
Design of a Novel and Potent Multi-Epitope Chimeric Vaccine against Human Papillomavirus (HPV): An Immunoinformatics Approach
Abstract
:1. Introduction
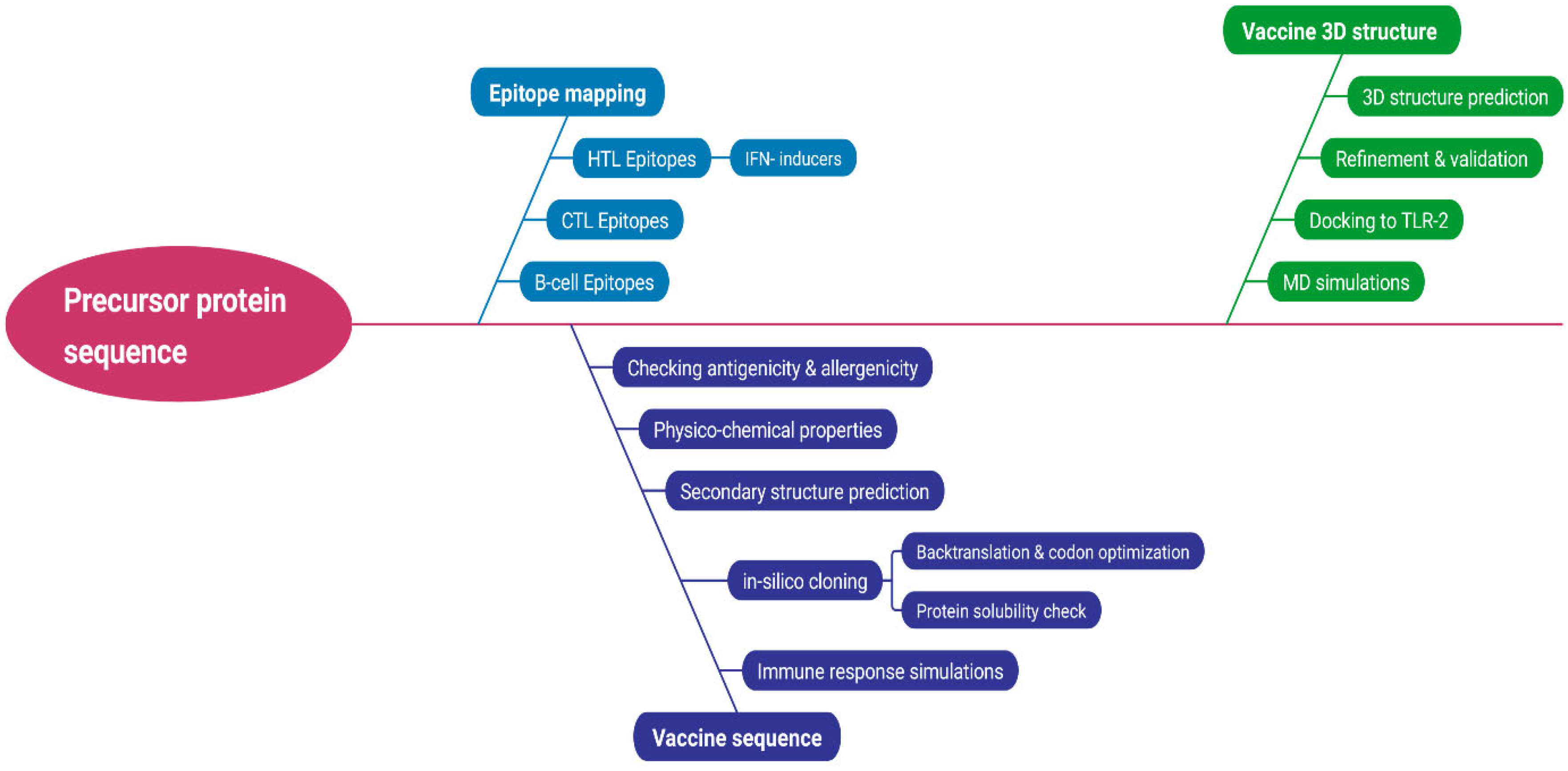
2. Methodology
2.1. Sequence Retrieval and Prioritization
2.2. Epitope Mapping
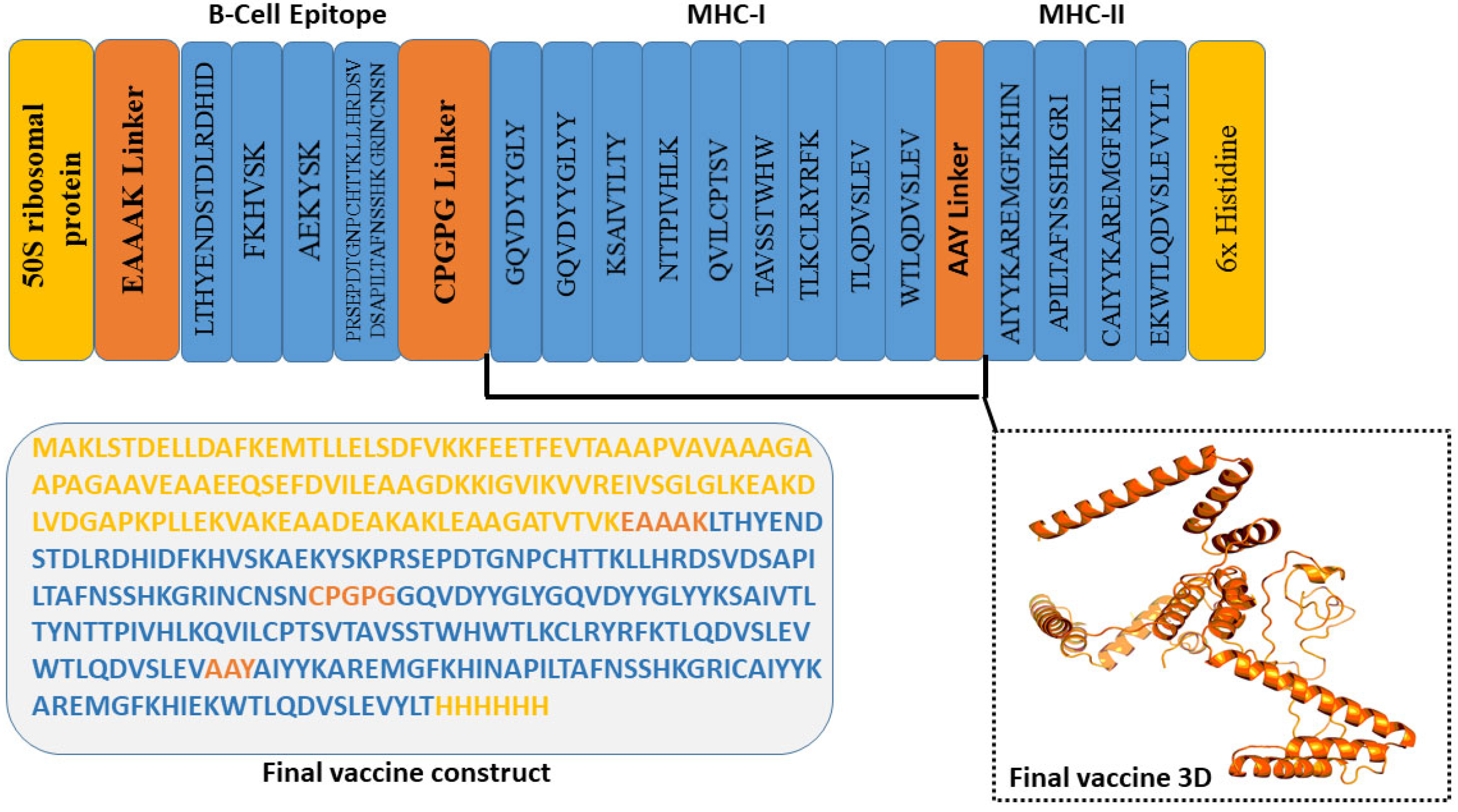
2.3. Vaccine Construct Design and Structure Prediction
2.4. Molecular Docking
2.5. Systematic Analysis of the Construct
2.6. Codon Optimization for Expression Analysis of the Vaccine Peptide
2.7. Immune Response Simulation
3. Result and Analysis
3.1. Sequence Retrieval, Phylogenetic Analysis, and Prioritization
3.2. Physicochemical Characterization
3.3. Linear B-Cell Epitope
3.4. Prediction of Helper T Lymphocytes (HTL) Epitope
3.5. Prediction of Linear B Cell Lymphocyte (LBL) Epitope
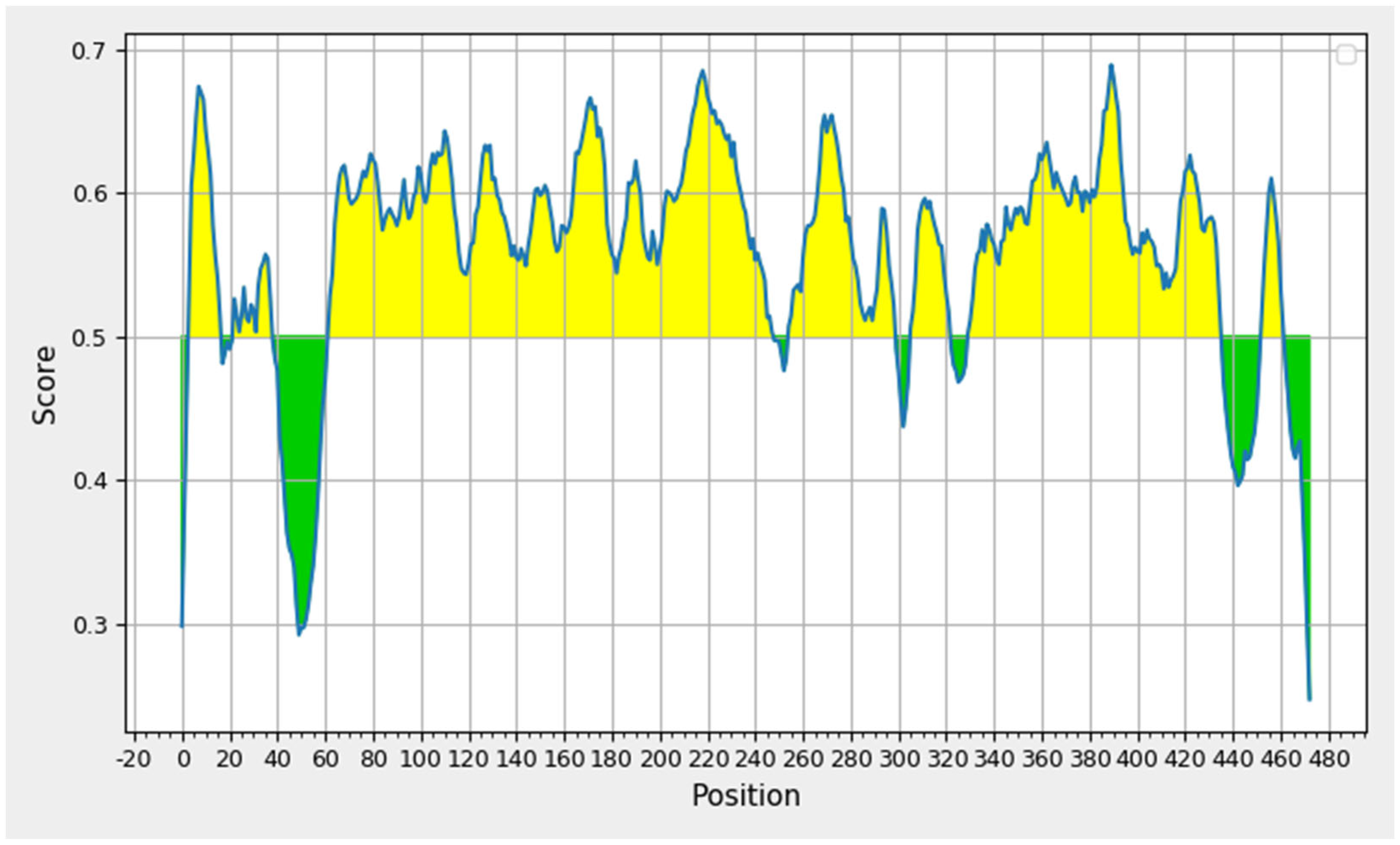
3.6. Secondary Structure Prediction and Population Coverage
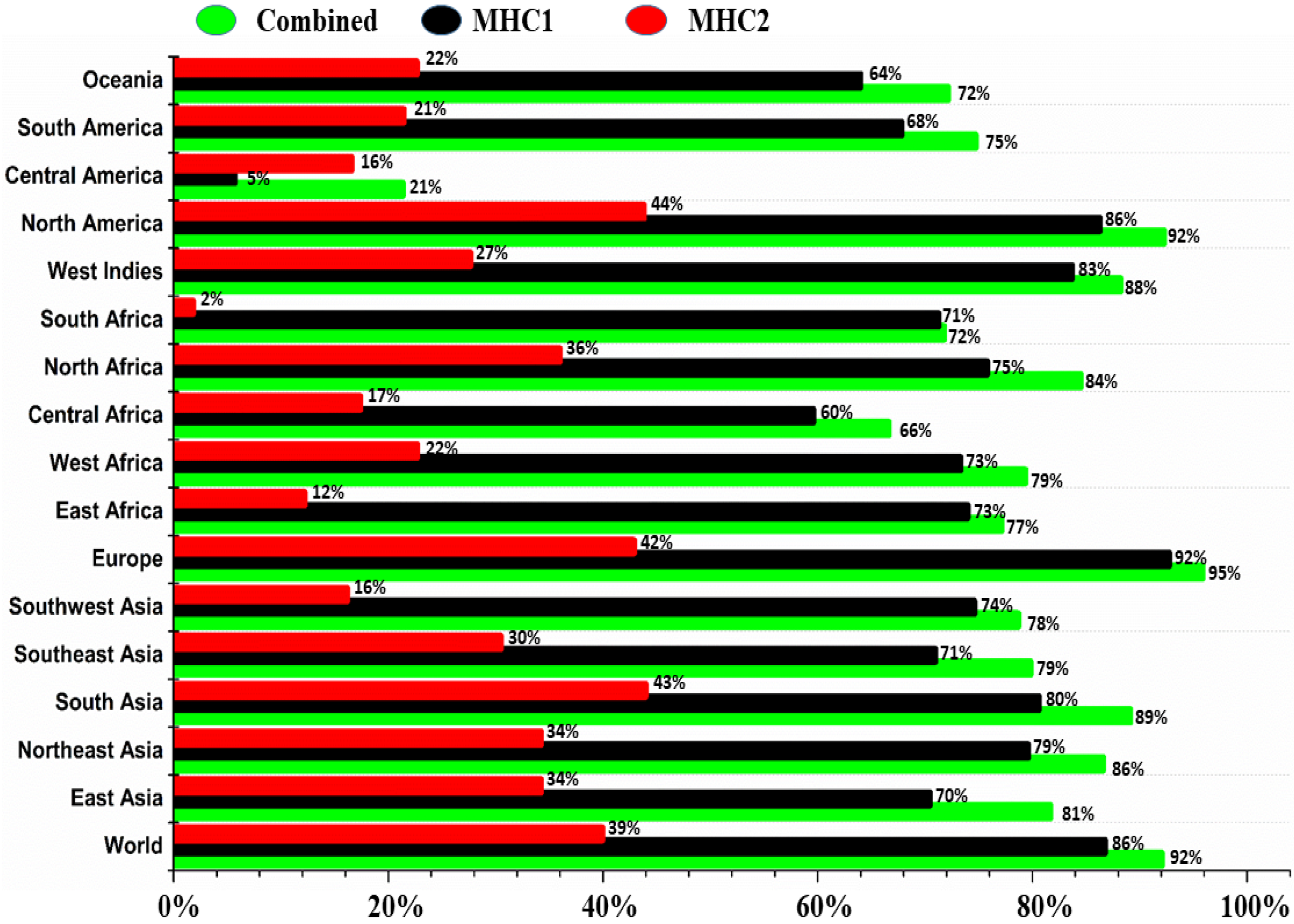
3.7. Population Coverage Analysis
3.8. The Proposed MEV Exhibited Admirable Qualities
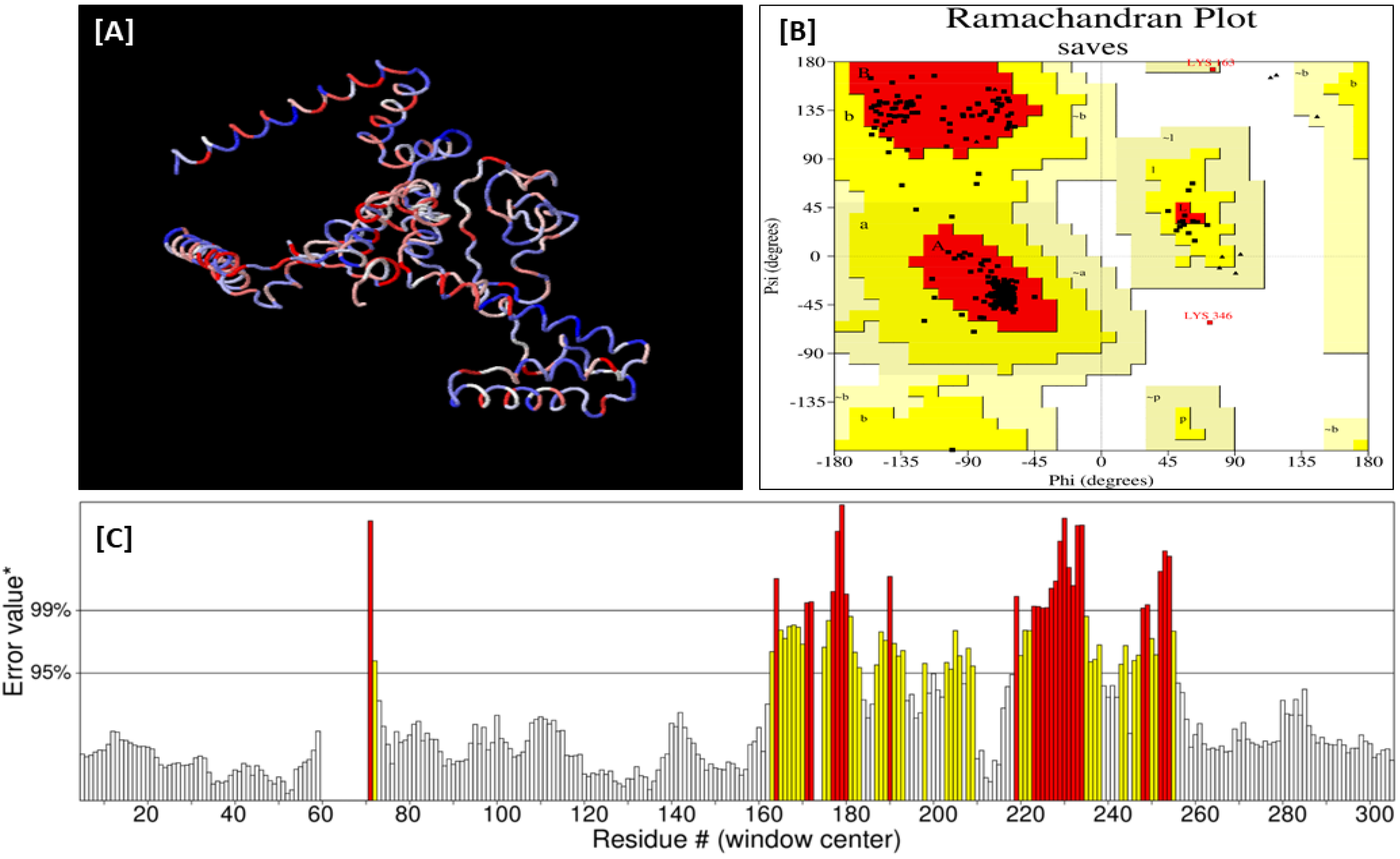
3.9. Vaccine 3D Structure Refinement, Validation, and Solubility Prediction
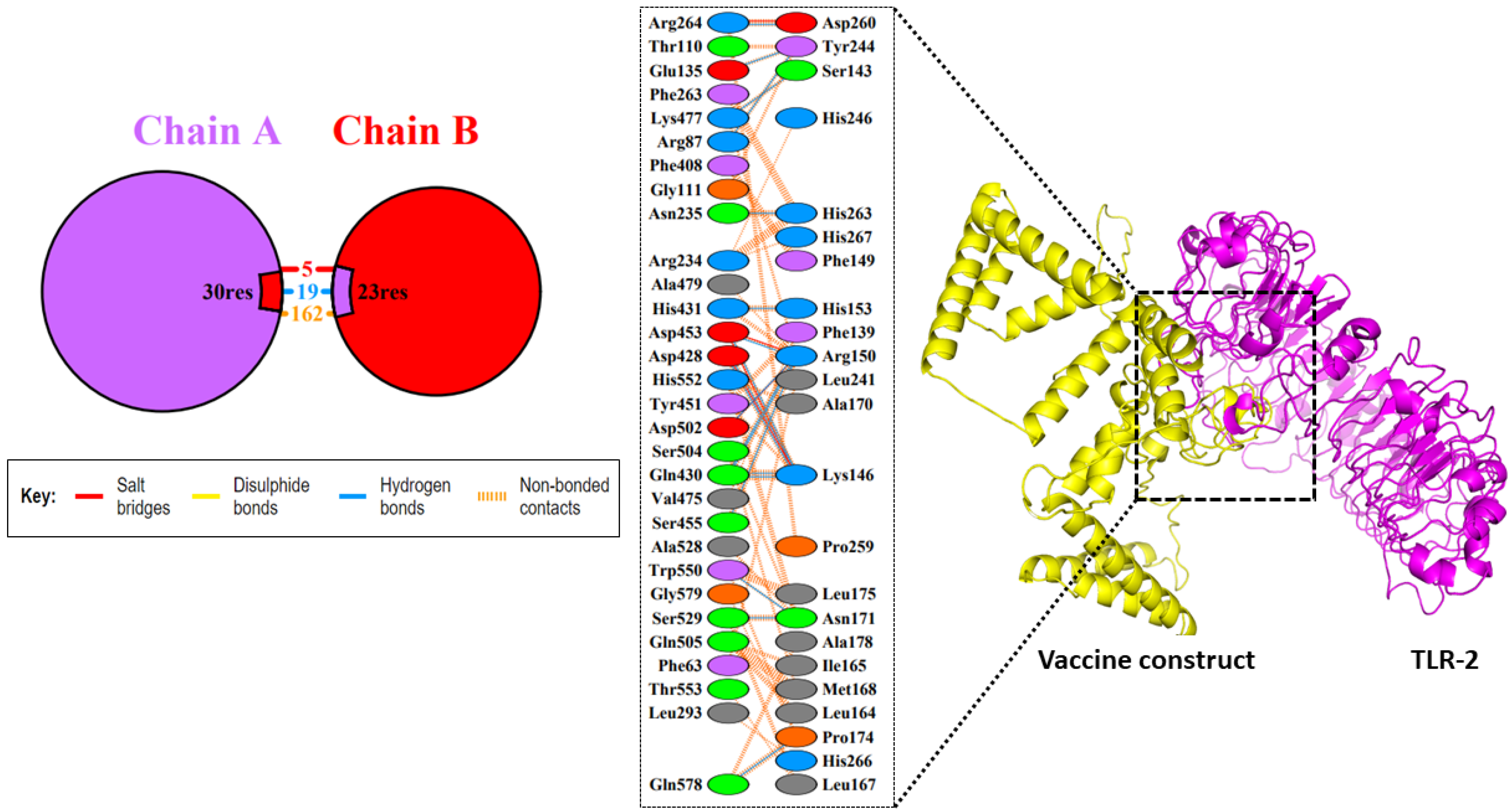
3.10. Molecular Docking Studies between the Vaccine Construct and the TLR2 Receptor
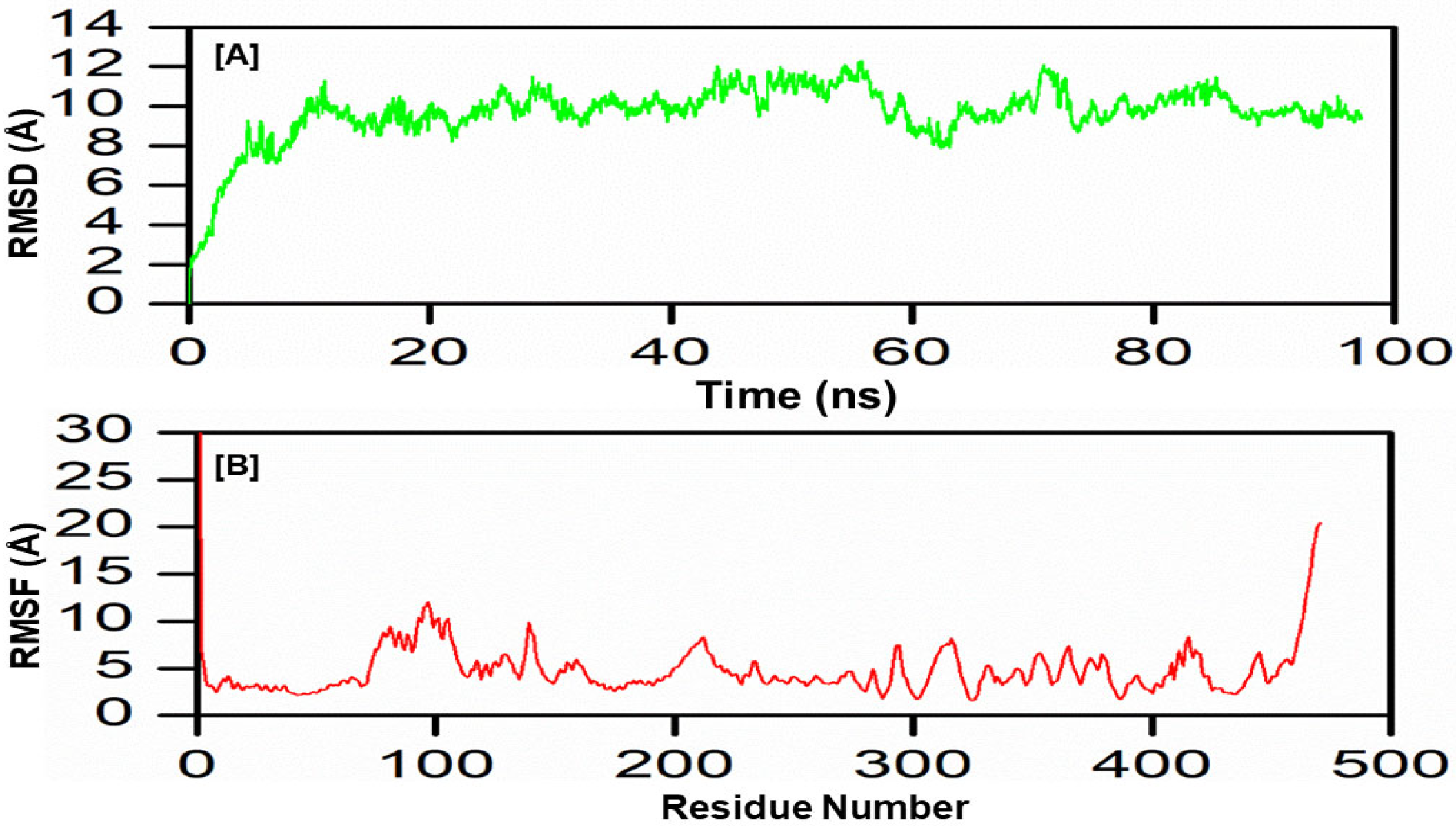
3.11. Molecular Dynamics Simulation
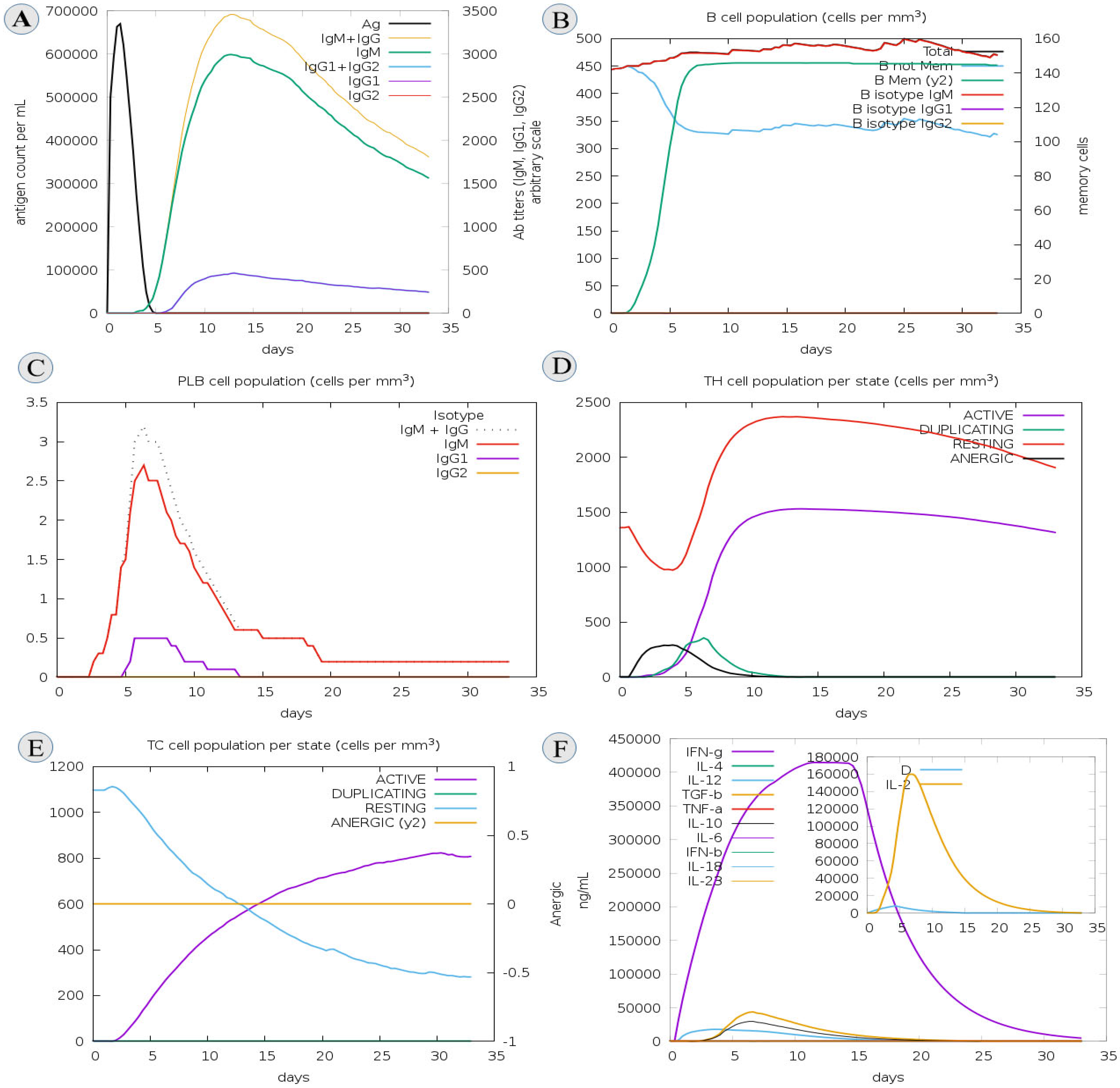
3.12. C-IMM Simulation
3.13. Vector Preparation and Cloning
4. Discussion
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 1–6. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Minneci, F.; Nugent, T.C.O.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013, 41, W349–W357. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.; Marcatili, P.; et al. IEDB-AR: Immune epitope database—Analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 1–15. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef]
