
Updates in Glioblastoma Immunotherapy: An Overview of the Current Clinical and Translational Scenario
 , , , ,
, , , ,  ,
,  and
and 
Abstract
:1. Introduction
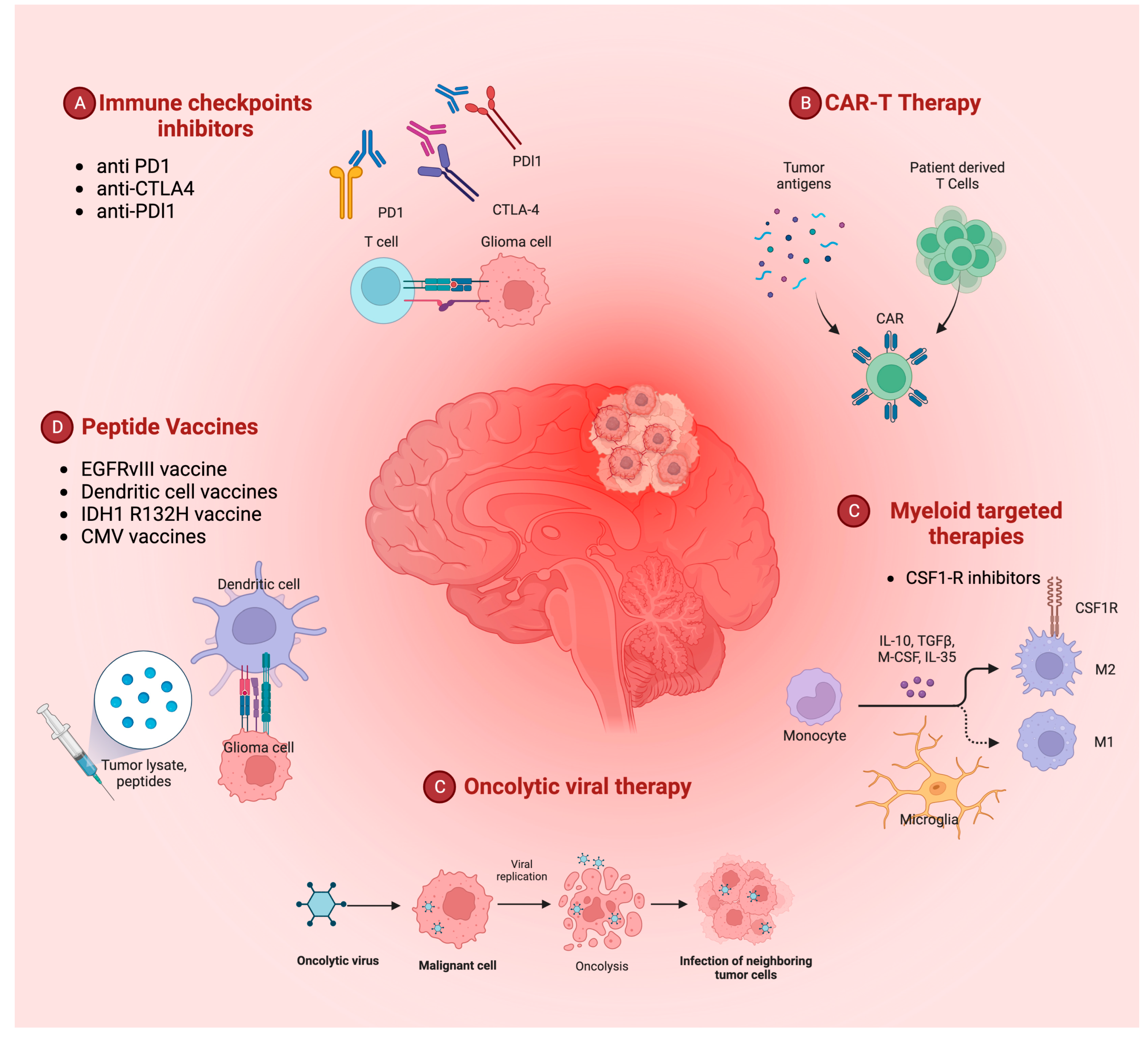
2. Immunotherapy Hints in Glioblastoma Treatment
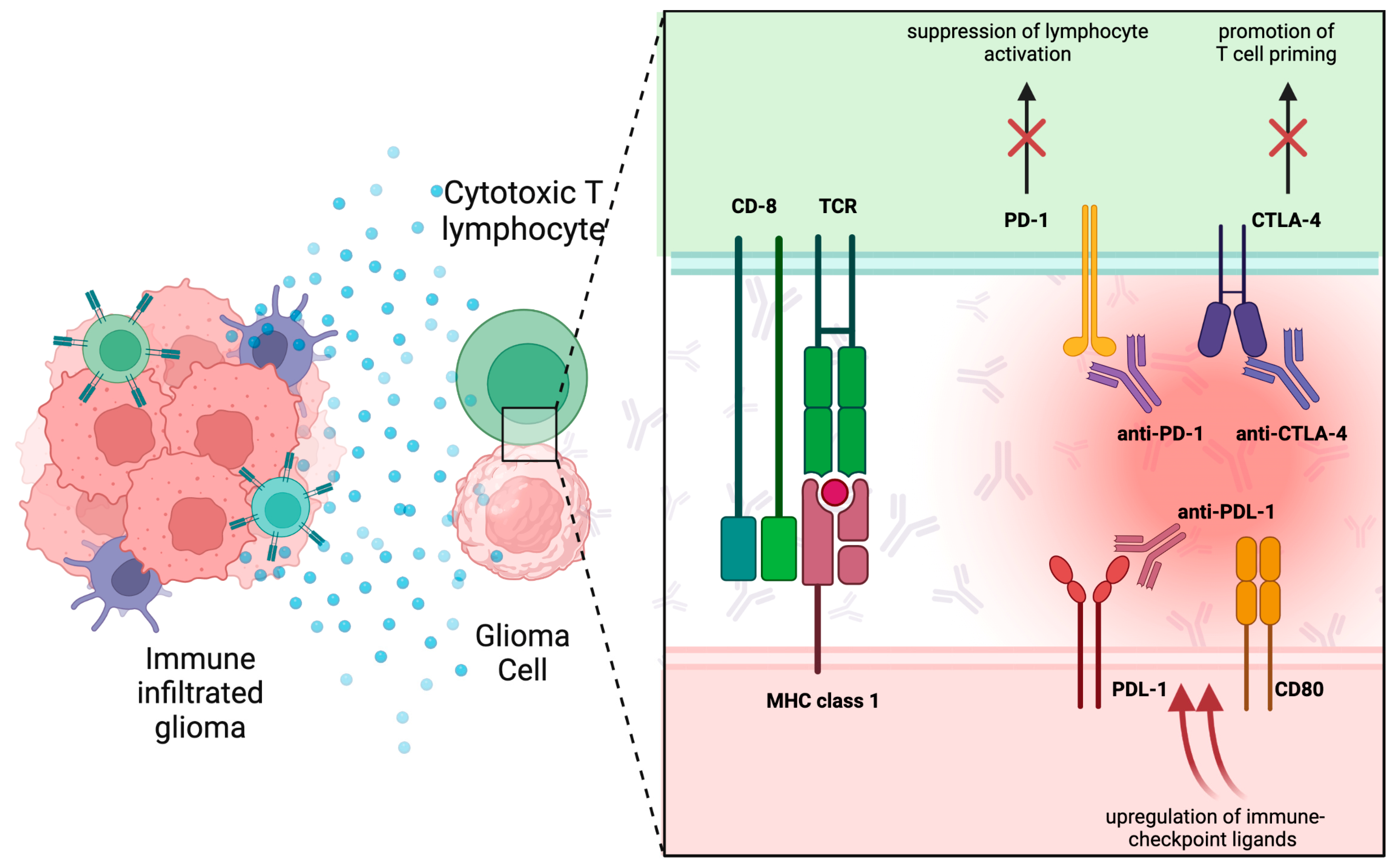
3. Immune Checkpoint Inhibitors
4. Peptide Vaccines
4.1. EGFR vIII Vaccine
4.2. IDH1 R132H Vaccine
4.3. Cytomegalovirus Vaccine
4.4. Dendritic Cell Vaccines
5. CAR-T Therapy
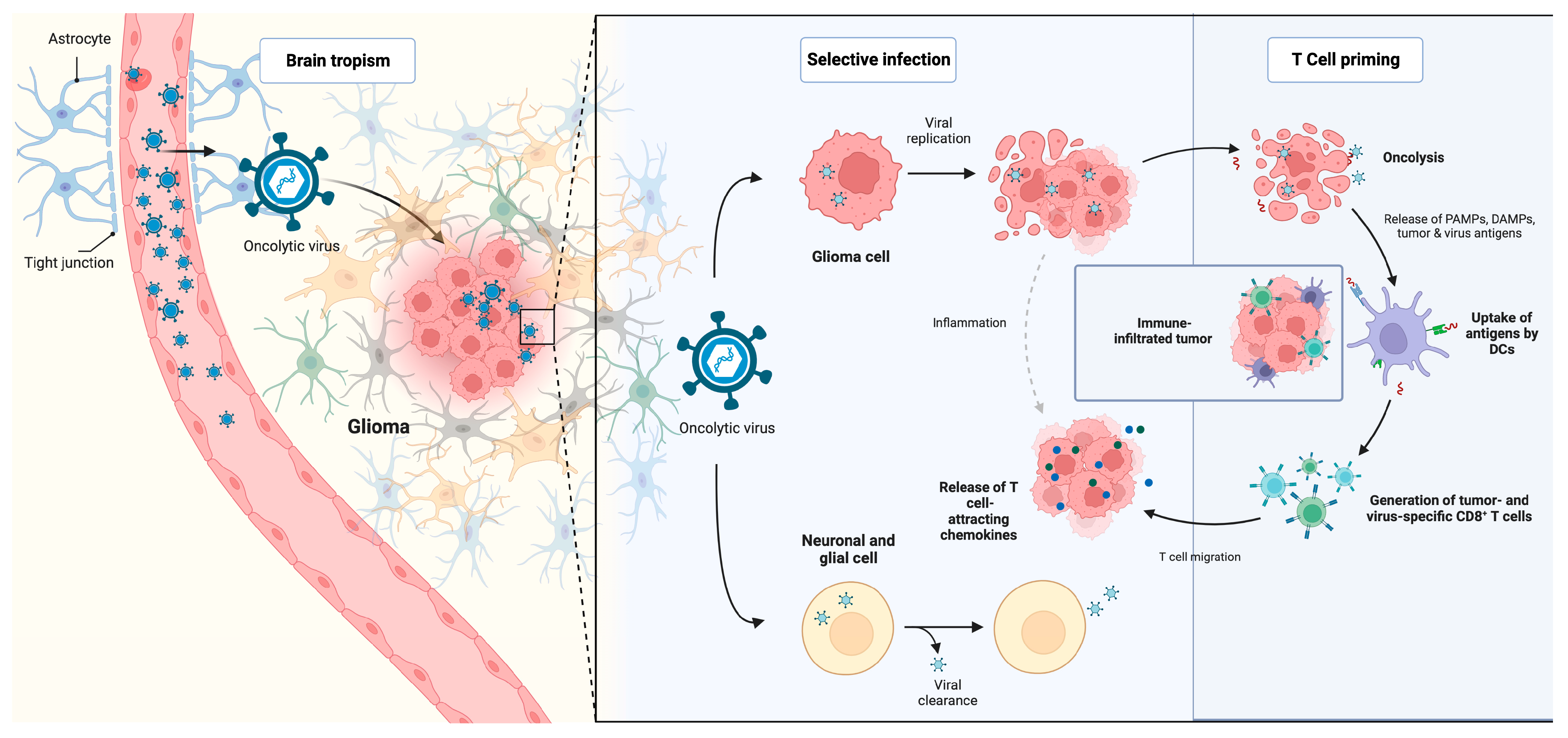
6. Oncolytic Viruses
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Eng. J Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Bruno, F.; Pellerino, A.; Pronello, E.; Palmiero, R.; Bertero, L.; Mantovani, C.; Bianconi, A.; Melcarne, A.; Garbossa, D.; Rudà, R. Elderly Gliobastoma Patients: The Impact of Surgery and Adjuvant Treatments on Survival: A Single Institution Experience. Brain Sci. 2022, 12, 632. [Google Scholar] [CrossRef]
- Zeppa, P.; De Marco, R.; Monticelli, M.; Massara, A.; Bianconi, A.; Di Perna, G.; Crasto, S.G.; Cofano, F.; Melcarne, A.; Lanotte, M.M.; et al. Fluorescence-Guided Surgery in Glioblastoma: 5-ALA, SF or Both? Differences between Fluorescent Dyes in 99 Consecutive Cases. Brain Sci. 2022, 12, 555. [Google Scholar] [CrossRef]
- Specchia, F.M.C.; Monticelli, M.; Zeppa, P.; Bianconi, A.; Zenga, F.; Altieri, R.; Pugliese, B.; Di Perna, G.; Cofano, F.; Tartara, F.; et al. Let Me See: Correlation between 5-ALA Fluorescence and Molecular Pathways in Glioblastoma: A Single Center Experience. Brain Sci. 2021, 11, 795. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.; Pesaresi, A.; Bianconi, A.; Zotta, M.; Deandreis, D.; Morana, G.; Zeppa, P.; Melcarne, A.; Garbossa, D.; Cofano, F. A Systematic Review of Amino Acid PET Imaging in Adult-Type High-Grade Glioma Surgery: A Neurosurgeon’s Perspective. Cancers 2022, 15, 90. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Royce, C.; Stroud, I.; Garren, N.; Mackey, M.; Butman, J.; Camphausen, K.; et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 740–745. [Google Scholar] [CrossRef]
- Simonetti, G.; Trevisan, E.; Silvani, A.; Gaviani, P.; Botturi, A.; Lamperti, E.; Beecher, D.; Bertero, L.; Bosa, C.; Salmaggi, A. Safety of bevacizumab in patients with malignant gliomas: A systematic review. Neurol. Sci. 2014, 35, 83–89. [Google Scholar] [CrossRef]
- Bianconi, A.; Prior, A.; Zona, G.; Fiaschi, P. Anticoagulant therapy in high grade gliomas: A systematic review on state of the art and future perspectives. J. Neurosurg. Sci. 2023, 67, 236–240. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.; Livingstone, E.; Roesch, A.; Schilling, B.; Schadendorf, D. Immunotherapy in melanoma: Recent advances and future directions. Eur. J. Surg. Oncol. EJSO 2017, 43, 604–611. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Sanchez-Perez, L.; Sampson, J.; Verla, T.; Reap, E.; Choi, B.; Fecci, P. Immunotherapy for malignant glioma. Surg. Neurol. Int. 2015, 6 (Suppl. 1), S68–S77. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Bunse, L.; Bunse, T.; Krämer, C.; Chih, Y.C.; Platten, M. Clinical and Translational Advances in Glioma Immunotherapy. Neurotherapeutics 2022, 19, 1799–1817. [Google Scholar] [CrossRef]
- Formica, V.; Leary, A.; Cunningham, D.; Chua, Y.J. 5-Fluorouracil can cross brain-blood barrier and cause encephalopathy: Should we expect the same from capecitabine? A case report on capecitabine-induced central neurotoxicity progressing to coma. Cancer Chemother. Pharmacol. 2006, 58, 276–278. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro-Oncology 2012, 14, 958–978. [Google Scholar] [CrossRef]
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia 2002, 40, 252–259. [Google Scholar] [CrossRef]
- Badie, B.; Schartner, J. Role of microglia in glioma biology. Microsc. Res. Tech. 2001, 54, 106–113. [Google Scholar] [CrossRef]
