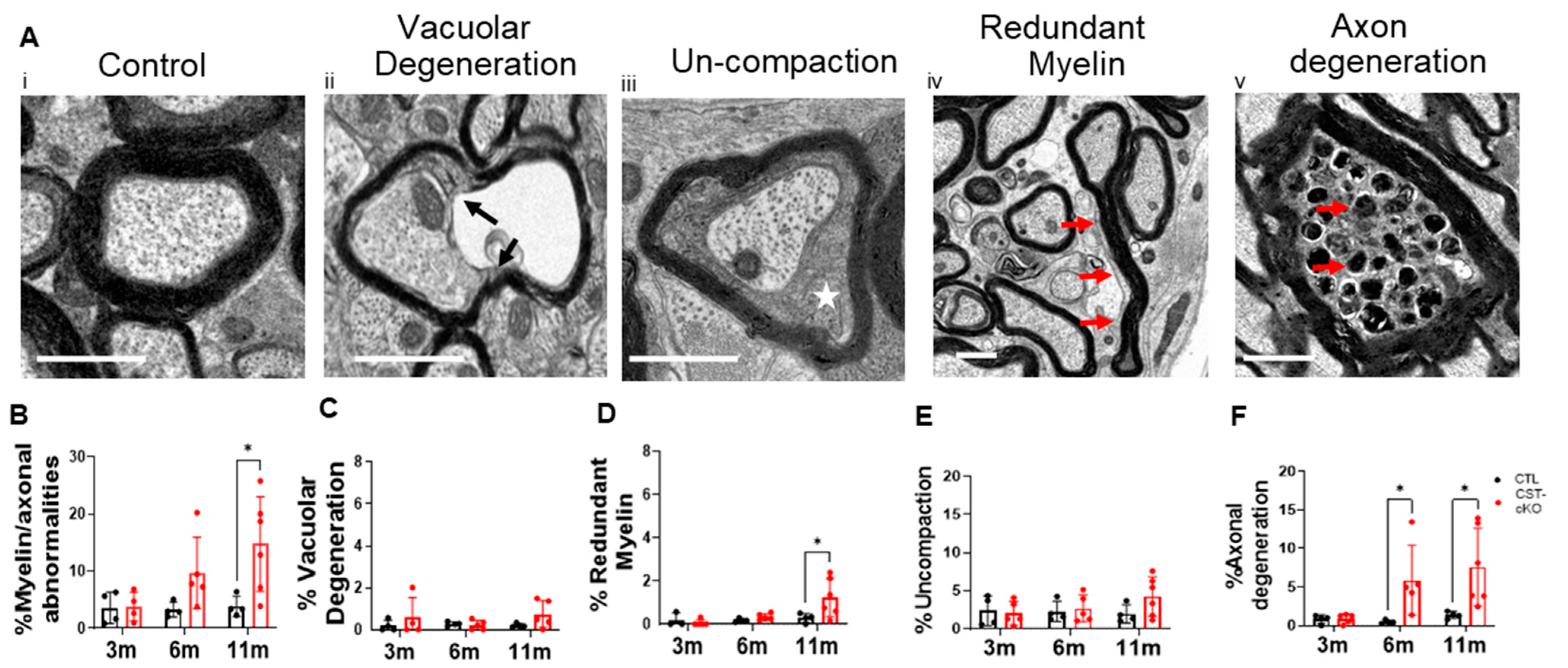
3.3. Myelin Thinning and Axon Ultrastructural Pathology
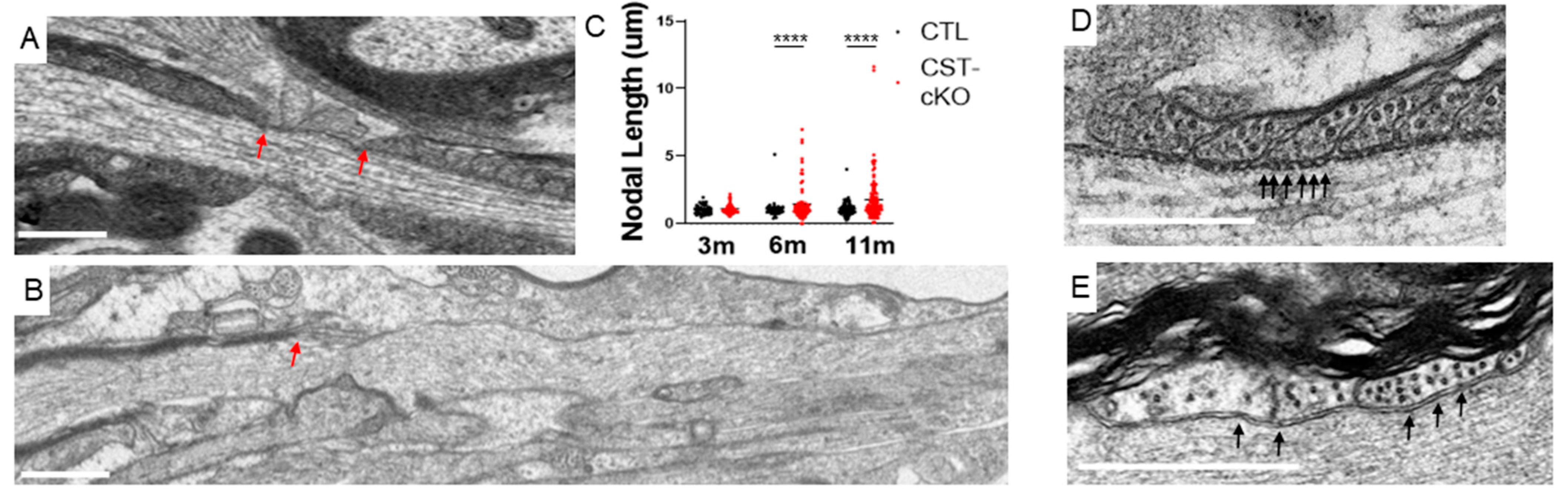
3.4. Compromised Nodal Structure
3.5. Compromised Molecular Organization of the Nodal Regions
3.6. Disruption of Membrane Stability
4. Discussion
4.1. What Proteins Rely on Sulfatide for Myelin Sheath Tethering?
4.2. Consequence of Myelin Protein Instability
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Caspr1 | Contactin-Associated protein 1 |
| cKO | conditional knock out |
| CNP | 2′:3′-cyclic nucleotide 3′-phosphodiesterase |
| CNS | Central Nervous System |
| CST | Ceramide Sulfotransferase |
| CTL | Control |
| EM | Electron Microscopy |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| KO | Knock-Out |
| Kv1.1 | Voltage-Gated Potassium Channel Kv1.1 |
| m | month |
| MAG | Myelin-Associated Glycoprotein |
| MBP | Myelin Basic Protein |
| MOG | Myelin Oligodendrocyte Glycoprotein |
| MS | Multiple Sclerosis |
| Nav1.6 | Voltage-gated sodium channel Nav1.6 |
| NAWM | Normal Appearing White Matter |
| Nfasc155 | Neurofascin 155 |
| Nfasc155H | Neurofascin 155 high |
| Nfasc155L | Neurofascin 155 L |
| PI | Post-Injection |
| PLP | Proteolipid Protein |
| Sulfatide | 3-O-sulfogalactosylceramide |
| TAG-1 | Transient Axonal Glycoproteintype-1 |
Appendix A

References