- Flower, D. Eudora Welty and Racism. JSTOR 2007, 60, 325–332. [Google Scholar]
- Zaharieva, N.; Dimitrov, I.; Flower, D.R.; Doytchinova, I. VaxiJen Dataset of Bacterial Immunogens: An Update. Curr. Comput. Aided-Drug Des. 2019, 15, 398–400. [Google Scholar] [CrossRef]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef]
- Shamriz, S.; Ofoghi, H.; Moazami, N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput. Biol. Med. 2016, 76, 24–29. [Google Scholar] [CrossRef]
- Shahab, M.; Hayat, C.; Sikandar, R.; Zheng, G.; Akter, S. In silico designing of a multi-epitope vaccine against Burkholderia pseudomallei: Reverse vaccinology and immunoinformatics. J. Genet. Eng. Biotechnol. 2022, 20, 100. [Google Scholar] [CrossRef]
- Garg, V.K.; Avashthi, H.; Tiwari, A.; Jain, P.A.; Ramkete, P.W.; Kayastha, A.M.; Singh, V.K. MFPPI–multi FASTA ProtParam interface. Bioinformation 2016, 12, 74. [Google Scholar] [CrossRef]
- Buchan, D.W.; Jones, D.T. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35 (Suppl. S2), W407–W410. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. PROCHECK: Validation of protein-structure coordinates. Int. Tables Crystallogr. 2006, F, 722–725. [Google Scholar]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Clementel, D.; Del Conte, A.; Monzon, A.M.; Camagni, G.F.; Minervini, G.; Piovesan, D.; Tosatto, S.C. RING 3.0: Fast generation of probabilistic residue interaction networks from structural ensembles. Nucleic Acids Res. 2022, 50, W651–W656. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Purohit, R. Targeting the protein-protein interface pocket of Aurora-A-TPX2 complex: Rational drug design and validation. J. Biomol. Struct. Dyn. 2021, 39, 3882–3891. [Google Scholar] [CrossRef]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33 (Suppl. S2), W526–W531. [Google Scholar] [CrossRef]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef]
- Sami, S.A.; Marma, K.K.S.; Mahmud, S.; Khan, A.N.; Albogami, S.; El-Shehawi, A.M.; Rakib, A.; Chakraborty, A.; Mohiuddin, M.; Dhama, K.; et al. Designing of a Multi-epitope Vaccine against the Structural Proteins of Marburg Virus Exploiting the Immunoinformatics Approach. ACS Omega 2021, 6, 32043–32071. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Rehman, A.; Tusleem, K.; Ashfaq, U.A.; Qasim, M.; Zhu, X.; Fatima, I.; Shahid, F.; Chen, L.-L. Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. PLoS ONE 2020, 15, e0244176. [Google Scholar]
- Khan, S.; Khan, A.; Rehman, A.U.; Ahmad, I.; Ullah, S.; Khan, A.A.; Ali, S.S.; Afridi, S.G.; Wei, D.-Q. Immunoinformatics and structural vaccinology driven prediction of multi-epitope vaccine against Mayaro virus and validation through in-silico expression. Infect. Genet. Evol. 2019, 73, 390–400. [Google Scholar] [CrossRef]
- Hayat, C.; Shahab, M.; Khan, S.A.; Liang, C.; Duan, X.; Khan, H.; Zheng, G.; Ul-Haq, Z. Design of a novel multiple epitope-based vaccine: An immunoinformatics approach to combat monkeypox. J. Biomol. Struct. Dyn. 2022, 1–12. [Google Scholar] [CrossRef]
- Saha, S.; Vashishtha, S.; Kundu, B.; Ghosh, M. In-silico design of an immunoinformatics based multi-epitope vaccine against Leishmania donovani. BMC Bioinform. 2022, 23, 1–28. [Google Scholar] [CrossRef]
- Sharma, R.; Rajput, V.S.; Jamal, S.; Grover, A.; Grover, S. An immunoinformatics approach to design a multi-epitope vaccine against Mycobacterium tuberculosis exploiting secreted exosome proteins. Sci. Rep. 2021, 11, 13836. [Google Scholar] [CrossRef]