- Assem, M.; Sibenaller, Z.; Agarwal, S.; Al-Keilani, M.S.; Alqudah, M.A.Y.; Ryken, T.C. Enhancing diagnosis, prognosis, and therapeutic outcome prediction of gliomas using genomics. OMICS 2012, 16, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, A.; Aruta, G.; Rizzo, F.; Salvati, L.F.; Zeppa, P.; Garbossa, D.; Cofano, F. Systematic Review on Tumor Microenvironment in Glial Neoplasm: From Understanding Pathogenesis to Future Therapeutic Perspectives. Int. J. Mol. Sci. 2022, 23, 4166. [Google Scholar] [CrossRef] [PubMed]
- Franson, A.; McClellan, B.L.; Varela, M.L.; Comba, A.; Syed, M.F.; Banerjee, K.; Zhu, Z.; Gonzalez, N.; Candolfi, M.; Lowenstein, P.; et al. Development of immunotherapy for high-grade gliomas: Overcoming the immunosuppressive tumor microenvironment. Front. Med. Lausanne 2022, 9, 966458. [Google Scholar] [CrossRef] [PubMed]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma: Fig. 1. Neuro Oncol. 2015, 17 (Suppl. 7), vii9–vii14. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas Promote Immunosuppression through Induction of B7-H1 Expression in Tumor-Associated Macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef]
- Huettner, C.; Paulus, W.; Roggendorf, W. Messenger RNA expression of the immunosuppressive cytokine IL-10 in human gliomas. Am. J. Pathol. 1995, 146, 317–322. [Google Scholar]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef]
- Melero, I.; Lasarte, J.J. Genetic basis for clinical response to CTLA-4 blockade. N. Engl. J. Med. 2015, 372, 783. [Google Scholar] [CrossRef]
- Mathios, D.; Kim, J.E.; Mangraviti, A.; Phallen, J.; Park, C.-K.; Jackson, C.M.; Garzon-Muvdi, T.; Kim, E.; Theodros, D.; Polanczyk, M.; et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 2016, 8, 370ra180. [Google Scholar] [CrossRef]
- Speranza, M.C.; Passaro, C.; Ricklefs, F.; Kasai, K.; Klein, S.R.; Nakashima, H.; Kaufmann, J.K.; Ahmed, A.-K.; O Nowicki, M.; Obi, P.; et al. Preclinical investigation of combined gene-mediated cytotoxic immunotherapy and immune checkpoint blockade in glioblastoma. Neuro Oncol. 2018, 20, 225–235. [Google Scholar] [CrossRef]
- Hardcastle, J.; Mills, L.; Malo, C.S.; Jin, F.; Kurokawa, C.; Geekiyanage, H.; Schroeder, M.; Sarkaria, J.; Johnson, A.J.; Galanis, E. Immunovirotherapy with measles virus strains in combination with anti-PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro Oncol. 2017, 19, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Hulse, R.E.; Swenson, W.G.; Kunkler, P.E.; White, D.M.; Kraig, R.P. Monomeric IgG Is Neuroprotective via Enhancing Microglial Recycling Endocytosis and TNF-α. J. Neurosci. 2008, 28, 12199–12211. [Google Scholar] [CrossRef] [PubMed]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef]
- Derer, A.; Spiljar, M.; Bäumler, M.; Hecht, M.; Fietkau, R.; Frey, B.; Gaipl, U.S. Chemoradiation Increases PD-L1 Expression in Certain Melanoma and Glioblastoma Cells. Front. Immunol. 2016, 7, 610. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell. Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.L.; Boddeke, H.W.G.M.; et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Ghouzlani, A.; Kandoussi, S.; Tall, M.; Reddy, K.P.; Rafii, S.; Badou, A. Immune Checkpoint Inhibitors in Human Glioma Microenvironment. Front. Immunol. 2021, 12, 2664. [Google Scholar] [CrossRef]
- Preusser, M.; Lim, M.; Hafler, D.A.; Reardon, D.A.; Sampson, J.H. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 2015, 11, 504–514. [Google Scholar] [CrossRef]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef]
- Van Bussel, M.T.J.; Beijnen, J.H.; Brandsma, D. Intracranial antitumor responses of nivolumab and ipilimumab: A pharmacodynamic and pharmacokinetic perspective, a scoping systematic review. BMC Cancer 2019, 19, 519. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, F.; Liu, Z.; Tang, H.; Wu, H.; Gong, Q.; Chen, J. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front. Pharmacol. 2017, 8, 242. [Google Scholar] [CrossRef] [PubMed]
- Disruption of the Immune-Checkpoint VISTA Gene Imparts a Proinflammatory Phenotype with Predisposition to the Development of Autoimmunity PNAS. Available online: https://www.pnas.org/doi/abs/10.1073/pnas.1407447111 (accessed on 1 January 2023).
