
Emerging Trends in the Field of Inflammation and Proteinopathy in ALS/FTD Spectrum Disorder

 , , , , ,
, , , , ,  , and
, and 
Abstract
:1. Introduction
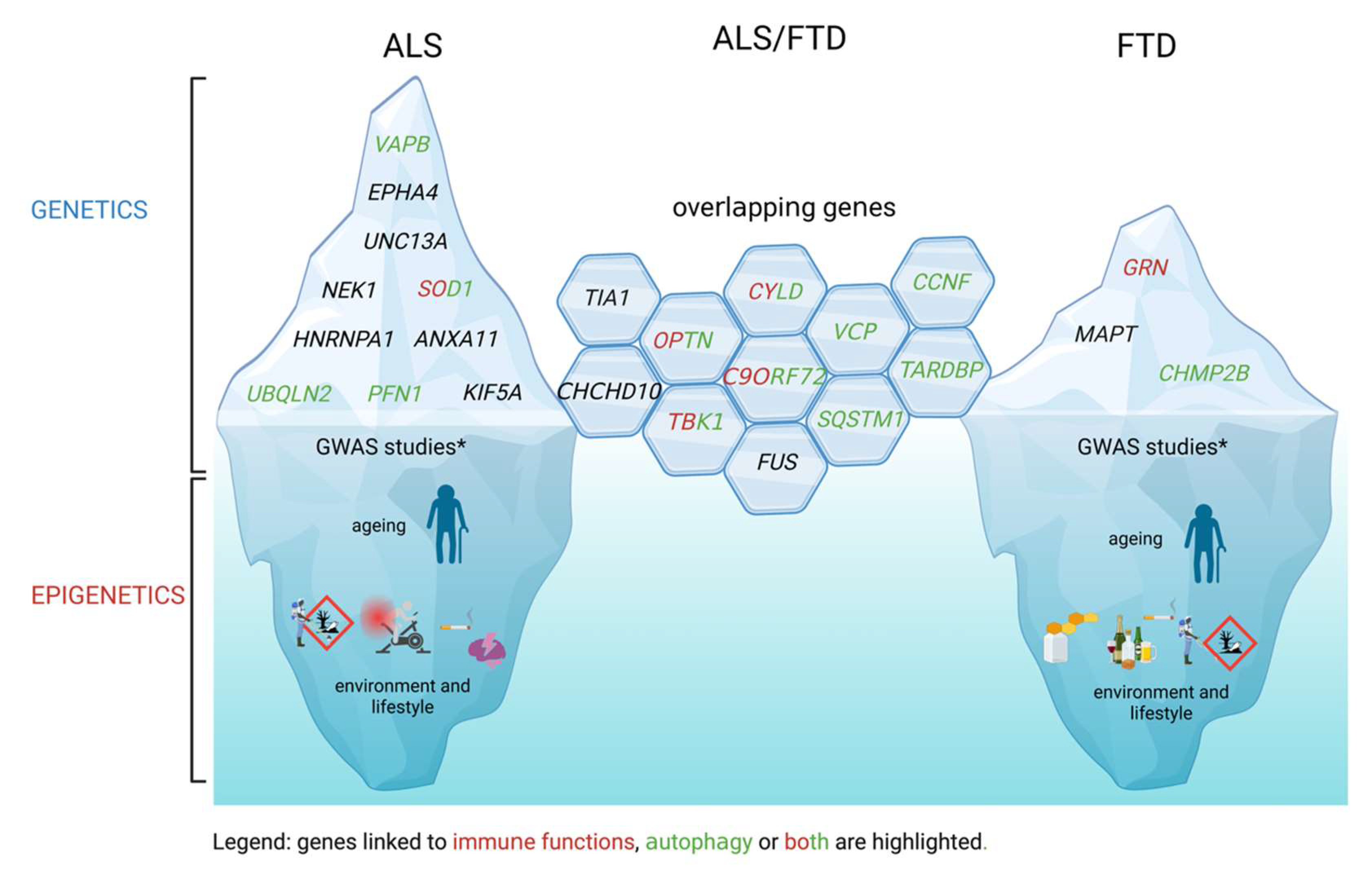
2. ALS and FTD Genetics, Epigenetics and Mechanisms
2.1. Mendelian Genetic Elements
2.2. GWAS Links
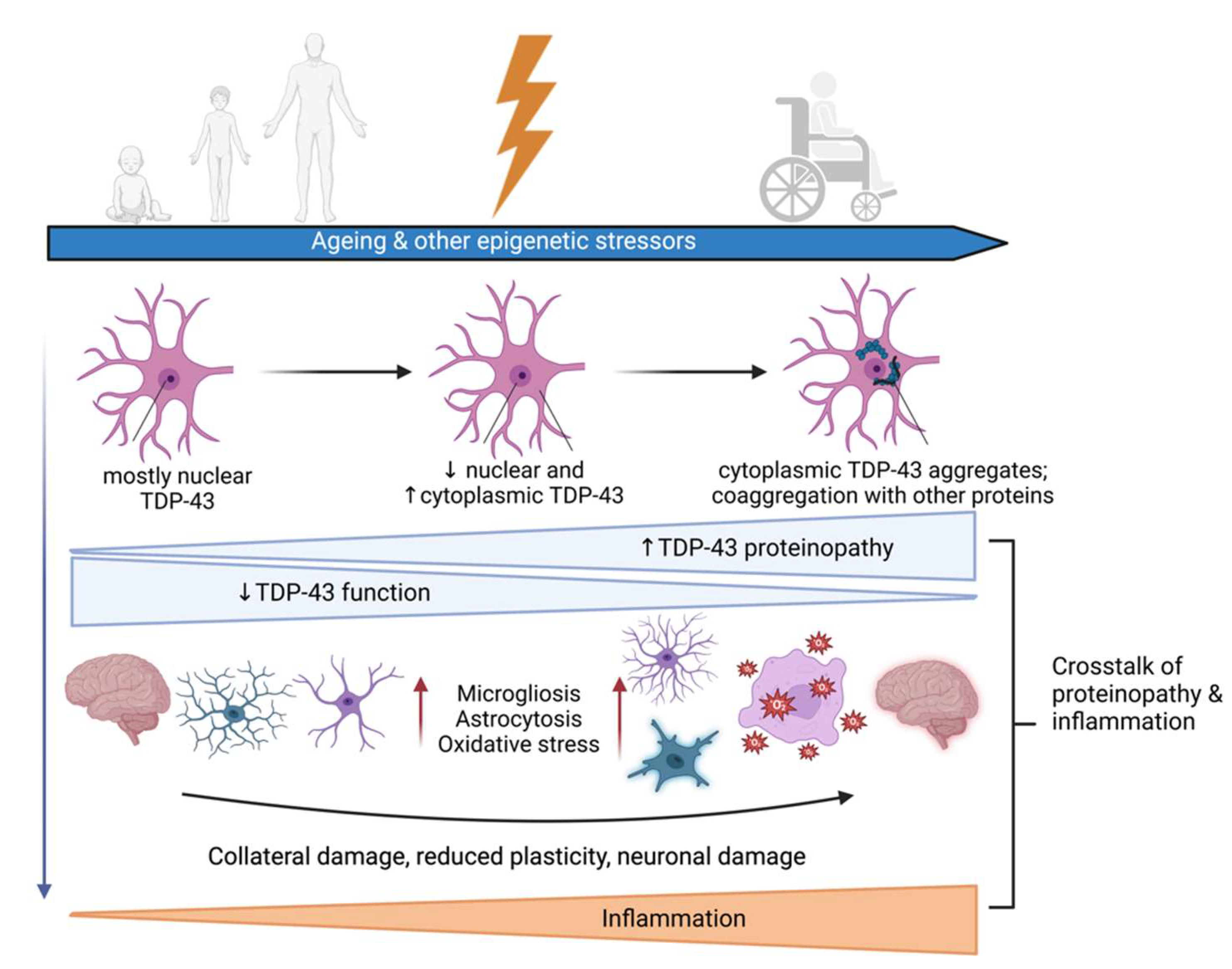
2.3. Environmental Factors and Epigenetics
3. Proteinopathy in ALS and FTD
4. Immunity in ALS and FTD
4.1. Immune System in Pathogenesis of Neurodegeneration: Brief Overview of Key Evidence
4.2. ALS as a Systemic Immune Disorder: Crosstalk between Immune Signaling and Metabolism
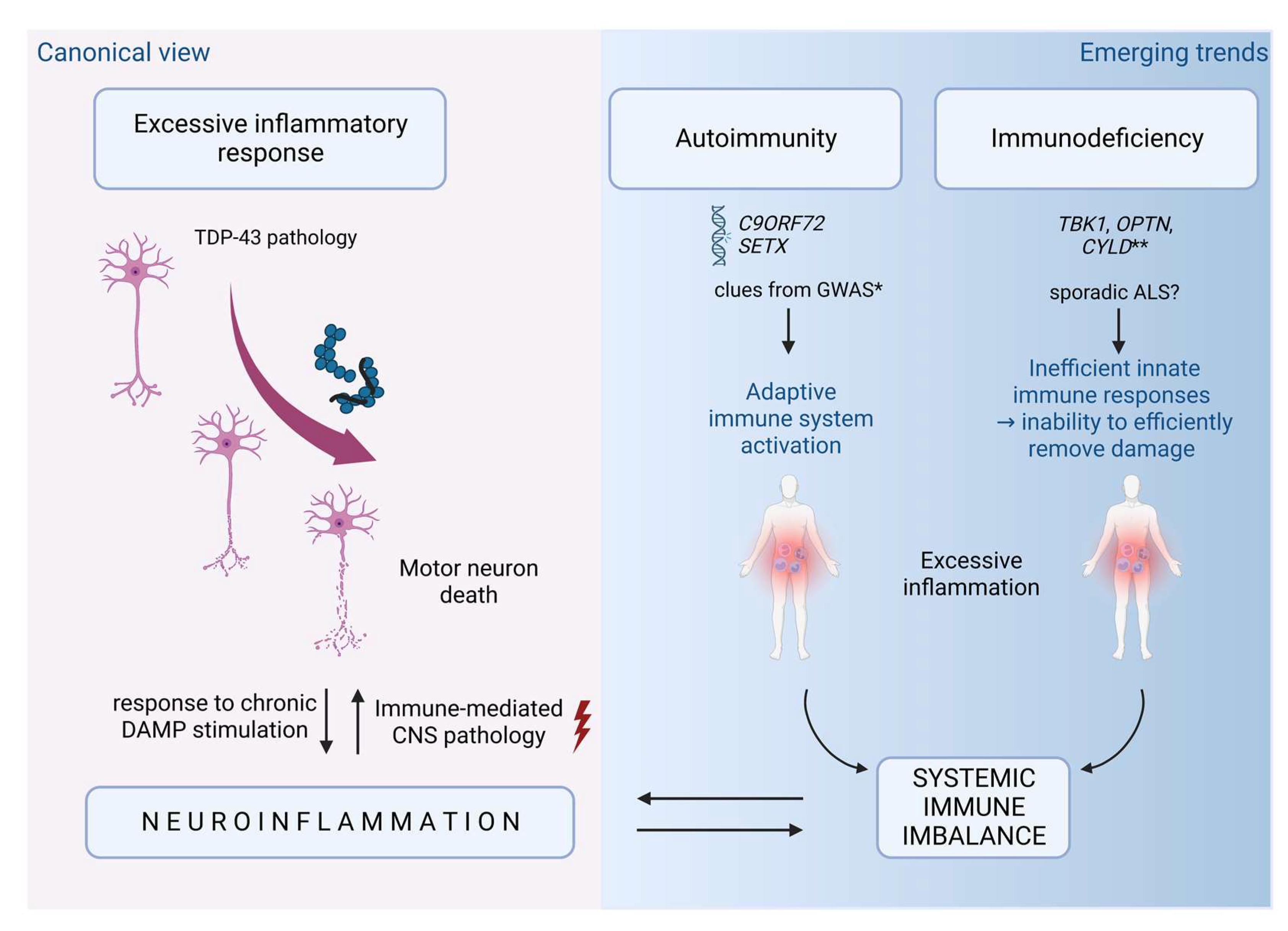
4.3. The Trilemma on the Origin of Immune Imbalance
4.3.1. Excessive Inflammatory Response
4.3.2. Immunodeficiency
4.3.3. Autoimmunity
5. Clinical Studies, Biomarkers and Emerging Therapies in ALS/FTD
5.1. Biomarkers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker | Patients | Notes | References |
|---|---|---|---|
| Neurofilaments | ALS/FTD | Correlation with axonal injury, pathophysiology, and disease progression rate (their levels correlate with shorter survival and more aggressive disease phenotypes); possible diagnostic and treatment markers (i.e., outcome in VALOR trial for SOD1 patients) | [222,223,224,225,226,227,228,229] |
| MAP2 | ALS | Increased CSF levels; possible motor neuron degeneration and disease-characterization marker | [230] |
| GFAP | FTD | Raised concentrations in GRN-related FTD; identification of different subgroups of FTD patients; astrogliosis marker; potential marker of proximity to onset | [231,232] |
| TREM2, CHIT1, YKL-40 | ALS/FTD | Increase in FTD forms associated with ALS; possible neurodegeneration and neuroinflammation markers in FTD | [234,235,236] |
| NPTX2 | GRN and C9orf72 mutation carriers | Reduced levels in patients | [234] |
| STMN-2 | ALS/FTD | Lower levels reported in post-mortem brain and spinal cord tissues of familial and sporadic ALS patients; possible diagnostic marker (not yet used as a marker in clinical trials) | [240,241,242,243] |
| TDP-43 | ALS/FTD | increased CSF levels; possible target engagement marker (not yet used as a marker in clinical trials) | [246,247] |
5.2. Experimental Therapies and Clinical Trials
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Modelling amyotrophic lateral sclerosis: Progress and possibilities. Dis. Model. Mech. 2017, 10, 537–549. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, R.; Haque, S.; Coley, K.; Shepheard, S.; Cooper-Knock, J.; Kirby, J. Multifaceted genes in amyotrophic lateral sclerosis-frontotemporal dementia. Front. Neurosci. 2020, 14, 684. [Google Scholar] [CrossRef] [PubMed]
- Broeck, L.V.; Callaerts, P.; Dermaut, B. TDP-43-mediated neurodegeneration: Towards a loss-of-function hypothesis? Trends Mol. Med. 2014, 20, 66–71. [Google Scholar] [CrossRef]
- Cascella, R.; Capitini, C.; Fani, G.; Dobson, C.M.; Cecchi, C.; Chiti, F. Quantification of the Relative Contributions of Loss-of-function and Gain-of-function Mechanisms in TAR DNA-binding Protein 43 (TDP-43) Proteinopathies. J. Biol. Chem. 2016, 291, 19437–19448. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: Mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Béland, L.-C.; Markovinovic, A.; Jakovac, H.; de Marchi, F.; Bilic, E.; Mazzini, L.; Kriz, J.; Munitic, I. Immunity in amyotrophic lateral sclerosis: Blurred lines between excessive inflammation and inefficient immune responses. Brain Commun. 2020, 2, fcaa124. [Google Scholar] [CrossRef]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, C.; Yuan, J.; Deng, Y. Peripheral immune system and neurodegenerative disease. Front. Aging Neurosci. 2022, 14, 970042. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.; Turner, M.R. Untangling neuroinflammation in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1303–1304. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Munitic, I.; Amedei, A.; Berry, J.D.; Feldman, E.L.; Aronica, E.; Nardo, G.; Van Weehaeghe, D.; Niccolai, E.; Prtenjaca, N. Interplay between immunity and amyotrophic lateral sclerosis: Clinical impact. Neurosci. Biobehav. Rev. 2021, 127, 958–978. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef]
- Greaves, C.V.; Rohrer, J.D. An update on genetic frontotemporal dementia. J. Neurol. 2019, 266, 2075–2086. [Google Scholar] [CrossRef]
- Lill, C.M.; Abel, O.; Bertram, L.; Al-Chalabi, A. Keeping up with genetic discoveries in amyotrophic lateral sclerosis: The ALSoD and ALSGene databases. Amyotroph. Lateral Scler. 2011, 12, 238–249. [Google Scholar] [CrossRef]
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Güneş, O.; Hall, P.; Hayhurst, J. The NHGRI-EBI GWAS Catalog: Knowledgebase and deposition resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; Van Swieten, J.C.; Myllykangas, L. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Smeyers, J.; Banchi, E.-G.; Latouche, M. C9ORF72: What it is, what it does, and why it matters. Front. Cell. Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Petrucelli, L. Disease mechanisms of C9ORF72 repeat expansions. Cold Spring Harb. Perspect. Med. 2018, 8, a024224. [Google Scholar] [CrossRef]
- Belzil, V.V.; Bauer, P.O.; Prudencio, M.; Gendron, T.F.; Stetler, C.T.; Yan, I.K.; Pregent, L.; Daughrity, L.; Baker, M.C.; Rademakers, R. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013, 126, 895–905. [Google Scholar] [CrossRef]
- O’rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- McCauley, M.E.; O’Rourke, J.G.; Yáñez, A.; Markman, J.L.; Ho, R.; Wang, X.; Chen, S.; Lall, D.; Jin, M.; Muhammad, A.K.M.G.; et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature 2020, 585, 96–101. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59. [Google Scholar] [CrossRef] [PubMed]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurother. J. Am. Soc. Exp. Neurother. 2015, 12, 364–375. [Google Scholar] [CrossRef]
- Weydt, P.; Yuen, E.C.; Ransom, B.R.; Möller, T. Increased cytotoxic potential of microglia from ALS-transgenic mice. Glia 2004, 48, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Markovinovic, A.; Cimbro, R.; Ljutic, T.; Kriz, J.; Rogelj, B.; Munitic, I. Optineurin in amyotrophic lateral sclerosis: Multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog. Neurobiol. 2017, 154, 1–20. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Pottier, C.; Bieniek, K.F.; Finch, N.; van de Vorst, M.; Baker, M.; Perkersen, R.; Brown, P.; Ravenscroft, T.; van Blitterswijk, M.; Nicholson, A.M.; et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015, 130, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Slowicka, K.; van Loo, G. Optineurin Functions for Optimal Immunity. Front. Immunol. 2018, 9, 769. [Google Scholar] [CrossRef] [PubMed]
- Toth, R.P.; Atkin, J.D. Dysfunction of Optineurin in Amyotrophic Lateral Sclerosis and Glaucoma. Front. Immunol. 2018, 9, 1017. [Google Scholar] [CrossRef]
- Markovinovic, A.; Ljutic, T.; Béland, L.-C.; Munitic, I. Optineurin Insufficiency Disbalances Proinflammatory and Anti-inflammatory Factors by Reducing Microglial IFN-β Responses. Neuroscience 2018, 388, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, S.; Oikawa, D.; Ishii, R.; Ayaki, T.; Takahashi, H.; Takeda, H.; Ishitani, R.; Kamei, K.; Takeyoshi, I.; Kawakami, H.; et al. Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat. Commun. 2016, 7, 12547. [Google Scholar] [CrossRef]
- Prtenjaca, N.; Dominovic, M.; Peradinovic, J.; Šajn, R.; Markovinovic, A.; Munitic, I. Optineurin dysfunction in amyotrophic lateral sclerosis: Why so puzzling? Period. Biol. 2020, 23–34. [Google Scholar] [CrossRef]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef]
- Schymick, J.C.; Yang, Y.; Andersen, P.M.; Vonsattel, J.P.; Greenway, M.; Momeni, P.; Elder, J.; Chiò, A.; Restagno, G.; Robberecht, W.; et al. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J. Neurol. Neurosurg. Psychiatry 2007, 78, 754–756. [Google Scholar] [CrossRef]
- Jian, J.; Konopka, J.; Liu, C. Insights into the role of progranulin in immunity, infection, and inflammation. J. Leukoc. Biol. 2013, 93, 199–208. [Google Scholar] [CrossRef]