- Dar, H.A.; Almajhdi, F.N.; Aziz, S.; Waheed, Y. Immunoinformatics-Aided Analysis of RSV Fusion and Attachment Glycoproteins to Design a Potent Multi-Epitope Vaccine. Vaccines 2022, 10, 1381. [Google Scholar] [CrossRef]
- Chakraborty, C.; Bhattacharya, M.; Sharma, A.R.; Dhama, K. Monkeypox virus vaccine evolution and global preparedness for vaccination. Int. Immunopharmacol. 2022, 113, 109346. [Google Scholar] [CrossRef]
- Shahab, M.; Alzahrani, A.K.; Duan, X.; Aslam, M.; Abida, A.; Imran, M.; Kamal, M.; Alam, T.; Zheng, G. An Immunoinformatics Approach to Design Novel and Potent Multi-Epitope-Based Vaccine to Target Lumpy Skin Disease. Biomedicines 2023, 11, 398. [Google Scholar] [CrossRef]
- Thorne, L.G.; Reuschl, A.K.; Zuliani-Alvarez, L.; Whelan, M.V.; Turner, J.; Noursadeghi, M.; Towers, G.J. SARS-CoV-2 sensing by RIG-I and MDA5 links epithelial infection to macrophage inflammation. EMBO J. 2021, 40, e107826. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No. | Protein Name | Sequence | VaxiJen Score | Antigenicity |
|---|---|---|---|---|
| AAA92891.1 | HPV-16 L2 capsid protein | MRHKRSAKRTKRASATQLYKTCKQAGTCPPDIIPKVEGKTIADQILQYGSMGVFFGGLGIGTGSGTGGRTGYIPLGTRPPTATDTLAPVRPPLTVDPVGPSDPSIVSLVEETSFIDAGAPTPVPSIPPDVSGFSITTSTDTTPAILDINNTVTTVTTHNNPTFTDPSVLQPPTPAETGGHFTLSSSTISTHNYEEIPMDTFIVSTNPNTVTSSTPIPGSRPVARLGLYSRTTQQVKVVDPAFVTTPTKLITYDNPAYEGIDVDNTLYFPSNDNSINIAPDPDFLDIVALHRPALTSRRTGIRYSRIGNKQTLRTRSGKSIGAKVHYYYDLSTINPAEEIELQTITPSTYTTPSHAASPTSINNGLYDIYADDFITDTFTTPVPSIPSTSLSGYIPANTTIPFGGAYNIPLVSGPDIPINTTDQTPSLIPIVPGSPQYTIIADGGDFYLHPSYYMLRKRRKRLPYFFSDVSLAA | 0.6019 | ANTIGEN |