- Flgel, A.; Bradl, M.; Kreutzberg, G.W.; Graeber, M.B. Transformation of donor-derived bone marrow precursors into host microglia during autoimmune CNS inflammation and during the retrograde response to axotomy. J. Neurosci. Res. 2001, 66, 74–82. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Lesterhuis, W.J.; Salmons, J.; Nowak, A.; Rozali, E.N.; Khong, A.; Dick, I.M.; Harken, J.A.; Robinson, B.W.; Lake, R.A. Synergistic Effect of CTLA-4 Blockade and Cancer Chemotherapy in the Induction of Anti-Tumor Immunity. PLoS ONE 2013, 8, e61895. [Google Scholar] [CrossRef]
- Kataoka, K.; Shiraishi, Y.; Takeda, Y.; Sakata, S.; Matsumoto, M.; Nagano, S.; Maeda, T.; Nagata, Y.; Kitanaka, A.; Mizuno, S.; et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature 2016, 534, 402–406. [Google Scholar] [CrossRef]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.-G.; Teh, C.; Firth, M.; et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 2016, 17, 816–824. [Google Scholar] [CrossRef]
- Sharon, E.; Streicher, H.; Goncalves, P.; Chen, H.X. Immune checkpoint inhibitors in clinical trials. Chin. J. Cancer 2014, 33, 434–444. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Pardoll, D.M. Immune checkpoint inhibitors: Making immunotherapy a reality for the treatment of lung cancer. Cancer Immunol. Res. 2013, 1, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- Topalian, L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell. 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.-S.; Kang, G. Immunotherapy in advanced melanoma: A network meta-analysis. Immunotherapy 2017, 9, 471–479. [Google Scholar] [CrossRef]
- Gomes, F.; Serra-Bellver, P.; Lorigan, P. The role of nivolumab in melanoma. Future Oncol. 2018, 14, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Forsyth, P.A.; Algazi, A.; Hamid, O.; Hodi, F.S.; Moschos, S.J.; Khushalani, N.I.; Lewis, K.; Lao, C.D.; Postow, M.A.; et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N. Engl. J. Med. 2018, 379, 722–730. [Google Scholar] [CrossRef]
- Ribas, A. Overcoming immunologic tolerance to melanoma: Targeting CTLA-4 with tremelimumab (CP-675,206). Oncologist 2008, 13 (Suppl. 4), 10–15. [Google Scholar] [CrossRef]
- Somaiah, N.; Conley, A.P.; Parra, E.R.; Lin, H.; Amini, B.; Soto, L.S.; Salazar, R.; Barreto, C.; Chen, H.; Gite, S.; et al. Durvalumab plus tremelimumab in advanced or metastatic soft tissue and bone sarcomas: A single-centre phase 2 trial. Lancet Oncol. 2022, 23, 1156–1166. [Google Scholar] [CrossRef]
- Schoenfeld, D.; Giobbie-Hurder, A.; Ranasinghe, S.; Kao, K.Z.; Lako, A.; Tsuji, J.; Liu, Y.; Brennick, R.C.; Gentzler, R.D.; Lee, C.; et al. Durvalumab plus tremelimumab alone or in combination with low-dose or hypofractionated radiotherapy in metastatic non-small-cell lung cancer refractory to previous PD(L)-1 therapy: An open-label, multicentre, randomised, phase 2 trial. Lancet Oncol. 2022, 23, 279–291. [Google Scholar] [CrossRef]
- Boussiotis, V.A. Somatic mutations and immunotherapy outcome with CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2230–2232. [Google Scholar] [CrossRef]
- Boussiotis, V.A.; Chatterjee, P.; Li, L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 2014, 20, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Long, G.V.; Atkinson, V.; Lo, S.; Sandhu, S.; Guminski, A.D.; Brown, M.P.; Wilmott, J.S.; Edwards, J.; Gonzalez, M.; Scolyer, R.A.; et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: A multicentre randomised phase 2 study. Lancet Oncol. 2018, 19, 672–681. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Menzer, C.; Gruellich, C.; Hassel, J.C. Editorial on the use of immunotherapy in renal-cell carcinoma—Promising results in combination therapy with ipilimumab and nivolumab. Transl. Cancer Res. 2017, 6 (Suppl. 7), S1208–S1211. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.-J.; Weber, J.S.; et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA-J. Am. Med. Assoc. 