- Yin, F.; Banerjee, R.; Thomas, B.; Zhou, P.; Qian, L.; Jia, T.; Ma, X.; Ma, Y.; Iadecola, C.; Beal, M.F.; et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J. Exp. Med. 2010, 207, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-E.; Bird, T.D.; Bekris, L.M.; Montine, T.J.; Leverenz, J.B.; Steinbart, E.; Galloway, N.M.; Feldman, H.; Woltjer, R.; Miller, C.A.; et al. The spectrum of mutations in progranulin: A collaborative study screening 545 cases of neurodegeneration. Arch. Neurol. 2010, 67, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Gijselinck, I.; Van Mossevelde, S.; van der Zee, J.; Sieben, A.; Philtjens, S.; Heeman, B.; Engelborghs, S.; Vandenbulcke, M.; De Baets, G.; Bäumer, V.; et al. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology 2015, 85, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, L.; Zhang, S.-Y.; Casanova, J.-L.; Sancho-Shimizu, V. Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol. Med. 2016, 22, 511–527. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.-M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef]
- Dobson-Stone, C.; Hallupp, M.; Shahheydari, H.; Ragagnin, A.M.G.; Chatterton, Z.; Carew-Jones, F.; Shepherd, C.E.; Stefen, H.; Paric, E.; Fath, T.; et al. CYLD is a causative gene for frontotemporal dementia—Amyotrophic lateral sclerosis. Brain 2020, 143, 783–799. [Google Scholar] [CrossRef]
- Wu, J.J.; Cai, A.; Greenslade, J.E.; Higgins, N.R.; Fan, C.; Le, N.T.; Tatman, M.; Whiteley, A.M.; Prado, M.A.; Dieriks, B.V.; et al. ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. Proc. Natl. Acad. Sci. USA 2020, 117, 15230–15241. [Google Scholar] [CrossRef]
- Morimoto, N.; Nagai, M.; Ohta, Y.; Miyazaki, K.; Kurata, T.; Morimoto, M.; Murakami, T.; Takehisa, Y.; Ikeda, Y.; Kamiya, T.; et al. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 2007, 1167, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Goode, A.; Butler, K.; Long, J.; Cavey, J.; Scott, D.; Shaw, B.; Sollenberger, J.; Gell, C.; Johansen, T.; Oldham, N.J.; et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 2016, 12, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Kurashige, T.; Kuramochi, M.; Ohsawa, R.; Yamashita, Y.; Shioi, G.; Morino, H.; Kamada, M.; Ayaki, T.; Ito, H.; Sotomaru, Y. Optineurin defects cause TDP43-pathology with autophagic vacuolar formation. Neurobiol. Dis. 2021, 148, 105215. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef]
- Pourcelot, M.; Zemirli, N.; Silva Da Costa, L.; Loyant, R.; Garcin, D.; Vitour, D.; Munitic, I.; Vazquez, A.; Arnoult, D. The Golgi apparatus acts as a platform for TBK1 activation after viral RNA sensing. BMC Biol. 2016, 14, 69. [Google Scholar] [CrossRef]
- Meena, N.P.; Zhu, G.; Mittelstadt, P.R.; Giardino Torchia, M.L.; Pourcelot, M.; Arnoult, D.; Ashwell, J.D.; Munitic, I. The TBK1-binding domain of optineurin promotes type I interferon responses. FEBS Lett. 2016, 590, 1498–1508. [Google Scholar] [CrossRef]
- Ciura, S.; Sellier, C.; Campanari, M.-L.; Charlet-Berguerand, N.; Kabashi, E. The most prevalent genetic cause of ALS-FTD, C9orf72 synergizes the toxicity of ATXN2 intermediate polyglutamine repeats through the autophagy pathway. Autophagy 2016, 12, 1406–1408. [Google Scholar] [CrossRef]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.K.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef]
- Sellier, C.; Campanari, M.-L.; Julie Corbier, C.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Boivin, M.; Pfister, V.; Gaucherot, A.; Ruffenach, F.; Negroni, L.; Sellier, C.; Charlet-Berguerand, N. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 2020, 39, e100574. [Google Scholar] [CrossRef] [PubMed]
- Božič, J.; Motaln, H.; Janež, A.P.; Markič, L.; Tripathi, P.; Yamoah, A.; Aronica, E.; Lee, Y.-B.; Heilig, R.; Fischer, R.; et al. Interactome screening of C9orf72 dipeptide repeats reveals VCP sequestration and functional impairment by polyGA. Brain 2022, 145, 684–699. [Google Scholar] [CrossRef]
- He, L.; Chen, L.; Li, L. The TBK1-OPTN Axis Mediates Crosstalk Between Mitophagy and the Innate Immune Response: A Potential Therapeutic Target for Neurodegenerative Diseases. Neurosci. Bull. 2017, 33, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Guo, H.; Dreser, A.; Yamoah, A.; Sechi, A.; Jesse, C.M.; Katona, I.; Doukas, P.; Nikolin, S.; Ernst, S.; et al. Pathomechanisms of ALS8: Altered autophagy and defective RNA binding protein (RBP) homeostasis due to the VAPB P56S mutation. Cell Death Dis. 2021, 12, 466. [Google Scholar] [CrossRef]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef]
- Burk, K.; Pasterkamp, R.J. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 2019, 137, 859–877. [Google Scholar] [CrossRef]
- Zhao, J.; Cooper, L.T.; Boyd, A.W.; Bartlett, P.F. Decreased signalling of EphA4 improves functional performance and motor neuron survival in the SOD1(G93A) ALS mouse model. Sci. Rep. 2018, 8, 11393. [Google Scholar] [CrossRef]
- Willemse, S.W.; Harley, P.; van Eijk, R.P.A.; Demaegd, K.C.; Zelina, P.; Pasterkamp, R.J.; van Damme, P.; Ingre, C.; van Rheenen, W.; Veldink, J.H.; et al. UNC13A in amyotrophic lateral sclerosis: From genetic association to therapeutic target. J. Neurol. Neurosurg. Psychiatry 2023. [Google Scholar] [CrossRef]
- Guo, W.; Vandoorne, T.; Steyaert, J.; Staats, K.A.; Van Den Bosch, L. The multifaceted role of kinases in amyotrophic lateral sclerosis: Genetic, pathological and therapeutic implications. Brain 2020, 143, 1651–1673. [Google Scholar] [CrossRef]
- Mead, R.J.; Shan, N.; Reiser, H.J.; Marshall, F.; Shaw, P.J. Amyotrophic lateral sclerosis: A neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discov. 2023, 22, 185–212. [Google Scholar] [CrossRef]
- Tak, Y.J.; Park, J.-H.; Rhim, H.; Kang, S. ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. Int. J. Mol. Sci. 2020, 21, 7525. [Google Scholar] [CrossRef] [PubMed]
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2018, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Kapeli, K.; Martinez, F.J.; Yeo, G.W. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum. Genet. 2017, 136, 1193–1214. [Google Scholar] [CrossRef] [PubMed]
- Nahm, M.; Lim, S.M.; Kim, Y.-E.; Park, J.; Noh, M.-Y.; Lee, S.; Roh, J.E.; Hwang, S.-M.; Park, C.-K.; Kim, Y.H.; et al. ANXA11 mutations in ALS cause dysregulation of calcium homeostasis and stress granule dynamics. Sci. Transl. Med. 2020, 12, eaax3993. [Google Scholar] [CrossRef] [PubMed]
- Teyssou, E.; Chartier, L.; Roussel, D.; Perera, N.D.; Nemazanyy, I.; Langui, D.; Albert, M.; Larmonier, T.; Saker, S.; Salachas, F.; et al. The Amyotrophic Lateral Sclerosis M114T PFN1 Mutation Deregulates Alternative Autophagy Pathways and Mitochondrial Homeostasis. Int. J. Mol. Sci. 2022, 23, 5694. [Google Scholar] [CrossRef]
- Kim, W.; Kim, D.-Y.; Lee, K.-H. RNA-Binding Proteins and the Complex Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 2598. [Google Scholar] [CrossRef]
- Baron, D.M.; Fenton, A.R.; Saez-Atienzar, S.; Giampetruzzi, A.; Sreeram, A.; Keagle, P.J.; Doocy, V.R.; Smith, N.J.; Danielson, E.W.; Andresano, M.; et al. ALS-associated KIF5A mutations abolish autoinhibition resulting in a toxic gain of function. Cell Rep. 2022, 39, 110598. [Google Scholar] [CrossRef]
- Chisholm, C.G.; Lum, J.S.; Farrawell, N.E.; Yerbury, J.J. Ubiquitin homeostasis disruption, a common cause of proteostasis collapse in amyotrophic lateral sclerosis? Neural Regen. Res. 2022, 17, 2218–2220. [Google Scholar] [CrossRef]
- Rossi, S.; Cozzolino, M. Dysfunction of RNA/RNA-Binding Proteins in ALS Astrocytes and Microglia. Cells 2021, 10, 3005. [Google Scholar] [CrossRef]
- Muyderman, H.; Chen, T. Mitochondrial dysfunction in amyotrophic lateral sclerosis—A valid pharmacological target? Br. J. Pharmacol. 2014, 171, 2191–2205. [Google Scholar] [CrossRef]
- Lehmer, C.; Schludi, M.H.; Ransom, L.; Greiling, J.; Junghänel, M.; Exner, N.; Riemenschneider, H.; van der Zee, J.; Van Broeckhoven, C.; Weydt, P.; et al. A novel CHCHD10 mutation implicates a Mia40-dependent mitochondrial import deficit in ALS. EMBO Mol. Med. 2018, 10, e8558. [Google Scholar] [CrossRef]
- Torres, P.; Cabral-Miranda, F.; Gonzalez-Teuber, V.; Hetz, C. Proteostasis deregulation as a driver of C9ORF72 pathogenesis. J. Neurochem. 2021, 159, 941–957. [Google Scholar] [CrossRef]
- Ryan, S.; Rollinson, S.; Hobbs, E.; Pickering-Brown, S. C9orf72 dipeptides disrupt the nucleocytoplasmic transport machinery and cause TDP-43 mislocalisation to the cytoplasm. Sci. Rep. 2022, 12, 4799. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Mehta, A.R.; Nirujogi, R.S.; Cooper, J.; James, O.G.; Nanda, J.; Longden, J.; Burr, K.; McDade, K.; Salzinger, A.; et al. Cell-autonomous immune dysfunction driven by disrupted autophagy in C9orf72-ALS iPSC-derived microglia contributes to neurodegeneration. Sci. Adv. 2023, 9, eabq0651. [Google Scholar] [CrossRef] [PubMed]