| AAO15711.1 | HPV-16 putative minor capsid protein L2 | MRHKRSAKRTKRASATQLYKTCKQAGTCPPDIIPKVEGKTIADQILQYGSMGVFFGGLGIGTGSGTGGRTGYIPLGTRPPTATDTLAPVRPPLTVDPVGPSDPSIVSLVEETSFIDAGAPTPVPSIPPDVSGFSITTSTDTTPAILDINNTVTTVTTHNNPTFTDPSVLQPPTPAETGGHFTLSSSTISTHNYEEIPMDTFIVSTNPNTVTSSTPIPGSRPVARLGLYSRTTQQVKVVDPAFVTTPTKLITYDNPAYEGIDVDNTLYFASNDNSINIAPDPDFLDIVALHRPALTSRRTGIRYSRIGNKQTLRTRSGKSIGAKVHYYYDLSTINPAEEIELQTITPSTYTTPSHAASPTSINNGLYDIYADDFITDTFTTPVPSIPSTSLSGYIPANTTIPFGGAYNIPLVSGPDIPINTTDQTPSLIPIVPGSPQYTIIADGGDFYLHPSYYMLRKRRKRLPYFFSDVSLAA | 0.6078 | ANTIGEN |
| AAO85414.1 | HPV-16 | MRHKRSAKRTKRASATQLYKTCKQAGTCPPDIIPKVEGKTIADQILQYGSMGVFFGGLGIGTGSGTGGRTGYIPLGTRPPTATDTLAPVRPPLTVDPVGPSDPSIVSLVEETSFIDAGAPTPVPSIPPDVSGFSITTSTDTTPAILDINNTVTTVTTHNNPTFTDPSVLQPPTPAETGGHFTLSSSTISTHNYEEIPMDTFIVSTNPNTVTSSTPIPGSRPVARLGLYSRTTQQVKVVDPAFVTAPTKLITYDNPAYEGIDVDNTFYFPSNDNSINIAPDPDFLDIVALHRPALTSRRTGIRYSRIGNKQTLRTRSGKSIGAKVHYYYDLSTINPAEEIELQTITPSTYTTTSHAASPTSINNGLYDIYADDFITDTVTTPVPAIPSTSLSGYIPANTTIPFGGAYNIPLVSGPDIPINTTDQTPSLIPIVPGSPQYTIIADGGDFYLHPSYYMLRKRRKRLPYFFSDVSLAA | 0.6153 | ANTIGEN |
| AAQ10718.1 | HPV-16 | MRHKRSAKRTKRASATQLYKTCKQAGTCPPDIIPKVEGKTIADQILQYGSMGVFFGGLGIGTGSGTGGRTGYIPLGTRPPTATDTLAPVRPPLTVDPVGPSDPSIVSLVEETSFIDAGAPTSVPSIPPDVSGFSITTSTDTTPAILDINNTVTTVTTHNNPTFTDPSVLQPPTPAETGGHFTLSSSTISTHNYEEIPMDTFIVSTNPNTVTSSTPIPGSRPVARLGLYSRTTQQVKVVDPAFVTTPTKLITYDNPAYEGIDVDNTLYFPSNDNSINIAPDPDFLDIVALHRPALTSRRTGIRYSRIGNKQTLRTRSGKSIGAKVHYYYDLSTIDPAEEIELQTITPSTYTTTLHAASPTSINNGLYDIYADDFITDTSTTPVPSVPSTSLSGYIPANTTIPFGGAYNIPLVSGPDIPINITDQAPSLIPIVPGSPQYTIIADAGDFYLHPSYYMLRKRRKRLPYFFSDVSLAA | 0.6352 | ANTIGEN |