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.-W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Nave, K.A. Myelin dynamics: Protecting and shaping neuronal functions. Curr. Opin. Neurobiol. 2017, 47, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef]
- Moore, J.W.; Joyner, R.W.; Brill, M.H.; Waxman, S.D.; Najar-Joa, M. Simulations of conduction in uniform myelinated fibers. Relative sensitivity to changes in nodal and internodal parameters. Biophys. J. 1978, 21, 147–160. [Google Scholar] [CrossRef]
- Hayashi, A.; Kaneko, N.; Tomihira, C.; Baba, H. Sulfatide decrease in myelin influences formation of the paranodal axo-glial junction and conduction velocity in the sciatic nerve. Glia 2013, 61, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.Y.; Zhou, L.; Zhang, C.-L.; Messing, A. Analysis of potassium channel functions in mammalian axons by gene knockouts. J. Neurocytol. 1999, 28, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Kister, A.; Kister, I. Overview of myelin, major myelin lipids, and myelin-associated proteins. Front. Chem. 2022, 10, 1041961. [Google Scholar] [CrossRef]
- Morell, P.; Quarles, R.H. Characteristic Composition of Myelin. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Published Online; 1999. Available online: https://www.ncbi.nlm.nih.gov/books/NBK28221/ (accessed on 12 August 2019).