- Herhaus, L. TBK1 (TANK-binding kinase 1)-mediated regulation of autophagy in health and disease. Matrix Biol. 2021, 100–101, 84–98. [Google Scholar] [CrossRef]
- Benson, B.C.; Shaw, P.J.; Azzouz, M.; Highley, J.R.; Hautbergue, G.M. Proteinopathies as Hallmarks of Impaired Gene Expression, Proteostasis and Mitochondrial Function in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2021, 15, 783624. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Wang, H.; Hao, Z.; Fu, C.; Hu, Q.; Gao, F.; Ren, H.; Chen, D.; Han, J.; Ying, Z.; et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J. 2016, 35, 121–142. [Google Scholar] [CrossRef]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.W.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef]
- Chou, C.-C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef]
- Zajicek, A.S.; Ruan, H.; Dai, H.; Skolfield, M.C.; Phillips, H.L.; Burnette, W.J.; Javidfar, B.; Sun, S.-C.; Akbarian, S.; Yao, W.-D. Cylindromatosis drives synapse pruning and weakening by promoting macroautophagy through Akt-mTOR signaling. Mol. Psychiatry 2022, 27, 2414–2424. [Google Scholar] [CrossRef]
- Deng, Z.; Lim, J.; Wang, Q.; Purtell, K.; Wu, S.; Palomo, G.M.; Tan, H.; Manfredi, G.; Zhao, Y.; Peng, J.; et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 2020, 16, 917–931. [Google Scholar] [CrossRef]
- Lee, A.; Rayner, S.L.; Gwee, S.S.L.; De Luca, A.; Shahheydari, H.; Sundaramoorthy, V.; Ragagnin, A.; Morsch, M.; Radford, R.; Galper, J.; et al. Pathogenic mutation in the ALS/FTD gene, CCNF, causes elevated Lys48-linked ubiquitylation and defective autophagy. Cell. Mol. Life Sci. 2018, 75, 335–354. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95, 808–816.e9. [Google Scholar] [CrossRef]
- Ljubenkov, P.A.; Miller, Z.; Mumford, P.; Zhang, J.; Allen, I.E.; Mitic, L.; Staffaroni, A.; Heuer, H.; Rojas, J.C.; Cobigo, Y.; et al. Peripheral Innate Immune Activation Correlates With Disease Severity in GRN Haploinsufficiency. Front. Neurol. 2019, 10, 1004. [Google Scholar] [CrossRef] [PubMed]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Sun, X.; Yue, W.; Duan, Y.; Hu, R.; Zhang, K.; Ni, J.; Cui, J.; Wang, Q.; Chen, Y.; et al. CHMP2B regulates TDP-43 phosphorylation and cytotoxicity independent of autophagy via CK1. J. Cell Biol. 2022, 221, e202103033. [Google Scholar] [CrossRef]
- Pottier, C.; Ren, Y.; Perkerson, R.B., 3rd; Baker, M.; Jenkins, G.D.; van Blitterswijk, M.; DeJesus-Hernandez, M.; van Rooij, J.G.J.; Murray, M.E.; Christopher, E.; et al. Genome-wide analyses as part of the international FTLD-TDP whole-genome sequencing consortium reveals novel disease risk factors and increases support for immune dysfunction in FTLD. Acta Neuropathol. 2019, 137, 879–899. [Google Scholar] [CrossRef]
- Ravnik-Glavač, M.; Goričar, K.; Vogrinc, D.; Koritnik, B.; Lavrenčič, J.G.; Glavač, D.; Dolžan, V. Genetic Variability of Inflammation and Oxidative Stress Genes Affects Onset, Progression of the Disease and Survival of Patients with Amyotrophic Lateral Sclerosis. Genes 2022, 13, 757. [Google Scholar] [CrossRef]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS genetics: Gains, losses, and implications for future therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef]
- Van Es, M.A.; Veldink, J.H.; Saris, C.G.J.; Blauw, H.M.; van Vught, P.W.J.; Birve, A.; Lemmens, R.; Schelhaas, H.J.; Groen, E.J.N.; Huisman, M.H.B.; et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 2009, 41, 1083–1087. [Google Scholar] [CrossRef]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.e6. [Google Scholar] [CrossRef]
- Lipstein, N.; Verhoeven-Duif, N.M.; Michelassi, F.E.; Calloway, N.; van Hasselt, P.M.; Pienkowska, K.; van Haaften, G.; van Haelst, M.M.; van Empelen, R.; Cuppen, I.; et al. Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder. J. Clin. Investig. 2017, 127, 1005–1018. [Google Scholar] [CrossRef]
- Diekstra, F.P.; van Vught, P.W.J.; van Rheenen, W.; Koppers, M.; Pasterkamp, R.J.; van Es, M.A.; Schelhaas, H.J.; de Visser, M.; Robberecht, W.; Van Damme, P.; et al. UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 630.e3–630.e8. [Google Scholar] [CrossRef]
- Brown, A.-L.; Wilkins, O.G.; Keuss, M.J.; Hill, S.E.; Zanovello, M.; Lee, W.C.; Bampton, A.; Lee, F.C.Y.; Masino, L.; Qi, Y.A.; et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 2022, 603, 131–137. [Google Scholar] [CrossRef]
- Ma, X.R.; Prudencio, M.; Koike, Y.; Vatsavayai, S.C.; Kim, G.; Harbinski, F.; Briner, A.; Rodriguez, C.M.; Guo, C.; Akiyama, T.; et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 2022, 603, 124–130. [Google Scholar] [CrossRef]
- Oskarsson, B.; Horton, D.K.; Mitsumoto, H. Potential Environmental Factors in Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 877–888. [Google Scholar] [CrossRef]
- Adani, G.; Filippini, T.; Garuti, C.; Malavolti, M.; Vinceti, G.; Zamboni, G.; Tondelli, M.; Galli, C.; Costa, M.; Vinceti, M.; et al. Environmental Risk Factors for Early-Onset Alzheimer’s Dementia and Frontotemporal Dementia: A Case-Control Study in Northern Italy. Int. J. Environ. Res. Public Health 2020, 17, 7941. [Google Scholar] [CrossRef]
- Henderson, R.D.; Kepp, K.P.; Eisen, A. ALS/FTD: Evolution, Aging, and Cellular Metabolic Exhaustion. Front. Neurol. 2022, 13, 890203. [Google Scholar] [CrossRef]
- Zhang, S.; Cooper-Knock, J.; Weimer, A.K.; Shi, M.; Moll, T.; Marshall, J.N.G.; Harvey, C.; Nezhad, H.G.; Franklin, J.; Souza, C.D.S.; et al. Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis. Neuron 2022, 110, 992–1008.e11. [Google Scholar] [CrossRef]
- Eitan, C.; Siany, A.; Barkan, E.; Olender, T.; van Eijk, K.R.; Moisse, M.; Farhan, S.M.K.; Danino, Y.M.; Yanowski, E.; Marmor-Kollet, H.; et al. Whole-genome sequencing reveals that variants in the Interleukin 18 Receptor Accessory Protein 3’UTR protect against ALS. Nat. Neurosci. 2022, 25, 433–445. [Google Scholar] [CrossRef]
- Lee, E.B.; Lee, V.M.-Y.; Trojanowski, J.Q. Gains or losses: Molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2011, 13, 38–50. [Google Scholar] [CrossRef]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef]
- Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L.; Krishnamurthy, S.; Paulucci-Holthauzen, A.A.; Sengupta, U.; Lasagna-Reeves, C.A.; Ahmad, Y.; Jackson, G.R.; Kayed, R. Amyloid-β oligomers as a template for secondary amyloidosis in Alzheimer’s disease. Neurobiol. Dis. 2014, 71, 14–23. [Google Scholar] [CrossRef]
- Yamashita, S.; Sakashita, N.; Yamashita, T.; Tawara, N.; Tasaki, M.; Kawakami, K.; Komohara, Y.; Fujiwara, Y.; Kamikawa, M.; Nakagawa, T.; et al. Concomitant accumulation of α-synuclein and TDP-43 in a patient with corticobasal degeneration. J. Neurol. 2014, 261, 2209–2217. [Google Scholar] [CrossRef]
- Koga, S.; Lin, W.-L.; Walton, R.L.; Ross, O.A.; Dickson, D.W. TDP-43 pathology in multiple system atrophy: Colocalization of TDP-43 and α-synuclein in glial cytoplasmic inclusions. Neuropathol. Appl. Neurobiol. 2018, 44, 707–721. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Nihei, Y.; Ito, D.; Suzuki, N. Roles of ataxin-2 in pathological cascades mediated by TAR DNA-binding protein 43 (TDP-43) and Fused in Sarcoma (FUS). J. Biol. Chem. 2012, 287, 41310–41323. [Google Scholar] [CrossRef]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G.; et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef]
- Gomez-Deza, J.; Lee, Y.-B.; Troakes, C.; Nolan, M.; Al-Sarraj, S.; Gallo, J.-M.; Shaw, C.E. Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutant amyotrophic lateral sclerosis and are unlikely to cause their degeneration. Acta Neuropathol. Commun. 2015, 3, 38. [Google Scholar] [CrossRef]
- Saberi, S.; Stauffer, J.E.; Jiang, J.; Garcia, S.D.; Taylor, A.E.; Schulte, D.; Ohkubo, T.; Schloffman, C.L.; Maldonado, M.; Baughn, M.; et al. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 135, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, M.; Takahashi, T.; Yamazaki, Y.; Kurashige, T.; Yamawaki, T.; Matsumoto, M. Cyclin-dependent kinase 5 immunoreactivity for granulovacuolar degeneration. Neuroreport 2012, 23, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Endo, R.; Takashima, N.; Nekooki-Machida, Y.; Komi, Y.; Hui, K.K.-W.; Takao, M.; Akatsu, H.; Murayama, S.; Sawa, A.; Tanaka, M. TAR DNA-Binding Protein 43 and Disrupted in Schizophrenia 1 Coaggregation Disrupts Dendritic Local Translation and Mental Function in Frontotemporal Lobar Degeneration. Biol. Psychiatry 2018, 84, 509–521. [Google Scholar] [CrossRef] [PubMed]