2016, 315, 1600–1609. [Google Scholar] [CrossRef]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). The Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; De Vries, H.E. Overcoming the blood–brain tumor barrier for effective glioblastoma treatment. Drug. Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Margolin, K.; Ernstoff, M.S.; Hamid, O.; Lawrence, D.; McDermott, D.; Puzanov, I.; Wolchok, J.D.; Clark, J.I.; Sznol, M.; Logan, T.F.; et al. Ipilimumab in patients with melanoma and brain metastases: An open-label, phase 2 trial. Lancet Oncol. 2012, 13, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Choroid plexus–CSF system. Neurology 2016, 86, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef]
- Omuro, A.; Reardon, D.A.; Sampson, J.H.; Baehring, J.; Sahebjam, S.; Cloughesy, T.F.; Chalamandaris, A.-G.; Von Potter, V.; Butowski, N.; Lim, M. Nivolumab plus radiotherapy with or without temozolomide in newly diagnosed glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neurooncol Adv. 2022, 4, vdac025. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; A Reardon, D.; Cloughesy, T.; Sumrall, A.; Baehring, J.; Bent, M.v.D.; Bähr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol. 2023, 25, 123–134. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022, 24, 1935–1949. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2019, 26, 39–46. [Google Scholar] [CrossRef]
- Galstyan, A.; Markman, J.L.; Shatalova, E.S.; Chiechi, A.; Korman, A.J.; Patil, R.; Klymyshyn, D.; Tourtellotte, W.G.; Israel, L.L.; Braubach, O.; et al. Blood–brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat. Commun. 2019, 10, 3850. [Google Scholar] [CrossRef]
- Swartz, A.M.; Batich, K.A.; Fecci, P.E.; Sampson, J.H. Peptide vaccines for the treatment of glioblastoma. J. Neurooncol. 2015, 123, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Suki, D.; Yang, D.; Shi, W.; Aldape, K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J. Transl. Med. 2005, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Aldape, K.D.; Archer, G.E.; Coan, A.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011, 13, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Bender, S.; Jones, D.T.W.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Bigner, S.H.; Humphrey, P.A.; Wong, A.J.; Vogelstein, B.; Mark, J.; Friedman, H.S.; Bigner, D.D. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 1990, 50, 8017–8022. [Google Scholar] [PubMed]
- Vogt, N.; Lefèvre, S.-H.; Apiou, F.; Dutrillaux, A.-M.; Cör, A.; Leuraud, P.; Poupon, M.-F.; Dutrillaux, B.; Debatisse, M.; Malfoy, B. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc. Natl. Acad. Sci. USA 2004, 101, 11368–11373. [Google Scholar] [CrossRef]
- Wikstrand, G.J.; Reist, C.J.; Archer, G.E.; Zalutsky, M.R.; Bigner, D.D. The class III variant of the epidermal growth factor receptor (EGFRvIII): Characterization and utilization as an immunotherapeutic target. J. Neurovirol. 1998, 4, 148–158. [Google Scholar] [CrossRef]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro Oncol. 2015, 17, 854–861. [Google Scholar] [CrossRef]
- Sampson, J.H.; Crotty, L.E.; Lee, S.; Archer, G.E.; Ashley, D.M.; Wikstrand, C.J.; Hale, L.P.; Small, C.; Dranoff, G.; Friedman, A.H.; et al. Unarmed, tumor-specific monoclonal antibody effectively treats brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 7503–7508. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Crotty, L.E.; E Archer, G.; Hess, K.R.; Wikstrand, C.J.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin. Cancer Res. 2003, 9, 4247–4254. [Google Scholar] [PubMed]