| AAQ10726.1 | HPV-16 | MRHKRSAKRTKRASATQLYKTCKQAGTCPPDIIPKVEGKTIADQILQYGSMGVFFGGLGIGTGSGTGGRTGYIPLGTRPPTATDTLAPVRPPLTVDPVGPSDPSIVSLVEETSFIDAGAPTPVPSIPPDVSGFSITTSTDTTPAILDINNTVTTVTTHNNPTFTDPSVLQPPTPAETGGHFTLSSSTISTHNYEEIPMDTFIVSTNPNTVTSSTPIPGSRPVARLGLYSRTTQQVKVVDPAFVTTPTKLITYDNPAYEGIDVDNTLYFPSNDNSINIAPDPDFLDIVALHRPALTSRRTGIRYSRIGNKQTLRTRSGKSIGAKVHYYYDLSTINPAEEIELQTITPSTYTTASHAASPTSINNGLYDIYADDFITDTSTTPVPSIPSTSLSGYIPANTTIPFGGAYNIPLVSGPDIPINTTDQTPSLIPIVPGSPQYTIIADGGDFYLHPSYYMLRKRRKRLPYFFSDVSLAA | 0.6197 | ANTIGEN |
| AAV91650.1 | HPV-16 | MRHKRSAKRTKRASATQLYKTCKQAGTCPPDIIPKVEGKTIADQILQYGSMGVFFGGLGIGTGSGTGGRTGYIPLGTRPPTATDTLAPVRPPLTVDPVGPSDPSIVSLVEETSFIDAGAPTPVPSIPPDVSGFSITTSTDTTPAILDINNTVTTVTTHNNPTFTDPSVLQPPTPAETGGHFTLSSSTISTHNYEEIPMDTFIVSTNPNTVTSSTPIPGSRPVARLGLYSRTTQQVKVVDPAFVTAPTKLITYDNPAYEGIDVDNTFYFPSNDNSINIAPDPDFLDIVALHRPALTSRRTGIRYSRIGNKQTLRTRSGKSIGAKVHYYYDLSTINPAEEIELQTITPSTYTPTSHAASPTSINNGLYDIYADDFITDTVTTPVPAIPSTSLSGYIPANTTIPFGGAYNIPLVSGPDIPINTTDQTPSLIPIVPGSPQYTIIADGGDFYLHPSYYMLRKRRKRLPYFFSDVSLAA | 0.6090 | ANTIGEN |
| ALB35319.1 | HPV-16 | MRHKRSAKRTKRASATQLYKTCKQAGTCPPDIIPKVEGKTIADQILQYGSMGVFFGGLGIGTGSGTGGRTGYIPLGTRPPTATDTLAPVRPPLTVDPVGPSDPSIVSLVEETSFIDAGAPTSVPSIPPDVSGFSITTSTDTTPAILDINNTVTTVTTHNNPTFTDPSVLQPPTPAETGGHFTLSSSTISTHNYEEIPMDTFIVSTNPNTVTSSTPIPGSRPVARLGLYSRTTQQVKVVDPAFVTTPTKLITYDNPAYEGIDVDNTLYFSSNDNSINIAPDPDFLDIVALHRPALTSRRTGIRYSRIGNKQTLRTRSGKSIGAKVHYYYDFSTIDPAEEIELQTITPSTYTTTSHAASPTSINNGLYDIYADDFITDTSTTPVPSVPSTSLSGYIPANTTIPFGGAYNIPLVSGPDIPINITDQAPSLIPIVPGSPQYTIIADAGDFYLHPSYYMLRKRRKRLPYFFSDVSLAA | 0.6401 | ANTIGEN |
| AAV91690.1 | HPV-16 | MRHKRSAKRTKRASATQLYKTCKQAGTCPPDIIPKVEGKTIADQILQYGSMGVFFGGLGIGTGSGTGGRTGYIPLGTRPPTATDTLAPVRPPLTVDPVGPSDPSIVSLVEETSFIDAGAPTSVPSIPPDVSGFSITTSTDTTPAILDINNTVTTVTTHNNPTFTDPSVLQPPTPAETGGHFTLSSSTISTHNYEEIPMDTFIVSTNPNTVTSSTPIPGSRPVARLGLYSRTTQQVKVVDPAFITTPTKLITYDNPAYEGIDVDNTLYFSSNDNSINIAPDPDFLDIVALHRPALTSRRTGIRYSRIGNKQTLRTRSGKSIGAKVHYYYDFSTIDPAEEIELQTITPSTYTTTSHAASPTSINNGLYDIYADDFITDTSTTPVPSVPSTSLSGYIPANTTIPFGGAYNIPLVSGPDIPINITDQAPSLIPIVPGSPQYTIIADAGDFYLHPSYYMLRKRRKRLPYFFSDVSLAA | 0.6393 | ANTIGEN |