- De Santis, R.; Alfano, V.; de Turris, V.; Colantoni, A.; Santini, L.; Garone, M.G.; Antonacci, G.; Peruzzi, G.; Sudria-Lopez, E.; Wyler, E.; et al. Mutant FUS and ELAVL4 (HuD) Aberrant Crosstalk in Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 27, 3818–3831.e5. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Jagaraj, C.J.; Vidal, M.; Ragagnin, A.M.G.; Perri, E.R.; Konopka, A.; Toth, R.P.; Galper, J.; Blair, I.P.; Thomas, C.J.; et al. ERp57 is protective against mutant SOD1-induced cellular pathology in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2018, 27, 1311–1331. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, Y.; Shimazawa, M.; Ohuchi, K.; Ito, J.; Takahashi, H.; Tsuruma, K.; Kakita, A.; Hara, H. GPNMB ameliorates mutant TDP-43-induced motor neuron cell death. J. Neurosci. Res. 2017, 95, 1647–1665. [Google Scholar] [CrossRef]
- Davidson, Y.S.; Robinson, A.C.; Flood, L.; Rollinson, S.; Benson, B.C.; Asi, Y.T.; Richardson, A.; Jones, M.; Snowden, J.S.; Pickering-Brown, S.; et al. Heterogeneous ribonuclear protein E2 (hnRNP E2) is associated with TDP-43-immunoreactive neurites in Semantic Dementia but not with other TDP-43 pathological subtypes of Frontotemporal Lobar Degeneration. Acta Neuropathol. Commun. 2017, 5, 54. [Google Scholar] [CrossRef]
- Douville, R.; Liu, J.; Rothstein, J.; Nath, A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol. 2011, 69, 141–151. [Google Scholar] [CrossRef]
- Berjaoui, S.; Povedano, M.; Garcia-Esparcia, P.; Carmona, M.; Aso, E.; Ferrer, I. Complex Inflammation mRNA-Related Response in ALS Is Region Dependent. Neural Plast. 2015, 2015, 573784. [Google Scholar] [CrossRef]
- Gleixner, A.M.; Verdone, B.M.; Otte, C.G.; Anderson, E.N.; Ramesh, N.; Shapiro, O.R.; Gale, J.R.; Mauna, J.C.; Mann, J.R.; Copley, K.E.; et al. NUP62 localizes to ALS/FTLD pathological assemblies and contributes to TDP-43 insolubility. Nat. Commun. 2022, 13, 3380. [Google Scholar] [CrossRef]
- Hortobágyi, T.; Troakes, C.; Nishimura, A.L.; Vance, C.; van Swieten, J.C.; Seelaar, H.; King, A.; Al-Sarraj, S.; Rogelj, B.; Shaw, C.E. Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders. Acta Neuropathol. 2011, 121, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T.; Mizuno, Y.; Fujita, Y.; Takatama, M.; Nakazato, Y.; Okamoto, K. Optineurin in neurodegenerative diseases. Neuropathology 2011, 31, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Rubino, E.; Rainero, I.; Chiò, A.; Rogaeva, E.; Galimberti, D.; Fenoglio, P.; Grinberg, Y.; Isaia, G.; Calvo, A.; Gentile, S. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 2012, 79, 1556–1562. [Google Scholar] [CrossRef]
- Tanji, K.; Zhang, H.-X.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. p62/sequestosome 1 binds to TDP-43 in brains with frontotemporal lobar degeneration with TDP-43 inclusions. J. Neurosci. Res. 2012, 90, 2034–2042. [Google Scholar] [CrossRef]
- McGurk, L.; Lee, V.M.; Trojanowksi, J.Q.; Van Deerlin, V.M.; Lee, E.B.; Bonini, N.M. Poly-A binding protein-1 localization to a subset of TDP-43 inclusions in amyotrophic lateral sclerosis occurs more frequently in patients harboring an expansion in C9orf72. J. Neuropathol. Exp. Neurol. 2014, 73, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Hasegawa, M. Profilin 1 mutants form aggregates that induce accumulation of prion-like TDP-43. Prion 2016, 10, 283–289. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nonaka, T.; Suzuki, G.; Kametani, F.; Hasegawa, M. Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum. Mol. Genet. 2016, 25, 1420–1433. [Google Scholar] [CrossRef]
- Seyfried, N.T.; Gozal, Y.M.; Donovan, L.E.; Herskowitz, J.H.; Dammer, E.B.; Xia, Q.; Ku, L.; Chang, J.; Duong, D.M.; Rees, H.D.; et al. Quantitative analysis of the detergent-insoluble brain proteome in frontotemporal lobar degeneration using SILAC internal standards. J. Proteome Res. 2012, 11, 2721–2738. [Google Scholar] [CrossRef]
- Liu, G.; Coyne, A.N.; Pei, F.; Vaughan, S.; Chaung, M.; Zarnescu, D.C.; Buchan, J.R. Endocytosis regulates TDP-43 toxicity and turnover. Nat. Commun. 2017, 8, 2092. [Google Scholar] [CrossRef]
- Collins, M.; Riascos, D.; Kovalik, T.; An, J.; Krupa, K.; Krupa, K.; Hood, B.L.; Conrads, T.P.; Renton, A.E.; Traynor, B.J.; et al. The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 2012, 124, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Collins, M.; An, J.; Geiser, R.; Tegeler, T.; Tsantilas, K.; Garcia, K.; Pirrotte, P.; Bowser, R. Immunoprecipitation and mass spectrometry defines an extensive RBM45 protein-protein interaction network. Brain Res. 2016, 1647, 79–93. [Google Scholar] [CrossRef]
- Mashiko, T.; Sakashita, E.; Kasashima, K.; Tominaga, K.; Kuroiwa, K.; Nozaki, Y.; Matsuura, T.; Hamamoto, T.; Endo, H. Developmentally Regulated RNA-binding Protein 1 (Drb1)/RNA-binding Motif Protein 45 (RBM45), a Nuclear-Cytoplasmic Trafficking Protein, Forms TAR DNA-binding Protein 43 (TDP-43)-mediated Cytoplasmic Aggregates. J. Biol. Chem. 2016, 291, 14996–15007. [Google Scholar] [CrossRef]
- Keller, B.A.; Volkening, K.; Droppelmann, C.A.; Ang, L.C.; Rademakers, R.; Strong, M.J. Co-aggregation of RNA binding proteins in ALS spinal motor neurons: Evidence of a common pathogenic mechanism. Acta Neuropathol. 2012, 124, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Droppelmann, C.A.; Keller, B.A.; Campos-Melo, D.; Volkening, K.; Strong, M.J. Rho guanine nucleotide exchange factor is an NFL mRNA destabilizing factor that forms cytoplasmic inclusions in amyotrophic lateral sclerosis. Neurobiol. Aging 2013, 34, 248–262. [Google Scholar] [CrossRef]
- Farhan, S.M.K.; Gendron, T.F.; Petrucelli, L.; Hegele, R.A.; Strong, M.J. ARHGEF28 p.Lys280Metfs40Ter in an amyotrophic lateral sclerosis family with a C9orf72 expansion. Neurol. Genet. 2017, 3, e190. [Google Scholar] [CrossRef]
- Shang, J.; Yamashita, T.; Nakano, Y.; Morihara, R.; Li, X.; Feng, T.; Liu, X.; Huang, Y.; Fukui, Y.; Hishikawa, N.; et al. Aberrant distributions of nuclear pore complex proteins in ALS mice and ALS patients. Neuroscience 2017, 350, 158–168. [Google Scholar] [CrossRef]
- Takeuchi, R.; Toyoshima, Y.; Tada, M.; Tanaka, H.; Shimizu, H.; Shiga, A.; Miura, T.; Aoki, K.; Aikawa, A.; Ishizawa, S.; et al. Globular Glial Mixed Four Repeat Tau and TDP-43 Proteinopathy with Motor Neuron Disease and Frontotemporal Dementia. Brain Pathol. 2016, 26, 82–94. [Google Scholar] [CrossRef]
- Smith, V.D.; Bachstetter, A.D.; Ighodaro, E.; Roberts, K.; Abner, E.L.; Fardo, D.W.; Nelson, P.T. Overlapping but distinct TDP-43 and tau pathologic patterns in aged hippocampi. Brain Pathol. 2018, 28, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Liachko, N.F.; McMillan, P.J.; Strovas, T.J.; Loomis, E.; Greenup, L.; Murrell, J.R.; Ghetti, B.; Raskind, M.A.; Montine, T.J.; Bird, T.D.; et al. The tau tubulin kinases TTBK1/2 promote accumulation of pathological TDP-43. PLoS Genet. 2014, 10, e1004803. [Google Scholar] [CrossRef]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.L.; Warraich, S.T.; Yang, S.; Solski, J.A.; Fernando, R.; Rouleau, G.A.; Nicholson, G.A.; Blair, I.P. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 2527.e3–2527.e10. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Tamaki, Y.; Ayaki, T.; Shodai, A.; Kaji, S.; Morimura, T.; Banno, Y.; Nishitsuji, K.; Sakashita, N.; Maki, T.; et al. CUL2-mediated clearance of misfolded TDP-43 is paradoxically affected by VHL in oligodendrocytes in ALS. Sci. Rep. 2016, 6, 19118. [Google Scholar] [CrossRef] [PubMed]
- Gozal, Y.M.; Seyfried, N.T.; Gearing, M.; Glass, J.D.; Heilman, C.J.; Wuu, J.; Duong, D.M.; Cheng, D.; Xia, Q.; Rees, H.D. Aberrant septin 11 is associated with sporadic frontotemporal lobar degeneration. Mol. Neurodegener. 2011, 6, 82. [Google Scholar] [CrossRef]
- Mehta, P.R.; Brown, A.-L.; Ward, M.E.; Fratta, P. The era of cryptic exons: Implications for ALS-FTD. Mol. Neurodegener. 2023, 18, 16. [Google Scholar] [CrossRef]
- Bampton, A.; Gatt, A.; Humphrey, J.; Cappelli, S.; Bhattacharya, D.; Foti, S.; Brown, A.-L.; Asi, Y.; Low, Y.H.; Foiani, M.; et al. HnRNP K mislocalisation is a novel protein pathology of frontotemporal lobar degeneration and ageing and leads to cryptic splicing. Acta Neuropathol. 2021, 142, 609–627. [Google Scholar] [CrossRef]
- Ghetti, B.; Buratti, E.; Boeve, B.; Rademakers, R. Frontotemporal dementias. Adv. Exp. Med. Biol. 2021, 1281, 1. [Google Scholar]