- Sampson, J.H.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; Herndon, J.E.; Lally-Goss, D.; McGehee-Norman, S.; Paolino, A.; Reardon, D.A.; Friedman, A.H.; et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol. Cancer Ther. 2009, 8, 2773–2779. [Google Scholar] [CrossRef]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Ii, J.E.H.; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Saaid, A.; Monticelli, M.; Ricci, A.A.; Orlando, G.; Botta, C.; Zeppa, P.; Bianconi, A.; Osella-Abate, S.; Bruno, F.; Pellerino, A.; et al. Prognostic Analysis of the IDH1 G105G (rs11554137) SNP in IDH-Wildtype Glioblastoma. Genes 2022, 13, 1439. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Takamura, Y.; Ikeda, H.; Kanaseki, T.; Toyota, M.; Tokino, T.; Imai, K.; Houkin, K.; Sato, N. Regulation of MHC class II expression in glioma cells by class II transactivator (CIITA). Glia 2004, 45, 392–405. [Google Scholar] [CrossRef]
- Dey, M.; Ahmed, A.U.; Lesniak, M.S. Cytomegalovirus and glioma: Putting the cart before the horse. J. Neurol. Neurosurg. Psychiatry 2015, 86, 191–199. [Google Scholar] [CrossRef]
- Dziurzynski, K.; Chang, S.M.; Heimberger, A.B.; Kalejta, R.F.; Dallas, S.R.M.; Smit, M.; Soroceanu, L.; Cobbs, C.S.; the HCMV and Gliomas Symposium. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 2012, 14, 246–255. [Google Scholar] [CrossRef]
- Thompson, E.; Landi, D.; Lipp, E.; Balajonda, B.; Herndon, J.; Buckley, E.; Flahiff, C.; Jaggers, D.; Schroeder, K.; Randazzo, D.; et al. CTIM-21. Peptide Vaccine Directed to CMV PP65 for Treatment of Recurrent Malignant Glioma and Medulloblastoma in Children and Young Adults: Preliminary Results of a Phase I Trial. Neuro Oncol. 2020, 22 (Suppl. 2), ii37. [Google Scholar] [CrossRef]
- Brown, M.C.; Holl, E.K.; Boczkowski, D.; Dobrikova, E.; Mosaheb, M.; Chandramohan, V.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci. Transl. Med. 2017, 9, eaan4220. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Prins, R.M.; Wang, X.; Soto, H.; Young, E.; Lisiero, D.N.; Fong, B.; Everson, R.; Yong, W.H.; Lai, A.; Li, G.; et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J. Immunother. 2013, 36, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, F.; Bosio, A.G.; Pattarozzi, A.; Tonelli, M.; Bajetto, A.; Verduci, I.; Cianci, F.; Cannavale, G.; Palloni, L.M.G.; Francesconi, V.; et al. Chloride intracellular channel 1 activity is not required for glioblastoma development but its inhibition dictates glioma stem cell responsivity to novel biguanide derivatives. J. Exp. Clin. Cancer Res. 2022, 41, 53. [Google Scholar] [CrossRef]
- Vik-Mo, E.O.; Nyakas, M.; Mikkelsen, B.V.; Moe, M.C.; Due-Tønnesen, P.; Suso, E.M.I.; Sæbøe-Larssen, S.; Sandberg, C.; Brinchmann, J.E.; Helseth, E.; et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 2013, 62, 1499–1509. [Google Scholar] [CrossRef]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.-J.; Glantz, M.; Peereboom, D.M.; et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin. Cancer Res. 2019, 25, 5799–5807. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Kikuchi, T.; Homma, S.; Koido, S.; Ohkusa, T.; Tasaki, T.; Hayashi, K.; Komita, H.; Watanabe, N.; Suzuki, Y.; et al. Phase I/II trial of combination of temozolomide chemotherapy and immunotherapy with fusions of dendritic and glioma cells in patients with glioblastoma. Cancer Immunol. Immunother. 2016, 65, 1499–1509. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.-N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E., II; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef]
- Kim, C.H.; Woo, S.-J.; Park, J.-S.; Kim, H.-S.; Park, M.-Y.; Park, S.-D.; Hong, Y.-K.; Kim, T.-G. Enhanced antitumour immunity by combined use of temozolomide and TAT-survivin pulsed dendritic cells in a murine glioma. Immunology 2007, 122, 615–622. [Google Scholar] [CrossRef]
- Hamieh, M.; Mansilla-Soto, J.; Rivière, I.; Sadelain, M. Programming CAR T Cell Tumor Recognition: Tuned Antigen Sensing and Logic Gating. Cancer Discov. 2023, 13, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.-F.; Orange, J.S.; Sumazin, P.; Man, T.-K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed]