- O’Brien, J.S.; Sampson, E.L. Lipid composition of the normal human brain: Gray matter, white matter, and myelin. J. Lipid Res. 1965, 6, 537–544. [Google Scholar] [CrossRef]
- Grassi, S.; Prioni, S.; Cabitta, L.; Aureli, M.; Sonnino, S.; Prinetti, A. The Role of 3-O-Sulfogalactosylceramide, Sulfatide, in the Lateral Organization of Myelin Membrane. Neurochem. Res. 2016, 41, 130–143. [Google Scholar] [CrossRef]
- Bazan, N.G.; Colangelo, V.; Lukiw, W.J. Prostaglandins and other lipid mediators in Alzheimer’s disease. Prostaglandins Other Lipid Mediat. 2002, 68–69, 197–210. [Google Scholar] [CrossRef]
- Quarles, R.H.; Macklin, W.B.; Morell, P. Myelin Formation, Structure and Biochemistry. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects; Academic Press: Cambridge, MA, USA, 2006; p. 21. [Google Scholar]
- Zöller, I.; Meixner, M.; Hartmann, D.; Büssow, H.; Meyer, R.; Gieselmann, V.; Eckhardt, M. Absence of 2-Hydroxylated Sphingolipids Is Compatible with Normal Neural Development But Causes Late-Onset Axon and Myelin Sheath Degeneration. J. Neurosci. 2008, 28, 9741–9754. [Google Scholar] [CrossRef] [PubMed]
- Dupree, J.L.; Pomicter, A.D. Myelin, DIGs, and membrane rafts in the central nervous system. Prostaglandins Other Lipid Mediat. 2010, 91, 118–129. [Google Scholar] [CrossRef]
- Traka, M.; Dupree, J.L.; Popko, B.; Karagogeos, D. The neuronal adhesion protein TAG-1 is expressed by Schwann cells and oligodendrocytes and is localized to the juxtaparanodal region of myelinated fibers. J. Neurosci. 2002, 22, 3016–3024. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.; Goutebroze, L.; Denisenko, N.; Bessa, M.; Nifli, A.-P.; Havaki, S.; Iwakura, Y.; Fukamauchi, F.; Watanabe, K.; Soliven, B.; et al. Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J. Cell Biol. 2003, 162, 1161–1172. [Google Scholar] [CrossRef]
- Tsiotra, P.C.; Theodorakis, K.; Papamatheakis, J.; Karagogeos, D. The Fibronectin Domains of the Neural Adhesion Molecule TAX-1 Are Necessary and Sufficient for Homophilic Binding. J. Biol. Chem. 1996, 271, 29216–29222. [Google Scholar] [CrossRef] [PubMed]
- Kunz, B.; Lierheimer, R.; Rader, C.; Spirig, M.; Ziegler, U.; Sonderegger, P. Axonin-1/TAG-1 Mediates Cell-Cell Adhesion by a Cis-assisted Trans-interaction. J. Biol. Chem. 2002, 277, 4551–4557. [Google Scholar] [CrossRef]
- Poliak, S.; Salomon, D.; Elhanany, H.; Sabanay, H.; Kiernan, B.; Pevny, L.; Stewart, C.L.; Xu, X.; Chiu, S.-Y.; Shrager, P.; et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 2003, 162, 1149–1160. [Google Scholar] [CrossRef]
- Hivert, B.; Pinatel, D.; Labasque, M.; Tricaud, N.; Goutebroze, L.; Faivre-Sarrailh, C. Assembly of juxtaparanodes in myelinating DRG culture: Differential clustering of the Kv1/Caspr2 complex and scaffolding protein 4.1B. Glia 2016, 64, 840–852. [Google Scholar] [CrossRef]
- Tait, S.; Gunn-Moore, F.; Collinson, J.M.; Huang, J.; Lubetzki, C.; Pedraza, L.; Sherman, D.L.; Colman, D.R.; Brophy, P.J. An Oligodendrocyte Cell Adhesion Molecule at the Site of Assembly of the Paranodal Axo-Glial Junction. J. Cell Biol. 2000, 150, 657–666. [Google Scholar] [CrossRef]
- Chang, K.J.; Zollinger, D.R.; Susuki, K.; Sherman, D.L.; Makara, M.A.; Brophy, P.J.; Cooper, E.C.; Bennett, V.; Mohler, P.J.; Rasband, M.N. Glial ankyrins facilitate paranodal axoglial junction assembly. Nat. Neurosci. 2014, 17, 1673–1681. [Google Scholar] [CrossRef]
- Rosenbluth, J.; Petzold, C.; Peles, E. Dependence of paranodal junctional gap width on transverse bands. J. Comp. Neurol. 2012, 520, 2774–2784. [Google Scholar] [CrossRef] [PubMed]