- Neumann, M.; Lee, E.B.; Mackenzie, I.R. Frontotemporal Lobar Degeneration TDP-43-Immunoreactive Pathological Subtypes: Clinical and Mechanistic Significance. Adv. Exp. Med. Biol. 2021, 1281, 201–217. [Google Scholar] [CrossRef]
- De Rossi, P.; Lewis, A.J.; Furrer, J.; De Vos, L.; Demeter, T.; Zbinden, A.; Zhong, W.; Wiersma, V.I.; Scialo, C.; Weber, J.; et al. FTLD-TDP assemblies seed neoaggregates with subtype-specific features via a prion-like cascade. EMBO Rep. 2021, 22, e53877. [Google Scholar] [CrossRef]
- Li, Q.; Babinchak, W.M.; Surewicz, W.K. Cryo-EM structure of amyloid fibrils formed by the entire low complexity domain of TDP-43. Nat. Commun. 2021, 12, 1620. [Google Scholar] [CrossRef]
- Arseni, D.; Hasegawa, M.; Murzin, A.G.; Kametani, F.; Arai, M.; Yoshida, M.; Ryskeldi-Falcon, B. Structure of pathological TDP-43 filaments from ALS with FTLD. Nature 2022, 601, 139–143. [Google Scholar] [CrossRef]
- Cascella, R.; Bigi, A.; Riffert, D.G.; Gagliani, M.C.; Ermini, E.; Moretti, M.; Cortese, K.; Cecchi, C.; Chiti, F. A quantitative biology approach correlates neuronal toxicity with the largest inclusions of TDP-43. Sci. Adv. 2022, 8, eabm6376. [Google Scholar] [CrossRef]
- Cragnaz, L.; Klima, R.; Skoko, N.; Budini, M.; Feiguin, F.; Baralle, F.E. Aggregate formation prevents dTDP-43 neurotoxicity in the Drosophila melanogaster eye. Neurobiol. Dis. 2014, 71, 74–80. [Google Scholar] [CrossRef]
- Štalekar, M.; Yin, X.; Rebolj, K.; Darovic, S.; Troakes, C.; Mayr, M.; Shaw, C.E.; Rogelj, B. Proteomic analyses reveal that loss of TDP-43 affects RNA processing and intracellular transport. Neuroscience 2015, 293, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Šušnjar, U.; Škrabar, N.; Brown, A.-L.; Abbassi, Y.; Phatnani, H.; Cortese, A.; Cereda, C.; Bugiardini, E.; Cardani, R.; Meola, G.; et al. Cell environment shapes TDP-43 function with implications in neuronal and muscle disease. Commun. Biol. 2022, 5, 314. [Google Scholar] [CrossRef] [PubMed]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef]
- Schwartz, M.; Kipnis, J.; Rivest, S.; Prat, A. How do immune cells support and shape the brain in health, disease, and aging? J. Neurosci. 2013, 33, 17587–17596. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; McGeer, E.G. Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol. 1988, 76, 550–557. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G.; Kawamata, T.; Yamada, T.; Akiyama, H. Reactions of the immune system in chronic degenerative neurological diseases. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 1991, 18, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Troost, D.; van den Oord, J.J.; de Jong, J.M.; Swaab, D.F. Lymphocytic infiltration in the spinal cord of patients with amyotrophic lateral sclerosis. Clin. Neuropathol. 1989, 8, 289–294. [Google Scholar] [PubMed]
- Engelhardt, J.I.; Appel, S.H. IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch. Neurol. 1990, 47, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.I.; Tajti, J.; Appel, S.H. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch. Neurol. 1993, 50, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Alshikho, M.J.; Zürcher, N.R.; Loggia, M.L.; Cernasov, P.; Reynolds, B.; Pijanowski, O.; Chonde, D.B.; Izquierdo Garcia, D.; Mainero, C.; Catana, C. Integrated magnetic resonance imaging and [11C]-PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann. Neurol. 2018, 83, 1186–1197. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Boillée, S.; Velde, C.V.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillee, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-type microglia extend survival in PU. 1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Appel, S.H.; Chiu, I.M.; Chen, A.; Zheng, Y.; Kosaras, B.; Tsiftsoglou, S.A.; et al. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. Proc. Natl. Acad. Sci. USA 2008, 105, 64–79. [Google Scholar]
- Chiu, I.M.; Chen, A.; Zheng, Y.; Kosaras, B.; Tsiftsoglou, S.A.; Vartanian, T.K.; Brown, R.H.; Carroll, M.C. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 17913–17918. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Huang, A.; Wen, S.; Liao, B.; Appel, S.H. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 2011, 134, 1293–1314. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Zhao, W.; Wang, J.; Zhang, X.; Wen, S.; Neal, D.; Thonhoff, J.R.; Alsuliman, A.S.; Shpall, E.J.; Rezvani, K. ALS patients’ regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight 2017, 2, e89530. [Google Scholar] [CrossRef] [PubMed]
- Sheean, R.K.; McKay, F.C.; Cretney, E.; Bye, C.R.; Perera, N.D.; Tomas, D.; Weston, R.A.; Scheller, K.J.; Djouma, E.; Menon, P.; et al. Association of Regulatory T-Cell Expansion with Progression of Amyotrophic Lateral Sclerosis: A Study of Humans and a Transgenic Mouse Model. JAMA Neurol. 2018, 75, 681–689. [Google Scholar] [CrossRef]
- Yazdani, S.; Seitz, C.; Cui, C.; Lovik, A.; Pan, L.; Piehl, F.; Pawitan, Y.; Kläppe, U.; Press, R.; Samuelsson, K.; et al. T cell responses at diagnosis of amyotrophic lateral sclerosis predict disease progression. Nat. Commun. 2022, 13, 6733. [Google Scholar] [CrossRef]
- Thonhoff, J.R.; Beers, D.R.; Zhao, W.; Pleitez, M.; Simpson, E.P.; Berry, J.D.; Cudkowicz, M.E.; Appel, S.H. Expanded autologous regulatory T-lymphocyte infusions in ALS: A phase I, first-in-human study. Neurol. Neuroinflamm. 2018, 5, e465. [Google Scholar] [CrossRef]
- Camu, W.; Mickunas, M.; Veyrune, J.-L.; Payan, C.; Garlanda, C.; Locati, M.; Juntas-Morales, R.; Pageot, N.; Malaspina, A.; Andreasson, U. Repeated 5-day cycles of low dose aldesleukin in amyotrophic lateral sclerosis (IMODALS): A phase 2a randomised, double-blind, placebo-controlled trial. eBioMedicine 2020, 59, 102844. [Google Scholar] [CrossRef] [PubMed]
- Thonhoff, J.R.; Berry, J.D.; Macklin, E.A.; Beers, D.R.; Mendoza, P.A.; Zhao, W.; Thome, A.D.; Triolo, F.; Moon, J.J.; Paganoni, S.; et al. Combined Regulatory T-Lymphocyte and IL-2 Treatment Is Safe, Tolerable, and Biologically Active for 1 Year in Persons With Amyotrophic Lateral Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Appel, S.H. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J. Neuroimmune Pharmacol. 2013, 8, 888–899. [Google Scholar] [CrossRef]
- Graves, M.; Fiala, M.; Dinglasan, L.A.; Liu, N.; Sayre, J.; Chiappelli, F.; van Kooten, C.; Vinters, H. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2004, 5, 213–219. [Google Scholar] [CrossRef]
- Chiot, A.; Zaïdi, S.; Iltis, C.; Ribon, M.; Berriat, F.; Schiaffino, L.; Jolly, A.; de la Grange, P.; Mallat, M.; Bohl, D.; et al. Modifying macrophages at the periphery has the capacity to change microglial reactivity and to extend ALS survival. Nat. Neurosci. 2020, 23, 1339–1351. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Hooten, K.G.; Sieglaff, D.H.; Zhang, A.; Kalyana-Sundaram, S.; Traini, C.M.; Halsey, W.S.; Hughes, A.M.; Sathe, G.M. Characterization of gene expression phenotype in amyotrophic lateral sclerosis monocytes. JAMA Neurol. 2017, 74, 677–685. [Google Scholar] [CrossRef]
- Zondler, L.; Feiler, M.S.; Freischmidt, A.; Ruf, W.P.; Ludolph, A.C.; Danzer, K.M.; Weishaupt, J.H. Impaired activation of ALS monocytes by exosomes. Immunol. Cell Biol. 2017, 95, 207–214. [Google Scholar] [CrossRef]
- Zondler, L.; Müller, K.; Khalaji, S.; Bliederhäuser, C.; Ruf, W.P.; Grozdanov, V.; Thiemann, M.; Fundel-Clemes, K.; Freischmidt, A.; Holzmann, K.; et al. Peripheral monocytes are functionally altered and invade the CNS in ALS patients. Acta Neuropathol. 2016, 132, 391–411. [Google Scholar] [CrossRef]
- Nardo, G.; Pozzi, S.; Pignataro, M.; Lauranzano, E.; Spano, G.; Garbelli, S.; Mantovani, S.; Marinou, K.; Papetti, L.; Monteforte, M.; et al. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS ONE 2011, 6, e25545. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deng, W. Reverse engineering human neurodegenerative disease using pluripotent stem cell technology. Brain Res. 2016, 1638, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Gascon, R.; Miller, R.G.; Gelinas, D.F.; Mass, J.; Hadlock, K.; Jin, X.; Reis, J.; Narvaez, A.; McGrath, M.S. Evidence for systemic immune system alterations in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 2005, 159, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Picher-Martel, V.; Boutej, H.; Vézina, A.; Cordeau, P.; Kaneb, H.; Julien, J.-P.; Genge, A.; Dupré, N.; Kriz, J. Distinct Plasma Immune Profile in ALS Implicates sTNFR-II in pAMPK/Leptin Homeostasis. Int. J. Mol. Sci. 2023, 24, 5065. [Google Scholar] [CrossRef]