- Sabahi, M.; Jabbari, P.; Haghighi, M.A.; Soltani, S.; Soudi, S.; Rahmani, F.; Rezaei, N. Proposing a tandem AND-gate CAR T cell targeting glioblastoma multiforme. Med. Hypotheses 2020, 137, 109559. [Google Scholar] [CrossRef]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Cenciarelli, C. Current progress in chimeric antigen receptor T cell therapy for glioblastoma multiforme. Cancer Med. 2021, 10, 5019–5030. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Wollmann, G.; Ozduman, K.; Van Den Pol, A.N. Oncolytic virus therapy for glioblastoma multiforme: Concepts and candidates. Cancer J. 2012, 18, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Coffin, R.S. From virotherapy to oncolytic immunotherapy: Where are we now? Curr. Opin. Virol. 2015, 13, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Walton, R.W.; Brown, M.C.; Sacco, M.T.; Gromeier, M. Engineered Oncolytic Poliovirus PVSRIPO Subverts MDA5-Dependent Innate Immune Responses in Cancer Cells. J. Virol. 2018, 92, e00879-18. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Herndon, J.E.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Witlox, A.M.; van Beusechem, V.W.; Molenaar, B.; Bras, H.; Schaap, G.R.; Alemany, R.; Curiel, D.T.; Pinedo, H.M.; Wuisman, P.I.J.M.; Gerritsen, W.R. Conditionally Replicative Adenovirus with Tropism Expanded towards Integrins Inhibits Osteosarcoma Tumor Growth in Vitro and in Vivo. Clin. Cancer Res. 2004, 10, 61–67. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Lang, F.F.; Tran, N.D.; Puduvalli, V.K.; Elder, J.B.; Fink, K.L.; Conrad, C.A.; Yung, W.K.A.; Penas-Prado, M.; Gomez-Manzano, C.; Peterkin, J.; et al. Phase 1b open-label randomized study of the oncolytic adenovirus DNX-2401 administered with or without interferon gamma for recurrent glioblastoma. J. Clin. Oncol. 2017, 35, 2002. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Abbed, K.M.; Tatter, S.; Louis, D.N.; Hochberg, F.H.; Barker, F.; Kracher, J.; Grossman, S.A.; Fisher, J.D.; Carson, K.; et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol. Ther. 2004, 10, 958–966. [Google Scholar] [CrossRef]
- Wheeler, L.A.; Manzanera, A.G.; Bell, S.D.; Cavaliere, R.; McGregor, J.M.; Grecula, J.C.; Newton, H.B.; Lo, S.S.; Badie, B.; Portnow, J.; et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro Oncol. 2016, 18, 1137–1145. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Aguilar, L.K.; Bell, S.D.; Kaur, B.; Hardcastle, J.; Cavaliere, R.; McGregor, J.; Lo, S.; Ray-Chaudhuri, A.; Chakravarti, A.; et al. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J. Clin. Oncol. 2011, 29, 3611–3619. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Landolfi, J.; Hogan, D.J.; Bloomfield, S.; Carter, B.; Chen, C.C.; Elder, J.B.; Kalkanis, S.N.; Kesari, S.; Lai, A.; et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci. Transl. Med. 2016, 8, 341ra75. [Google Scholar] [CrossRef]
- Ostertag, D.; Amundson, K.K.; Espinoza, F.L.; Martin, B.; Buckley, T.; Da Silva, A.P.G.; Lin, A.H.; Valenta, D.T.; Perez, O.D.; Ibañez, C.E.; et al. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro Oncol. 2012, 14, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, K.; Kimura, T.; Logg, C.R.; Kasahara, N. Tumor-selective gene expression in a hepatic metastasis model after locoregional delivery of a replication-competent retrovirus vector. Clin. Cancer Res. 2006, 12, 7108–7116. [Google Scholar] [CrossRef] [PubMed]
- Perez, O.D.; Logg, C.R.; Hiraoka, K.; Diago, O.; Burnett, R.; Inagaki, A.; Jolson, D.; Amundson, K.; Buckley, T.; Lohse, D.; et al. Design and selection of Toca 511 for clinical use: Modified retroviral replicating vector with improved stability and gene expression. Mol. Ther. 2012, 20, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Huber, B.E.; Austin, E.A.; Richards, C.A.; Davis, S.T.; Good, S.S. Metabolism of 5-fluorocytosine to 5-fluorouracil in human colorectal tumor cells transduced with the cytosine deaminase gene: Significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase. Proc. Natl. Acad. Sci. USA 1994, 91, 8302–8306. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Parab, S.; Burnett, R.; Diago, O.; Ostertag, D.; Hofman, F.M.; Espinoza, F.L.; Martin, B.; Ibañez, C.E.; Kasahara, N.; et al. Intravenous administration of retroviral replicating vector, Toca 511, demonstrates therapeutic efficacy in orthotopic immune-competent mouse glioma model. Hum. Gene Ther. 2015, 26, 82–93. [Google Scholar] [CrossRef]