- Faivre-Sarrailh, C. Molecular organization and function of vertebrate septate-like junctions. Biochim. Biophys. Acta (BBA) Biomembr. 2020, 1862, 183211. [Google Scholar] [CrossRef]
- Rhodes, K.J.; Strassle, B.W.; Monaghan, M.M.; Bekele-Arcuri, Z.; Matos, M.F.; Trimmer, J.S. Association and Colocalization of the Kvβ1 and Kvβ2 β-Subunits with Kv1 α-Subunits in Mammalian Brain K+Channel Complexes. J. Neurosci. 1997, 17, 8246–8258. [Google Scholar] [CrossRef]
- Pomicter, A.D.; Shroff, S.M.; Fuss, B.; Sato-Bigbee, C.; Brophy, P.J.; Rasband, M.N.; Bhat, M.A.; Dupree, J.L. Novel forms of neurofascin 155 in the central nervous system: Alterations in paranodal disruption models and multiple sclerosis. Brain 2010, 133, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Pomicter, A.D.; DeLoyht, J.M.; Hackett, A.R.; Purdie, N.; Sato-Bigbee, C.; Henderson, S.C.; DuPree, J.L. Nfasc155H and MAG are specifically susceptible to detergent extraction in the absence of the myelin sphingolipid sulfatide. Neurochem. Res. 2013, 38, 2490–2502. [Google Scholar] [CrossRef] [PubMed]
- Dupree, J.L.; Coetzee, T.; Suzuki, K.; Popko, B. Myelin abnormalities in mice deficient in galactocerebroside and sulfatide. J. Neurocytol. 1998, 27, 649–659. [Google Scholar] [CrossRef]
- Dupree, J.L.; Girault, J.A.; Popko, B. Axo-glial interactions regulate the localization of axonal paranodal proteins. J. Cell Biol. 1999, 147, 1145–1152. [Google Scholar] [CrossRef]
- Marcus, J.; Honigbaum, S.; Shroff, S.; Honke, K.; Rosenbluth, J.; Dupree, J.L. Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia 2006, 53, 372–381. [Google Scholar] [CrossRef]
- Bieberich, E. Sphingolipids and lipid rafts: Novel concepts and methods of analysis. Chem. Phys. Lipids 2018, 216, 114–131. [Google Scholar] [CrossRef]
- Taylor, C.M.; Coetzee, T.; Pfeiffer, S.E. Detergent-insoluble glycosphingolipid/cholesterol microdomains of the myelin membrane. J. Neurochem. 2002, 81, 993–1004. [Google Scholar] [CrossRef]
- Schafer, D.P. Does Paranode Formation and Maintenance Require Partitioning of Neurofascin 155 into Lipid Rafts? J. Neurosci. 2004, 24, 3176–3185. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Pfeiffer, S.E. Myelin glycosphingolipid/cholesterol-enriched microdomains selectively sequester the non-compact myelin proteins CNP and MOG. J. Neurocytol. 1999, 28, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Rohrer, B.; Castello-Serrano, I.; Chan, S.H.; Wang, H.-Y.; Shurer, C.R.; Levental, K.R.; Levental, I. Rab3 mediates a pathway for endocytic sorting and plasma membrane recycling of ordered microdomains. Proc. Natl. Acad. Sci. USA 2023, 120, e2207461120. [Google Scholar] [CrossRef]
- Grassi, S.; Giussani, P.; Mauri, L.; Prioni, S.; Sonnino, S.; Prinetti, A. Lipid rafts and neurodegeneration: Structural and functional roles in physiologic aging and neurodegenerative diseases: Thematic Review Series: Biology of Lipid Rafts. J. Lipid Res. 2020, 61, 636–654. [Google Scholar] [CrossRef]
- Isik, O.A.; Cizmecioglu, O. Rafting on the Plasma Membrane: Lipid Rafts in Signaling and Disease. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2023. [Google Scholar] [CrossRef]
- Arvanitis, D.N.; Min, W.; Gong, Y.; Heng, Y.M.; Boggs, J.M. Two types of detergent-insoluble, glycosphingolipid/cholesterol-rich membrane domains from isolated myelin. J. Neurochem. 2005, 94, 1696–1710. [Google Scholar] [CrossRef]