- Leoni, E.; Bremang, M.; Mitra, V.; Zubiri, I.; Jung, S.; Lu, C.-H.; Adiutori, R.; Lombardi, V.; Russell, C.; Koncarevic, S.; et al. Combined Tissue-Fluid Proteomics to Unravel Phenotypic Variability in Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 4478. [Google Scholar] [CrossRef]
- McGill, R.B.; Steyn, F.J.; Ngo, S.T.; Thorpe, K.A.; Heggie, S.; Ruitenberg, M.J.; Henderson, R.D.; McCombe, P.A.; Woodruff, T.M. Monocytes and neutrophils are associated with clinical features in amyotrophic lateral sclerosis. Brain Commun. 2020, 2, fcaa013. [Google Scholar] [CrossRef]
- Yildiz, O.; Schroth, J.; Lombardi, V.; Pucino, V.; Bobeva, Y.; Yip, P.K.; Schmierer, K.; Mauro, C.; Tree, T.; Henson, S.M.; et al. The Expression of Active CD11b Monocytes in Blood and Disease Progression in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 23, 3370. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Steyn, F.J. The interplay between metabolic homeostasis and neurodegeneration: Insights into the neurometabolic nature of amyotrophic lateral sclerosis. Cell Regen. 2015, 4, 5. [Google Scholar] [CrossRef]
- Ferrer-Donato, A.; Contreras, A.; Fernandez, P.; Fernandez-Martos, C.M. The potential benefit of leptin therapy against amyotrophic lateral sclerosis (ALS). Brain Behav. 2022, 12, e2465. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Donato, A.; Contreras, A.; Frago, L.M.; Chowen, J.A.; Fernandez-Martos, C.M. Alterations in Leptin Signaling in Amyotrophic Lateral Sclerosis (ALS). Int. J. Mol. Sci. 2021, 22, 10305. [Google Scholar] [CrossRef] [PubMed]
- Bright, F.; Chan, G.; van Hummel, A.; Ittner, L.M.; Ke, Y.D. TDP-43 and inflammation: Implications for amyotrophic lateral sclerosis and frontotemporal dementia. Int. J. Mol. Sci. 2021, 22, 7781. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef]
- Swarup, V.; Phaneuf, D.; Dupré, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.-P. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB–mediated pathogenic pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef]
- Ohta, Y.; Tremblay, C.; Schneider, J.A.; Bennett, D.A.; Calon, F.; Julien, J.-P. Interaction of transactive response DNA binding protein 43 with nuclear factor κB in mild cognitive impairment with episodic memory deficits. Acta Neuropathol. Commun. 2014, 2, 37. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Jawaid, A.; Henstridge, C.M.; Valeri, A.; Merlini, M.; Robinson, J.L.; Lee, E.B.; Rose, J.; Appel, S.; Lee, V.M.-Y.; et al. TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss. Neuron 2017, 95, 297–308.e6. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.S.; Patel, P.; Dutta, K.; Julien, J.-P. Inflammation Induces TDP-43 Mislocalization and Aggregation. PLoS ONE 2015, 10, e0140248. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Rivest, S. MyD88-deficient bone marrow cells accelerate onset and reduce survival in a mouse model of amyotrophic lateral sclerosis. J. Cell Biol. 2007, 179, 1219–1230. [Google Scholar] [CrossRef]
- Komine, O.; Yamashita, H.; Fujimori-Tonou, N.; Koike, M.; Jin, S.; Moriwaki, Y.; Endo, F.; Watanabe, S.; Uematsu, S.; Akira, S.; et al. Innate immune adaptor TRIF deficiency accelerates disease progression of ALS mice with accumulation of aberrantly activated astrocytes. Cell Death Differ. 2018, 25, 2130–2146. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Goldacre, R.; Ramagopalan, S.; Talbot, K.; Goldacre, M.J. Autoimmune disease preceding amyotrophic lateral sclerosis: An epidemiologic study. Neurology 2013, 81, 1222–1225. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Puentes, F.; Lombardi, V.; Lu, C.-H.; Yildiz, O.; Fratta, P.; Isaacs, A.; Bobeva, Y.; Wuu, J.; Benatar, M.; Malaspina, A. Humoral response to neurofilaments and dipeptide repeats in ALS progression. Ann. Clin. Transl. Neurol. 2021, 8, 1831–1844. [Google Scholar] [CrossRef]
- Broce, I.; Karch, C.M.; Wen, N.; Fan, C.C.; Wang, Y.; Tan, C.H.; Kouri, N.; Ross, O.A.; Höglinger, G.U.; Muller, U.; et al. Immune-related genetic enrichment in frontotemporal dementia: An analysis of genome-wide association studies. PLoS Med. 2018, 15, e1002487. [Google Scholar] [CrossRef]
- Alipour, P.; Senkevich, K.; Ross, J.P.; Spiegelman, D.; Manousaki, D.; Dion, P.A.; Rouleau, G.A. Investigation of the causal relationship between ALS and autoimmune disorders: A Mendelian randomization study. BMC Med. 2022, 20, 382. [Google Scholar] [CrossRef]
- Li, C.Y.; Yang, T.M.; Ou, R.W.; Wei, Q.Q.; Shang, H.F. Genome-wide genetic links between amyotrophic lateral sclerosis and autoimmune diseases. BMC Med. 2021, 19, 27. [Google Scholar] [CrossRef]
- Campisi, L.; Chizari, S.; Ho, J.S.Y.; Gromova, A.; Arnold, F.J.; Mosca, L.; Mei, X.; Fstkchyan, Y.; Torre, D.; Beharry, C.; et al. Clonally expanded CD8 T cells characterize amyotrophic lateral sclerosis-4. Nature 2022, 606, 945–952. [Google Scholar] [CrossRef]
- Tondo, G.; De Marchi, F. From Biomarkers to Precision Medicine in Neurodegenerative Diseases: Where Are We? J. Clin. Med. 2022, 11, 4515. [Google Scholar] [CrossRef] [PubMed]
- Fournier, C.N. Considerations for Amyotrophic Lateral Sclerosis (ALS) Clinical Trial Design. Neurother. J. Am. Soc. Exp. Neurother. 2022, 19, 1180–1192. [Google Scholar] [CrossRef]
- Falzone, Y.M.; Russo, T.; Domi, T.; Pozzi, L.; Quattrini, A.; Filippi, M.; Riva, N. Current application of neurofilaments in amyotrophic lateral sclerosis and future perspectives. Neural Regen. Res. 2021, 16, 1985–1991. [Google Scholar] [CrossRef]
- Illán-Gala, I.; Alcolea, D.; Montal, V.; Dols-Icardo, O.; Muñoz, L.; De Luna, N.; Turón-Sans, J.; Cortés-Vicente, E.; Belén Sánchez-Saudinós, M.A.; Subirana, A.; et al. CSF sAPPβ, YKL-40, and NfL along the ALS-FTD spectrum. Neurology 2018, 91, E1619–E1628. [Google Scholar] [CrossRef] [PubMed]
- Verde, F.; Otto, M.; Silani, V. Neurofilament Light Chain as Biomarker for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2021, 15, 679199. [Google Scholar] [CrossRef] [PubMed]
- Benatar, M.; Wuu, J.; Lombardi, V.; Jeromin, A.; Bowser, R.; Andersen, P.M.; Malaspina, A. Neurofilaments in pre-symptomatic ALS and the impact of genotype. Amyotroph. Lateral Scler. Frontotemporal Degener. 2019, 20, 538–548. [Google Scholar] [CrossRef] [PubMed]
- van der Ende, E.L.; Meeter, L.H.; Poos, J.M.; Panman, J.L.; Jiskoot, L.C.; Dopper, E.G.P.; Papma, J.M.; de Jong, F.J.; Verberk, I.M.W.; Teunissen, C.; et al. Serum neurofilament light chain in genetic frontotemporal dementia: A longitudinal, multicentre cohort study. Lancet. Neurol. 2019, 18, 1103–1111. [Google Scholar] [CrossRef]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef]
- Behzadi, A.; Pujol-Calderón, F.; Tjust, A.E.; Wuolikainen, A.; Höglund, K.; Forsberg, K.; Portelius, E.; Blennow, K.; Zetterberg, H.; Andersen, P.M. Neurofilaments can differentiate ALS subgroups and ALS from common diagnostic mimics. Sci. Rep. 2021, 11, 22128. [Google Scholar] [CrossRef]
- Oeckl, P.; Weydt, P.; Thal, D.R.; Weishaupt, J.H.; Ludolph, A.C.; Otto, M. Proteomics in cerebrospinal fluid and spinal cord suggests UCHL1, MAP2 and GPNMB as biomarkers and underpins importance of transcriptional pathways in amyotrophic lateral sclerosis. Acta Neuropathol. 2020, 139, 119–134. [Google Scholar] [CrossRef]
- Cousins, K.A.Q.; Shaw, L.M.; Chen-Plotkin, A.; Wolk, D.A.; Van Deerlin, V.M.; Lee, E.B.; McMillan, C.T.; Grossman, M.; Irwin, D.J. Distinguishing Frontotemporal Lobar Degeneration Tau From TDP-43 Using Plasma Biomarkers. JAMA Neurol. 2022, 79, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Heller, C.; Foiani, M.S.; Moore, K.; Convery, R.; Bocchetta, M.; Neason, M.; Cash, D.M.; Thomas, D.; Greaves, C.V.; Woollacott, I.O.; et al. Plasma glial fibrillary acidic protein is raised in progranulin-associated frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 2020, 91, 263–270. [Google Scholar] [CrossRef]
- Bright, F.; Werry, E.L.; Dobson-Stone, C.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kassiou, M.; et al. Neuroinflammation in frontotemporal dementia. Nat. Rev. Neurol. 2019, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Barschke, P.; Oeckl, P.; Steinacker, P.; Al Shweiki, M.R.; Weishaupt, J.H.; Landwehrmeyer, G.B.; Anderl-Straub, S.; Weydt, P.; Diehl-Schmid, J.; Danek, A.; et al. Different CSF protein profiles in amyotrophic lateral sclerosis and frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. J. Neurol. Neurosurg. Psychiatry 2020, 91, 503–511. [Google Scholar] [CrossRef]