| AAV91674.1 | HPV-16 | MRHKRSAKRTKRASATQLYKTCKQAGTCPPDIIPKVEGKTIADQILQYGSMGVFFGGLGIGTGSGTGGRTGYIPLGTRPPTATDTLAPVRPPLTVDPVGPSDPSIVSLVEETSFIDAGAPTPVPSIPPDVSGFSITTSTDTTPAILDINNTVTTVTTHNNPTFTDPSVLQPPTPAETGGHFTLSSSTISTHNYEEIPMDTFIVSTNPNTVTSSTPIPGSRPVARLGLYSRTTQQVKVVDPAFVTAPTKLITYDNPAYEGIDVDNTFYFPSNDNSINIAPDPDFLDIVALHRPALTSRRTGIRYSRIGNKQTLRTRSGKSIGAKVHYYYDLSTINPAEEIELQTITPSTYTPTSHAASPTSINNGLYDIYADDFITDTVTTPVPAIPSTSLSGYIPANTTIPFGGAYNIPLVSGPDIPINTTDQTPSLIPIVPGSPQYTIIADGGDFYLHPSYYMLRKRRKRLPYFFSDVSLAA | 0.6090 | ANTIGEN |
| AAV91682.1 | HPV-16 | MRHKRSAKRTKRASATQLYKTCKQAGTCPPDIIPKVEGKTIADQILQYGSMGVFFGGLGIGTGSGTGGRTGYIPLGTRPPTATDTLAPVRPPLTVDPVGPSDPSIVSLVEETSFIDAGAPTSVPSIPPDVSGFSITTSTDTTPAILDINNTVTTVTTHNNPTFTDPSVLQPPTPAETGGHFTLSSSTISTHNYEEIPMDTFIVSTNPNTVTSSTPIPGSRPVARLGLYSRTTQQVKVVDPAFVTTPTKLITYDNPAYEGIDVDNTLYFSSNDNSINIAPDPDFLDIVALHRPALTSRRTGIRYSRIGNKQTLRTRSGKSIGAKVHYYYDFSTIDPAEEIELQTITPSTYTTTSHAASPTSINNGLYDIYADDFITDTSTTPVPSVPSTSLSGYIPANTTIPFGGAYNIPLVSGPDIPINITDQAPSLIPIVPGSPQYTIIADAGDFYLHPSYYMLRKRRKRLPYFFSDVSLAA | 0.6493 | ANTIGEN |
| Start | End | Peptide | Length |
|---|---|---|---|
| 4 | 17 | KRSAKRTKRASATQ | 16 |
| 63 | 68 | FKHVSK | 6 |
| 175 | 180 | AEKYSK | 6 |
| 196 | 239 | PRSEPDTGNPCHTTKLLHRDSVDSAPILTAFNSSHKGRINCNSN | 44 |
| Epitopes | Interacting Alleles | Antigenicity | Allergenicity | Toxicity |
|---|---|---|---|---|
| GQVDYYGLY | HLA-B*15:01,HLA-A*30:02 | 0.6511 | NO | NO |
| GQVDYYGLYY | HLA-B*15:01,HLA-A*01:01 | 0.549 | NO | NO |
| KSAIVTLTY | HLA-B*58:01,HLA-A*30:02, HLA-A*32:01,HLA-B*15:01 | 0.8082 | NO | NO |
| NTTPIVHLK | HLA-A*68:01,HLA-A*11:01 | 1.5790 | NO | NO |
| QVILCPTSV | HLA-A*68:02,HLA-A*02:03 | 0.528 | NO | NO |
| TAVSSTWHW | HLA-B*58:01, HLA-B*53:01,HLA-B*57:01 | 1.1083 | NO | NO |