- Yang, L.J.; Zeller, C.B.; Shaper, N.L.; Kiso, M.; Hasegawa, A.; Shapiro, R.E.; Schnaar, R.L. Gangliosides are neuronal ligands for myelin-associated glycoprotein. Proc. Natl. Acad. Sci. USA 1996, 93, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Charles, P.; Tait, S.; Faivre-Sarrailh, C.; Barbin, G.; Gunn-Moore, F.; Denisenko-Nehrbass, N.; Guennoc, A.-M.; Girault, J.-A.; Brophy, P.J.; Lubetzki, C. Neurofascin Is a Glial Receptor for the Paranodin/Caspr-Contactin Axonal Complex at the Axoglial Junction. Curr. Biol. 2002, 12, 217–220. [Google Scholar] [CrossRef]
- Schnaar, R.L. Brain gangliosides in axon-myelin stability and axon regeneration. FEBS Lett. 2010, 584, 1741–1747. [Google Scholar] [CrossRef]
- Janssen, B.J.C. Inside-out or outside-in, a new factor in MAG-mediated signaling in the nervous system: An Editorial for “High-affinity heterotetramer formation between the large myelin-associated glycoprotein and the dynein light chain DYNLL1” on page 764. J. Neurochem. 2018, 147, 712–714. [Google Scholar] [CrossRef]
- Honke, K.; Hirahara, Y.; Dupree, J.; Suzuki, K.; Popko, B.; Fukushima, K.; Fukushima, J.; Nagasawa, T.; Yoshida, N.; Wada, Y.; et al. Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc. Natl. Acad. Sci. USA 2002, 99, 4227–4232. [Google Scholar] [CrossRef]
- Qiu, S.; Palavicini, J.P.; Wang, J.; Gonzalez, N.S.; He, S.; Dustin, E.; Zou, C.; Ding, L.; Bhattacharjee, A.; Van Skike, C.E.; et al. Adult-onset CNS myelin sulfatide deficiency is sufficient to cause Alzheimer’s disease-like neuroinflammation and cognitive impairment. Mol. Neurodegener. 2021, 16, 64. [Google Scholar] [CrossRef]
- Palavicini, J.P.; Ding, L.; Pan, M.; Qiu, S.; Wang, H.; Shen, Q.; Dupree, J.L.; Han, X. Sulfatide Deficiency, an Early Alzheimer’s Lipidomic Signature, Causes Brain Ventricular Enlargement in the Absence of Classical Neuropathological Hallmarks. Int. J. Mol. Sci. 2022, 24, 233. [Google Scholar] [CrossRef] [PubMed]
- Doerflinger, N.H.; Macklin, W.B.; Popko, B. Inducible site-specific recombination in myelinating cells. Genesis 2003, 35, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Norton, W.T.; Cammer, W. Isolation and Characterization of Myelin. In Myelin; Morell, P., Ed.; Springer: New York, NY, USA, 1984; pp. 147–195. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Dupree, J.L.; Coetzee, T.; Blight, A.; Suzuki, K.; Popko, B. Myelin Galactolipids Are Essential for Proper Node of Ranvier Formation in the CNS. J. Neurosci. 1998, 18, 1642–1649. [Google Scholar] [CrossRef]
- Mason, J.L.; Langaman, C.; Morell, P.; Suzuki, K.; Matsushima, G.K. Episodic demyelination and subsequent remyelination within the murine central nervous system: Changes in axonal calibre. Neuropathol. Appl. Neurobiol. 2001, 27, 50–58. [Google Scholar] [CrossRef]
- Duncan, G.J.; Simkins, T.J.; Emery, B. Neuron-Oligodendrocyte Interactions in the Structure and Integrity of Axons. Front. Cell Dev. Biol. 2021, 9, 653101. [Google Scholar] [CrossRef]
- Dupree, J.L.; Feinstein, D.L. Influence of diet on axonal damage in the EAE mouse model of multiple sclerosis. J. Neuroimmunol. 2018, 322, 9–14. [Google Scholar] [CrossRef]
- Allt, G. Repair of segmental dehyelination in peripheral nerves: An electron microscope study. Brain 1969, 92, 639–646. [Google Scholar] [CrossRef]
- Clark, K.C.; Josephson, A.; Benusa, S.D.; Hartley, R.K.; Baer, M.; Thummala, S.; Joslyn, M.; Sword, B.A.; Elford, H.; Oh, U.; et al. Compromised axon initial segment integrity in EAE is preceded by microglial reactivity and contact. Glia 2016, 64, 1190–1209. [Google Scholar] [CrossRef]