- Woollacott, I.O.C.; Nicholas, J.M.; Heller, C.; Foiani, M.S.; Moore, K.M.; Russell, L.L.; Paterson, R.W.; Keshavan, A.; Schott, J.M.; Warren, J.D.; et al. Cerebrospinal Fluid YKL-40 and Chitotriosidase Levels in Frontotemporal Dementia Vary by Clinical, Genetic and Pathological Subtype. Dement. Geriatr. Cogn. Disord. 2020, 49, 56–76. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Steinacker, P.; Polischi, B.; Mammana, A.; Bartoletti-Stella, A.; Oeckl, P.; Baiardi, S.; Zenesini, C.; Huss, A.; Cortelli, P.; et al. CSF biomarkers of neuroinflammation in distinct forms and subtypes of neurodegenerative dementia. Alzheimers. Res. Ther. 2019, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Bonsi, R.; Fenoglio, C.; Serpente, M.; Cioffi, S.M.G.; Fumagalli, G.; Arighi, A.; Ghezzi, L.; Arcaro, M.; Mercurio, M.; et al. Inflammatory molecules in Frontotemporal Dementia: Cerebrospinal fluid signature of progranulin mutation carriers. Brain. Behav. Immun. 2015, 49, 182–187. [Google Scholar] [CrossRef]
- Vignaroli, F.; Mele, A.; Tondo, G.; De Giorgis, V.; Manfredi, M.; Comi, C.; Mazzini, L.; De Marchi, F. The Need for Biomarkers in the ALS–FTD Spectrum: A Clinical Point of View on the Role of Proteomics. Proteomes 2023, 11, 1. [Google Scholar] [CrossRef]
- Clarke, M.T.M.; Brinkmalm, A.; Foiani, M.S.; Woollacott, I.O.C.; Heller, C.; Heslegrave, A.; Keshavan, A.; Fox, N.C.; Schott, J.M.; Warren, J.D.; et al. CSF synaptic protein concentrations are raised in those with atypical Alzheimer’s disease but not frontotemporal dementia. Alzheimers. Res. Ther. 2019, 11, 105. [Google Scholar] [CrossRef]
- Melamed, Z.; López-Erauskin, J.; Baughn, M.W.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.W.; et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180–190. [Google Scholar] [CrossRef]
- Klim, J.R.; Williams, L.A.; Limone, F.; Guerra San Juan, I.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.J.; Gamage, K.K.; Kirchner, R.; et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 2019, 22, 167–179. [Google Scholar] [CrossRef]
- Gagliardi, D.; Pagliari, E.; Meneri, M.; Melzi, V.; Rizzo, F.; Comi, G.P.; Corti, S.; Taiana, M.; Nizzardo, M. Stathmins and Motor Neuron Diseases: Pathophysiology and Therapeutic Targets. Biomedicines 2022, 10, 711. [Google Scholar] [CrossRef]
- Prudencio, M.; Humphrey, J.; Pickles, S.; Brown, A.-L.; Hill, S.E.; Kachergus, J.M.; Shi, J.; Heckman, M.G.; Spiegel, M.R.; Cook, C.; et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J. Clin. Investig. 2020, 130, 6080–6092. [Google Scholar] [CrossRef] [PubMed]
- Scialò, C.; Tran, T.H.; Salzano, G.; Novi, G.; Caponnetto, C.; Chiò, A.; Calvo, A.; Canosa, A.; Moda, F.; Caroppo, P.; et al. TDP-43 real-time quaking induced conversion reaction optimization and detection of seeding activity in CSF of amyotrophic lateral sclerosis and frontotemporal dementia patients. Brain Commun. 2020, 2, fcaa142. [Google Scholar] [CrossRef] [PubMed]
- Riva, N.; Gentile, F.; Cerri, F.; Gallia, F.; Podini, P.; Dina, G.; Falzone, Y.M.; Fazio, R.; Lunetta, C.; Calvo, A.; et al. Phosphorylated TDP-43 aggregates in peripheral motor nerves of patients with amyotrophic lateral sclerosis. Brain 2022, 145, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Gambino, C.M.; Ciaccio, A.M.; Lo Sasso, B.; Giglio, R.V.; Vidali, M.; Agnello, L.; Ciaccio, M. The Role of TAR DNA Binding Protein 43 (TDP-43) as a CandiDate Biomarker of Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Diagnostics 2023, 13, 416. [Google Scholar] [CrossRef]
- Irwin, K.E.; Jasin, P.; Braunstein, K.E.; Sinha, I.; Bowden, K.D.; Moghekar, A.; Oh, E.S.; Raitcheva, D.; Bartlett, D.; Berry, J.D.; et al. A fluid biomarker reveals loss of TDP-43 splicing repression in pre-symptomatic ALS. bioRxiv Prepr. Serv. Biol. 2023. [Google Scholar] [CrossRef]
- Chiò, A.; Mazzini, L.; Mora, G. Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology 2020, 167, 107986. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V.; ALS/Riluzole Study Group. A controlled trial of riluzole in amyotrophic lateral sclerosis. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Andrews, J.A.; Jackson, C.E.; Heiman-Patterson, T.D.; Bettica, P.; Brooks, B.R.; Pioro, E.P. Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 509–518. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Andersen, P.M.; Chandran, S.; Chio, A.; Corcia, P.; Couratier, P.; Danielsson, O.; de Carvalho, M.; Desnuelle, C.; Grehl, T. July 2017 ENCALS statement on edaravone. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Shefner, J.M.; Andrews, J.A.; Genge, A.; Jackson, C.; Lechtzin, N.; Miller, T.M.; Cockroft, B.M.; Meng, L.; Wei, J.; Wolff, A.A.; et al. A Phase 2, Double-Blind, Randomized, Dose-Ranging Trial Of Reldesemtiv In Patients With ALS. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 287–299. [Google Scholar] [CrossRef]
- Rudnicki, S.A.; Andrews, J.A.; Genge, A.; Jackson, C.; Lechtzin, N.; Miller, T.M.; Cockroft, B.M.; Malik, F.I.; Meng, L.; Wei, J.; et al. Prescription and acceptance of durable medical equipment in FORTITUDE-ALS, a study of reldesemtiv in ALS: Post hoc analyses of a randomized, double-blind, placebo-controlled clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2022, 23, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, I.; Bayatti, N.; Brown, A.; Wang, D.; Mickunas, M.; Camu, W.; Veyrune, J.-L.; Payan, C.; Garlanda, C.; Locati, M.; et al. Amyotrophic lateral sclerosis transcriptomics reveals immunological effects of low-dose interleukin-2. Brain Commun. 2021, 3, fcab141. [Google Scholar] [CrossRef]
- Berry, J.D.; Cudkowicz, M.E.; Windebank, A.J.; Staff, N.P.; Owegi, M.; Nicholson, K.; McKenna-Yasek, D.; Levy, Y.S.; Abramov, N.; Kaspi, H. NurOwn, phase 2, randomized, clinical trial in patients with ALS: Safety, clinical, and biomarker results. Neurology 2019, 93, e2294–e2305. [Google Scholar] [CrossRef]
- De Marchi, F.; Mareschi, K.; Ferrero, I.; Cantello, R.; Fagioli, F.; Mazzini, L. Effect of mesenchymal stromal cell transplantation on long-term survival in amyotrophic lateral sclerosis. Cytotherapy, 2023; in press. [Google Scholar] [CrossRef]
- Francois-Moutal, L.; Scott, D.D.; Khanna, M. Direct targeting of TDP-43, from small molecules to biologics: The therapeutic landscape. RSC Chem. Biol. 2021, 2, 1158–1166. [Google Scholar] [CrossRef]
- Tsou, Y.-S.; Lai, J.-H.; Chen, K.-Y.; Chang, C.-F.; Huang, C.-C. Therapeutic Effect of Rapamycin on TDP-43-Related Pathogenesis in Ischemic Stroke. Int. J. Mol. Sci. 2022, 24, 676. [Google Scholar] [CrossRef]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P. Causative genes in amyotrophic lateral sclerosis and protein degradation pathways: A link to neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef] [PubMed]
- François-Moutal, L.; Scott, D.D.; Ambrose, A.J.; Zerio, C.J.; Rodriguez-Sanchez, M.; Dissanayake, K.; May, D.G.; Carlson, J.M.; Barbieri, E.; Moutal, A.; et al. Heat shock protein Grp78/BiP/HspA5 binds directly to TDP-43 and mitigates toxicity associated with disease pathology. Sci. Rep. 2022, 12, 8140. [Google Scholar] [CrossRef]
- Fang, T.; Je, G.; Pacut, P.; Keyhanian, K.; Gao, J.; Ghasemi, M. Gene Therapy in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2066. [Google Scholar] [CrossRef]
- Poulin-Brière, A.; Rezaei, E.; Pozzi, S. Antibody-Based Therapeutic Interventions for Amyotrophic Lateral Sclerosis: A Systematic Literature Review. Front. Neurosci. 2021, 15, 790114. [Google Scholar] [CrossRef] [PubMed]
- VandeVrede, L.; Boxer, A.L.; Polydoro, M. Targeting tau: Clinical trials and novel therapeutic approaches. Neurosci. Lett. 2020, 731, 134919. [Google Scholar] [CrossRef] [PubMed]
- Boeve, B.F.; Boxer, A.L.; Kumfor, F.; Pijnenburg, Y.; Rohrer, J.D. Advances and controversies in frontotemporal dementia: Diagnosis, biomarkers, and therapeutic considerations. Lancet Neurol. 2022, 21, 258–272. [Google Scholar] [CrossRef]
- Brody, M.; Agronin, M.; Herskowitz, B.J.; Bookheimer, S.Y.; Small, G.W.; Hitchinson, B.; Ramdas, K.; Wishard, T.; McInerney, K.F.; Vellas, B.; et al. Results and insights from a phase I clinical trial of Lomecel-B for Alzheimer’s disease. Alzheimers Dement. 2023, 19, 261–273. [Google Scholar] [CrossRef]




| Disease | Genes | Affected Processes | References |
|---|---|---|---|
| ALS | VAPB | autophagy, RNA binding, protein homeostasis, mitochondrial functions, vesicle trafficking | [65,66,67] |