- Zadeh, G.; Bota, D.; Cachia, D.; Landolfi, J.; Schiff, D.; Vogelbaum, M.; Walbert, T.; Tran, D.; Chu, A.; Das, A.; et al. PC3-151 Toca 5: A Phase 2/3 Randomized, Open-Label Study of Toca 511, a Retroviral Replicating Vector, Combined with Toca FC versus Standard of Care in Patients Undergoing Planned Resection for Recurrent Glioblastoma (GBM) or Anaplastic Astrocytoma (AA) (NCT02414165). Can. J. Neurol. Sci. 2016, 43, S17. [Google Scholar] [CrossRef]
- Ji, N.; Weng, D.; Liu, C.; Gu, Z.; Chen, S.; Guo, Y.; Fan, Z.; Wang, X.; Chen, J.; Zhao, Y.; et al. Adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of recurrent high-grade glioma. Oncotarget 2016, 7, 4369–4378. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zeng, G. Cancer and innate immune system interactions: Translational potentials for cancer immunotherapy. J. Immunother. 2012, 35, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Treatment | Target (s) | Type of Study | Year | Primary Endpoint | n° | Results | Identifier |
|---|---|---|---|---|---|---|---|
| NICs on Poly(β-L-malic acid) with covalently attached anti-CTLA 4 and anti PD-1 antibody | CTLA-4 PD-1 | Murine | 2019 | OS of mice bearing intracranial GBM treated with free mAbs or NICs alone or in combination. | Significant improvement of OS in mice trated with checkpoint inhibitor mAb attached to NIC | ||
| Nivolumab plus Ipilimumab | PD1 and CTLA-4 | Phase III | 2013 | Effectiveness and Safety of Nivolumab Compared to Bevacizumab and of Nivolumab With or Without Ipilimumab in GBM Patients | 529 | Median OS was 9.8 months with nivolumab versus 10.0 months with bevacizumab | NCT02017717 |
| Nivolumab | PD1 | Phase III | 2015 | OS in Nivolumab compared to TMZ with RT for newly-diagnosed GBM | 560 | Median OS was 13.40 months with nivolumab versus 14.88 months in TMZ | NCT02617589 (concluded) |
| Nivolumab | PD1 | Phase III | 2016 | OS in TMZ Plus RT combined with Nivolumab or placebo in newly diagnosed MGMT-Methylated GBM | 716 | Median PFS was 10.64 months with RT, TMZ plus Nivolumab versus 10.32 months in RT, TMZ Plus Placebo | NCT02667587 (ongoing) |
| VICTORI Rindopepimut | Vaccine anti-EGFR III | Phase I | 2009 | Rindopepimut toxicity in GBM patients with gross total resection and standard external beam RT | 15 | Minimal toxicity without symptoms of autoimmunity, without statistically significant improvement of outcome. | |
| ACTIVATE Rindopepimut | Vaccine anti-EGFR III | Phase II | 2010 | PFS and OS of vaccinated patients with newly diagnosed EGFRvIII-expressing GBM with minimal residual disease | 35 | OS and PFS of vaccinated patients were greater than that observed in a control group | NCT00643097 |
| ACT II Rindopepimut | Vaccine anti-EGFR III and TMZ | Phase II | 2011 | If TMZ-induced lymphopenia with standard or intensified dose would enhance immune responses to the anti-EGFRIII-vaccine | 22 | Humoral and cellular vaccine-induced immune responses are more enhanced by a intensified TMZ dose than the standard TMZ dose | |
| ACT III Rindopepimut | Vaccine anti-EGFR III and TMZ | Phase II | 2011 | Efficacy and safety of Rindopepimut in EGFRvIII-positive GBM with gross total resection and no evidence of progression after RT and TMZ | 65 | Vaccine well-tolerated. Improved PFS and OS | NCT00458601 |
| PERFORMANCE | PEP-CMV vaccination | Phase I | 2016 | Efficacy and safety of PEP-CMV vaccine | 27 | Vaccine generates an immune response No adverse events | NCT02864368 (terminated) |
| DCs vaccine | ATL-pulsed DCs vaccine | Phase I | 2011 | Vaccine safety and efficacy in inducing immunologic response in GBM after RT and TMZ. | 10 | Vaccinated patients with major immune response had improved survival, with no serious adverse events | |