- Benusa, S.D.; George, N.M.; Sword, B.A.; DeVries, G.H.; Dupree, J.L. Acute neuroinflammation induces AIS structural plasticity in a NOX2-dependent manner. J. Neuroinflammation 2017, 14, 116. [Google Scholar] [CrossRef] [PubMed]
- Duncan, I.D.; Radcliff, A.B.; Heidari, M.; Kidd, G.; August, B.K.; Wierenga, L.A. The adult oligodendrocyte can participate in remyelination. Proc. Natl. Acad. Sci. USA 2018, 115, E11807–E11816. [Google Scholar] [CrossRef]
- Ishibashi, T.; Dupree, J.L.; Ikenaka, K.; Hirahara, Y.; Honke, K.; Peles, E.; Popko, B.; Suzuki, K.; Nishino, H.; Baba, H. A Myelin Galactolipid, Sulfatide, Is Essential for Maintenance of Ion Channels on Myelinated Axon But Not Essential for Initial Cluster Formation. J. Neurosci. 2002, 22, 6507–6514. [Google Scholar] [CrossRef] [PubMed]
- Caritá, A.C.; Mattei, B.; Domingues, C.C.; de Paula, E.; Riske, K.A. Effect of Triton X-100 on Raft-Like Lipid Mixtures: Phase Separation and Selective Solubilization. Langmuir 2017, 33, 7312–7321. [Google Scholar] [CrossRef] [PubMed]
- Rodi, P.M.; Bocco Gianello, M.D.; Corregido, M.C.; Gennaro, A.M. Comparative study of the interaction of CHAPS and Triton X-100 with the erythrocyte membrane. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 859–866. [Google Scholar] [CrossRef]
- Ozgen, H.; Baron, W.; Hoekstra, D.; Kahya, N. Oligodendroglial membrane dynamics in relation to myelin biogenesis. Cell. Mol. Life Sci. 2016, 73, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Gielen, E.; Baron, W.; Vandeven, M.; Steels, P.; Hoekstra, D.; Ameloot, M. Rafts in oligodendrocytes: Evidence and structure–function relationship. Glia 2006, 54, 499–512. [Google Scholar] [CrossRef]
- Pronker, M.F.; Lemstra, S.; Snijder, J.; Heck, A.J.R.; Thies-Weesie, D.M.E.; Pasterkamp, R.J.; Janssen, B.J.C. Structural basis of myelin-associated glycoprotein adhesion and signalling. Nat. Commun. 2016, 7, 13584. [Google Scholar] [CrossRef] [PubMed]
- Cawley, J.L.; Jordan, L.R.; Wittenberg, N.J. Detection and Characterization of Vesicular Gangliosides Binding to Myelin-Associated Glycoprotein on Supported Lipid Bilayers. Anal. Chem. 2021, 93, 1185–1192. [Google Scholar] [CrossRef]
- Hinman, J.D.; Chen, C.D.; Oh, S.Y.; Hollander, W.; Abraham, C.R. Age-dependent accumulation of ubiquitinated 2′,3′-cyclic nucleotide 3′-phosphodiesterase in myelin lipid rafts. Glia 2008, 56, 118–133. [Google Scholar] [CrossRef]
- Braun, P.E.; Sandillon, F.; Edwards, A.; Matthieu, J.M.; Privat, A. Immunocytochemical localization by electron microscopy of 2′3′-cyclic nucleotide 3′-phosphodiesterase in developing oligodendrocytes of normal and mutant brain. J. Neurosci. 1988, 8, 3057–3066. [Google Scholar] [CrossRef]
- Brunner, C.; Lassmann, H.; Waehneldt, T.V.; Matthieu, J.M.; Linington, C. Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2′,3′-cyclic nucleotide 3′-phosphodiesterase in the CNS of adult rats. J. Neurochem. 1989, 52, 296–304. [Google Scholar] [CrossRef]
- Maier, O.; Hoekstra, D.; Baron, W. Polarity Development in Oligodendrocytes: Sorting and Trafficking of Myelin Components. J. Mol. Neurosci. 2008, 35, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Bernier, L.; Andrews, S.B.; Colman, D.R. Cellular and Subcellular Distribution of 2′,3′-Cyclic Nucleotide 3′-Phosphodiesterase and Its mRNA in the Rat Central Nervous System. J. Neurochem. 1988, 51, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, H.C.; Sprinkle, T.J.; Agrawal, D. In vivo phosphorylation of 2′,3′-cyclic nucleotide 3′-phosphohydrolase (CNP): CNP in brain myelin is phosphorylated by forskolin- and phorbol ester-sensitive protein kinases. Neurochem. Res. 1994, 19, 721–728. [Google Scholar] [CrossRef]