| TLKCLRYRFK | HLA-A*31:01, HLA-A*03:01,HLA-A*11:01 | 1.2871 | NO | NO |
| TLQDVSLEV | HLA-A*02:03, HLA-A*02:01,HLA-A*02:06 | 1.3187 | NO | NO |
| WTLQDVSLEV | HLA-A*02:06, HLA-A*02:01,HLA-A*02:03,HLA-A*68:02 | 1.6596 | NO | NO |
| Epitopes | Interacting Alleles | Antigenicity | Allergenicity | Toxicity |
|---|---|---|---|---|
| AIYYKAREMGFKHIN | HLA-DRB5*01:01,HLA-DRB1*09:01 | 1.4971 | NO | NO |
| APILTAFNSSHKGRI | HLA-DRB1*07:01, HLA-DRB1*15:01,HLA-DRB5*01:01 | 0.5116 | NO | NO |
| CAIYYKAREMGFKHI | HLA-DRB1*04:01, HLA-DRB1*07:01 | 1.3288 | NO | NO |
| EKWTLQDVSLEVYLT | HLA-DPA1*02:01/ HLA-DRB1*01:01,HLA-DPA1*03:01/DPB1*04:02 | 0.9309 | NO | NO |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahab, M.; Guo, D.; Zheng, G.; Zou, Y. Design of a Novel and Potent Multi-Epitope Chimeric Vaccine against Human Papillomavirus (HPV): An Immunoinformatics Approach. Biomedicines 2023, 11, 1493. https://doi.org/10.3390/biomedicines11051493
Shahab M, Guo D, Zheng G, Zou Y. Design of a Novel and Potent Multi-Epitope Chimeric Vaccine against Human Papillomavirus (HPV): An Immunoinformatics Approach. Biomedicines. 2023; 11(5):1493. https://doi.org/10.3390/biomedicines11051493
Chicago/Turabian StyleShahab, Muhammad, Dejia Guo, Guojun Zheng, and Yening Zou. 2023. "Design of a Novel and Potent Multi-Epitope Chimeric Vaccine against Human Papillomavirus (HPV): An Immunoinformatics Approach" Biomedicines 11, no. 5: 1493. https://doi.org/10.3390/biomedicines11051493
APA StyleShahab, M., Guo, D., Zheng, G., & Zou, Y. (2023). Design of a Novel and Potent Multi-Epitope Chimeric Vaccine against Human Papillomavirus (HPV): An Immunoinformatics Approach. Biomedicines, 11(5), 1493. https://doi.org/10.3390/biomedicines11051493