| DCs vaccine | ATL-pulsed DCs vaccine versus GAA peptide-pulsed DCs vaccine | Phase I | 2013 | Comparison of safety, feasibility and immune responses of ATL-pulsed DC vaccine, with GAA peptide-pulsed DCs vaccine | 34 | More activated NK cells in GAA patients. Correlation between decreased Treg ratios (post/pre vaccination) and OS in both trials. | |
| DCs vaccine | TSC derived mRNA- Transfected DCs vaccine | Phase I Phase II | 2009 | Safety, immunological response, time to disease progression and survival time in vaccinated GBM patients | 20 | No adverse autoimmune events or other side effects. PFS was 2.9 times longer in vaccinated patients | NCT00846456 (completed) |
| DCs vaccine ICT-107 | Autologous DCs pulsed with six synthetic peptide epitopes targeting GBM tumor/stem cell-associated antigens MAGE-1, HER-2, AIM-2, TRP-2, gp100, and IL13Rα2 | Phase II | 2017 | ICT-107 tested efficacy, safety, QoL and immune response | 124 | No adverse autoimmune events. PFS significantly improved in ICT-107-treated patients with maintenance of QoL. HLA-A2 subgroup showed increased ICT-107 activity clinically and immunologically. | NCT01280552 (completed) |
| DCs vaccine | DC cells pulsed with CMV-pp65 RNA vaccine | Phase I | 2017 | Pp65-specific cellular responses and the effects on long-term PFS and OS | 11 | Long-term PFS (25.3 months) and OS (41.1 months) in vaccinated patients | |
| CAR-T therapy | Autologous anti-EGFRvIII CAR T cells | Phase I | 2014 | Safety and feasibility of CAR T-EGFRvIII | 11 | No incidence of cytokine-release syndrome or neurotoxicity. OS not affected by therapy | NCT02209376 (terminated) |
| CAR-T therapy | HER2-specific CAR-modified virus-specific T cells | Phase I | 2019 | Dose-Escalation Trial | 16 | Infusions well tolerated, with no dose-limiting toxic effects | NCT01109095 (completed) |
| Oncolytic viruses therapy | Recombinant oncolytic Polio/Rhinovirus PVSRIPO | Phase I | 2021 | Dose-finding and safety Study in recurrent GBM | 61 | Intratumoral reinfusion of PVSRIPO via CED is safe, and encouraging efficacy results have been observed | NCT01491893 (completed) |
| Oncolytic viruses DNX-2401 + Pembrolizumab | Genetically modified oncolytic adenovirus+ Anti-PD1 | Phase II | 2021 | Objective response rate and OS | 49 | Not disclosed | NCT02798406 (completed) |
| Oncolytic viruses Toca 511 | Vocimagene amiretrorepvec vector for a yeast cytosine deaminase gene which converts the prodrug Toca FC into the antimetabolite 5-fluorouracil | Phase I | 2016 | Safety, efficacy, and molecular profiling of Toca 511 OS | 45 | Excellent tolerability OS for recurrent high grade glioma was 13.6 months, statistically improved relative to an external control |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianconi, A.; Palmieri, G.; Aruta, G.; Monticelli, M.; Zeppa, P.; Tartara, F.; Melcarne, A.; Garbossa, D.; Cofano, F. Updates in Glioblastoma Immunotherapy: An Overview of the Current Clinical and Translational Scenario. Biomedicines 2023, 11, 1520. https://doi.org/10.3390/biomedicines11061520
Bianconi A, Palmieri G, Aruta G, Monticelli M, Zeppa P, Tartara F, Melcarne A, Garbossa D, Cofano F. Updates in Glioblastoma Immunotherapy: An Overview of the Current Clinical and Translational Scenario. Biomedicines. 2023; 11(6):1520. https://doi.org/10.3390/biomedicines11061520
Chicago/Turabian StyleBianconi, Andrea, Giuseppe Palmieri, Gelsomina Aruta, Matteo Monticelli, Pietro Zeppa, Fulvio Tartara, Antonio Melcarne, Diego Garbossa, and Fabio Cofano. 2023. "Updates in Glioblastoma Immunotherapy: An Overview of the Current Clinical and Translational Scenario" Biomedicines 11, no. 6: 1520. https://doi.org/10.3390/biomedicines11061520
APA StyleBianconi, A., Palmieri, G., Aruta, G., Monticelli, M., Zeppa, P., Tartara, F., Melcarne, A., Garbossa, D., & Cofano, F. (2023). Updates in Glioblastoma Immunotherapy: An Overview of the Current Clinical and Translational Scenario. Biomedicines, 11(6), 1520. https://doi.org/10.3390/biomedicines11061520