- Braun, P.E.; De Angelis, D.; Shtybel, W.W.; Bernier, L. Isoprenoid modification permits 2′,3′-cyclic nucleotide 3′-phosphodiesterase to bind to membranes. J. Neurosci. Res. 1991, 30, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Dyer, C.A.; Benjamins, J.A. Organization of oligodendroglial membrane sheets. I: Association of myelin basic protein and 2′,3′-cyclic nucleotide 3′-phosphohydrolase with cytoskeleton. J. Neurosci. Res. 1989, 24, 201–211. [Google Scholar] [CrossRef]
- Knapp, P.E.; Skoff, R.P.; Sprinkle, T.J. Differential expression of galactocerebroside, myelin basic protein, and 2′,3′-cyclic nucleotide 3′-phosphohydrolase during development of oligodendrocytes in vitro. J. Neurosci. Res. 1988, 21, 249–259. [Google Scholar] [CrossRef]
- Wilson, R.; Brophy, P.J. Role for the oligodendrocyte cytoskeleton in myelination. J. Neurosci. Res. 1989, 22, 439–448. [Google Scholar] [CrossRef]
- Snaidero, N.; Velte, C.; Myllykoski, M.; Raasakka, A.; Ignatev, A.; Werner, H.B.; Erwig, M.S.; Möbius, W.; Kursula, P.; Nave, K.-A.; et al. Antagonistic Functions of MBP and CNP Establish Cytosolic Channels in CNS Myelin. Cell Rep. 2017, 18, 314–323. [Google Scholar] [CrossRef]
- Rasband, M.N.; Tayler, J.; Kaga, Y.; Yang, Y.; Lappe-Siefke, C.; Nave, K.-A.; Bansal, R. CNP is required for maintenance of axon–glia interactions at nodes of Ranvier in the CNS. Glia 2005, 50, 86–90. [Google Scholar] [CrossRef]
- Lappe-Siefke, C.; Goebbels, S.; Gravel, M.; Nicksch, E.; Lee, J.; Braun, P.E.; Griffiths, I.R.; Nave, K.-A. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 2003, 33, 366–374. [Google Scholar] [CrossRef]
- Edgar, J.M.; McLaughlin, M.; Werner, H.B.; McCulloch, M.C.; Barrie, J.A.; Brown, A.; Faichney, A.B.; Snaidero, N.; Nave, K.-A.; Griffiths, I.R. Early ultrastructural defects of axons and axon–glia junctions in mice lacking expression of Cnp1. Glia 2009, 57, 1815–1824. [Google Scholar] [CrossRef]
- Brown, D.A.; Rose, J.K. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 1992, 68, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Kinoshita, M.; Matsumori, N. Impact of sphingomyelin acyl chain heterogeneity upon properties of raft-like membranes. Biochim. Biophys. Acta Biomembr. 2022, 1864, 184036. [Google Scholar] [CrossRef] [PubMed]
- Pillai, A.M.; Thaxton, C.; Pribisko, A.L.; Cheng, J.G.; Dupree, J.L.; Bhat, M.A. Spatiotemporal Ablation of Myelinating Glia-Specific Neurofascin (NfascNF155) in Mice Reveals Gradual Loss of Paranodal Axoglial Junctions and Concomitant Disorganization of Axonal Domains. J. Neurosci. Res. 2009, 87, 1773–1793. [Google Scholar] [CrossRef] [PubMed]
- Thaxton, C.; Pillai, A.M.; Pribisko, A.L.; Dupree, J.L.; Bhat, M.A. Nodes of Ranvier act as barriers to restrict invasion of flanking paranodal domains in myelinated axons. Neuron 2011, 69, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Mierzwa, A.J.; Arevalo, J.C.; Schiff, R.; Chao, M.V.; Rosenbluth, J. Role of transverse bands in maintaining paranodal structure and axolemmal domain organization in myelinated nerve fibers: Effect on longevity in dysmyelinated mutant mice. J. Comp. Neurol. 2010, 518, 2841–2853. [Google Scholar] [CrossRef]
- Susuki, K.; Chang, K.J.; Zollinger, D.R.; Liu, Y.; Ogawa, Y.; Eshed-Eisenbach, Y.; Dours-Zimmermann, M.T.; Oses-Prieto, J.A.; Burlingame, A.L.; Seidenbecher, C.I.; et al. Three mechanisms assemble central nervous system nodes of Ranvier. Neuron 2013, 78, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Amor, V.; Feinberg, K.; Eshed-Eisenbach, Y.; Vainshtein, A.; Frechter, S.; Grumet, M.; Rosenbluth, J.; Peles, E. Long-Term Maintenance of Na+ Channels at Nodes of Ranvier Depends on Glial Contact Mediated by Gliomedin and NrCAM. J. Neurosci. 2014, 34, 5089–5098. [Google Scholar] [CrossRef]
- Susuki, K.; Zollinger, D.R.; Chang, K.J.; Zhang, C.; Huang, C.Y.-M.; Tsai, C.-R.; Galiano, M.R.; Liu, Y.; Benusa, S.; Yermakov, L.M.; et al. Glial βII Spectrin Contributes to Paranode Formation and Maintenance. J. Neurosci. 2018, 38, 6063–6075. [Google Scholar] [CrossRef] [PubMed]
- Salzer, J.L. Polarized domains of myelinated axons. Neuron 2003, 40, 297–318. [Google Scholar] [CrossRef] [PubMed]
- Arancibia-Cárcamo, I.L.; Ford, M.C.; Cossell, L.; Ishida, K.; Tohyama, K.; Attwell, D. Node of Ranvier length as a potential regulator of myelinated axon conduction speed. Elife 2017, 6, e23329. [Google Scholar] [CrossRef] [PubMed]
- Lubetzki, C.; Sol-Foulon, N.; Desmazières, A. Nodes of Ranvier during development and repair in the CNS. Nat. Rev. Neurol. 2020, 16, 426–439. [Google Scholar] [CrossRef]
- Yu, F.; Fan, Q.; Tian, Q.; Ngamsombat, C.; Machado, N.; Bireley, J.; Russo, A.; Nummenmaa, A.; Witzel, T.; Wald, L.; et al. Imaging G-Ratio in Multiple Sclerosis Using High-Gradient Diffusion MRI and Macromolecular Tissue Volume. AJNR Am. J. Neuroradiol. 2019, 40, 1871–1877. [Google Scholar] [CrossRef]
- Howell, O.W.; Palser, A.; Polito, A.; Melrose, S.; Zonta, B.; Scheiermann, C.; Vora, A.J.; Brophy, P.J.; Reynolds, R. Disruption of neurofascin localization reveals early changes preceding demyelination and remyelination in multiple sclerosis. Brain 2006, 129, 3173–3185. [Google Scholar] [CrossRef]
- Suzuki, K.; Andrews, J.; Waltz, J.; Terry, R. Ultrastructural studies of multiple sclerosis. Lab. Investig. 1969, 20, 444–454. [Google Scholar]
- Smith, K.J. Sodium Channels and Multiple Sclerosis: Roles in Symptom Production, Damage and Therapy. Brain Pathol. 2007, 17, 230–242. [Google Scholar] [CrossRef]
- Freeman, S.A.; Desmazières, A.; Fricker, D.; Lubetzki, C.; Sol-Foulon, N. Mechanisms of sodium channel clustering and its influence on axonal impulse conduction. Cell. Mol. Life Sci. 2016, 73, 723–735. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dustin, E.; Suarez-Pozos, E.; Stotesberry, C.; Qiu, S.; Palavicini, J.P.; Han, X.; Dupree, J.L. Compromised Myelin and Axonal Molecular Organization Following Adult-Onset Sulfatide Depletion. Biomedicines 2023, 11, 1431. https://doi.org/10.3390/biomedicines11051431
Dustin E, Suarez-Pozos E, Stotesberry C, Qiu S, Palavicini JP, Han X, Dupree JL. Compromised Myelin and Axonal Molecular Organization Following Adult-Onset Sulfatide Depletion. Biomedicines. 2023; 11(5):1431. https://doi.org/10.3390/biomedicines11051431
Chicago/Turabian StyleDustin, Elizabeth, Edna Suarez-Pozos, Camryn Stotesberry, Shulan Qiu, Juan Pablo Palavicini, Xianlin Han, and Jeffrey L. Dupree. 2023. "Compromised Myelin and Axonal Molecular Organization Following Adult-Onset Sulfatide Depletion" Biomedicines 11, no. 5: 1431. https://doi.org/10.3390/biomedicines11051431
APA StyleDustin, E., Suarez-Pozos, E., Stotesberry, C., Qiu, S., Palavicini, J. P., Han, X., & Dupree, J. L. (2023). Compromised Myelin and Axonal Molecular Organization Following Adult-Onset Sulfatide Depletion. Biomedicines, 11(5), 1431. https://doi.org/10.3390/biomedicines11051431