| EPHA4 | motor neuron survival | [68] | |
| UNC13A | neurotransmission | [69] | |
| NEK1 | RNA metabolism, DNA repair, axonal polarity, neuronal morphology | [70] | |
| SOD1 | autophagy, mitophagy, RNA metabolism, protein homeostasis, mitochondrial and immune functions | [71,72,73] | |
| HNRNPA1 | protein folding, stress granule dynamics | [74] | |
| ANXA11 | calcium homeostasis, stress granule dynamics, axon morphology | [71,75] | |
| PFN1 | autophagy, RNA metabolism, stress granule dynamics | [76,77] | |
| KIF5A | trafficking and neuronal homeostasis | [78] | |
| UBQLN2 | autophagy, RNA metabolism | [50,71,79] | |
| ALS/FTD | VCP | autophagy, mitochondrial function | [80,81] |
| OPTN | autophagy, mitophagy, vesicular trafficking, immune signaling | [37] | |
| CHCHD10 | mitochondrial function | [82] | |
| C9ORF72 | autophagy, RNA metabolism, protein homeostasis, nucleocytoplasmic transport | [73,83,84,85] | |
| TBK1 | autophagy, mitophagy, protein homeostasis, mitochondrial function | [86,87] | |
| TARDBP | autophagy, nucleocytoplasmic transport, RNA metabolism, axonal transport | [88,89,90] | |
| CYLD | autophagy, immune signaling | [10,91] | |
| FUS | nucleocytoplasmic transport, DNA damage repair, RNA metabolism | [71] | |
| SQSTM1 | autophagy | [92] | |
| CCNF | autophagy, axon morphology | [71,93] | |
| TIA1 | stress granule dynamics | [94] | |
| FTD | GRN | immune signaling, lysosomal functions | [95] |
| MAPT | vesicular trafficking, lysosomal functions | [96] | |
| CHMP2B | autophagy | [97] |
| Co-Aggregating Proteins and Peptides | Notes | References |
|---|---|---|
| Amyloidogenic proteins | Using an anti-oligomer antibody, potential hybrid oligomers composed of amyloid-β, prion protein, α-synuclein, and TDP-43 phosphorylated at serine 409/410 were detected in AD brains. Colocalization of α-synuclein, tau, and TDP-43 has also been occasionally reported in patients suffering from CBD and MSA. | [115,116,117] |
| ATXN2 | ATXN2 and TDP-43 colocalize in cytoplasmic inclusions in FTD. ATXN2 and TDP-43 associated in a complex that is dependent on RNA and can act as a powerful disease modifier. In a mouse TDP-43 model, the decrease in ataxin-2 markedly increased survival and improved motor function. | [118,119,120] |
| C9orf72 DPRs | TDP-43 has been shown to colocalize with poly-GR and poly-PA inclusions. No colocalization was observed for poly-GP, poly-GA, or poly-PR immunoreactive inclusions. In the motor regions of C9 ALS cases, only poly-GR dendritic aggregations had significant colocalization with phosphorylated TDP-43. | [121,122] |
| CDK5 | CDK5-positive granules have been shown to overlap with pSmad2/3, ubiquitin, and phospho-TDP-43 in several AD patients. | [123] |
| DISC1 | Cytosolic TDP-43 and DISC1 co-aggregate in brains of both FTD mouse models and FTD patients and disrupt the activity-dependent local translation in dendrites. | [124] |
| HuD/ELAVL4 | ELAVL4 has been found as a neural-specific component of FUS-positive cytoplasmic aggregates, whereas in sporadic ALS patients, it colocalized with positive inclusions of phosphorylated TDP-43. | [125] |
| ERp57 | ERp57 colocalizes with phospho-TDP-43-positive inclusions present in sporadic ALS patients. | [126] |
| GPNMB | GPNMB aggregates colocalize with TDP-43 in the spinal cord of ALS patients. In NSC-34 cells, the expression level of GPNMB increased by overexpression of mutant M337V and A315T TDP-43. | [127] |
| hnRNP E2 | hnRNP E2 immunostaining colocalizes with TDP-43 pathological changes, but only in patients with semantic dementia and FTD type C TDP-43 histology. | [128] |
| HERV-K RT | The reverse transcriptase protein of this endogenous retrovirus was observed to localize to cortical neurons of ALS patients and strongly correlated with TDP-43 expression. | [129] |
| IL-10 | IL-10 colocalizes with TDP-43-positive cytoplasmic inclusions in anterior horn motor neurons in ALS patients. | [130] |
| Nup62 | Cytoplasmic NUP62-TDP-43 inclusions are frequently found in C9orf72 ALS/FTD as well as in sporadic ALS/FTD post-mortem CNS tissue. | [131] |
| OPTN | Mutations in the OPTN gene have been reported to be causative for familial ALS and FTD, but mutated optineurin has not been found in aggregates. In contrast, in sALS cases optineurin has been observed in cytoplasmic skein-like inclusions colocalizing with ubiquitin, TDP-43, and possibly FUS; similar optineurin positive inclusions have been reported in AD, PD, Creutzfeldt-Jakob and Pick disease. | [132,133] |
| p62/SQSTM1 | p62 physiologically binds to TDP-43 and is involved in degradation of the TDP-43 35-kDa fragment. It also colocalizes with TDP-43-positive cytoplasmic inclusions, ubiquitin, and UBQLN2 in patients with FTD/ALS. | [134,135] |
| PABP-1 | This protein colocalizes to mature TDP-43 inclusions (but not to pre-inclusions) in ALS motor neurons and is more prevalent in patients bearing C9ORF72 expansions. | [136] |
| PFN1 | Profilin mutations can induce aggregation of TDP-43 and PFN1 in inclusions positive for phosphorylated TDP-43 in ALS patients. | [137,138] |
| RBM14, PSF, NONO | These paraspeckle markers are found in insoluble TDP-43 artificial aggregates together with stress granule markers. In a proteomic study, PSF was found to be enriched in the TDP-43-positive detergent-insoluble proteome of four post-mortem FTD patients. | [139] |
| Rab5 | In yeast, TDP-43 foci colocalized frequently with endogenous Rab5 foci, suggesting a greater association of TDP-43 with endosomal-like compartments over autophagic compartments. | [140] |
| RBM45 | RBM45 colocalizes with TDP-43 in inclusion bodies and is especially present in ALS/FTD patients with C9ORF72 expansions. It was later shown that RBM45 forms homo-oligomers and physically associates with TDP-43 and FUS in the nucleus. | [141,142,143] |
| RGNEF | Mutations in the murine homologue of this protein cause altered NFL mRNA stability and lead to NF aggregate formation and motor neuronopathy. This protein can also interact by immunoprecipitation with FUS and p62. Mutations in RGNEF have been described in an ALS family. | [144,145,146] |
| RANGAP1 /NUP205 | Aberrant colocalizations of TDP-43 with the nuclear pore complex proteins RanGAP1 and NUP205 have been observed in motor neurons of ALS patients. | [147] |
| Tau | Occasional colocalization of TDP-43 with tau has been described in globular astrocytic inclusions (GAIs) of a Japanese patient affected by ALS/FTD. A recent survey of more than 200 AD patients has observed that almost 30% of TDP-positive cases colocalized with phosphorylated tau (detected using PHF-1 antibody). | [148,149] |
| TTBK1/TTBK2 | These kinases have been observed to colocalize with phosphorylated TDP-43 in human post-mortem tissues from both FTD and ALS cases. | [150] |
| UBQLN2 | In autopsy material of human spinal cord samples of UBQLN2 mutation carriers, its skein-like inclusions are positive for UBQLN2, ubiquitin, p62, TDP-43, FUS, and OPTN, but not SOD1. | [151,152] |
| VHL/CUL2 E3 complex | VHL preferentially recognizes misfolded forms of TDP-43 and promotes ubiquitin-mediated proteasomal degradation of fragmented forms of TDP-43. Phosphorylated TDP-43 and VHL are occasionally colocalized in cytoplasmic inclusions in oligodendrocytes in ALS. | [153] |
| VCP | In hippocampal dentate gyrus neurons of C9ORF72 patients, VCP inclusions have been reported to co-aggregate with phospho-TDP-43. | [63] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Marchi, F.; Franjkic, T.; Schito, P.; Russo, T.; Nimac, J.; Chami, A.A.; Mele, A.; Vidatic, L.; Kriz, J.; Julien, J.-P.; et al. Emerging Trends in the Field of Inflammation and Proteinopathy in ALS/FTD Spectrum Disorder. Biomedicines 2023, 11, 1599. https://doi.org/10.3390/biomedicines11061599
De Marchi F, Franjkic T, Schito P, Russo T, Nimac J, Chami AA, Mele A, Vidatic L, Kriz J, Julien J-P, et al. Emerging Trends in the Field of Inflammation and Proteinopathy in ALS/FTD Spectrum Disorder. Biomedicines. 2023; 11(6):1599. https://doi.org/10.3390/biomedicines11061599
Chicago/Turabian StyleDe Marchi, Fabiola, Toni Franjkic, Paride Schito, Tommaso Russo, Jerneja Nimac, Anna A. Chami, Angelica Mele, Lea Vidatic, Jasna Kriz, Jean-Pierre Julien, and et al. 2023. "Emerging Trends in the Field of Inflammation and Proteinopathy in ALS/FTD Spectrum Disorder" Biomedicines 11, no. 6: 1599. https://doi.org/10.3390/biomedicines11061599
APA StyleDe Marchi, F., Franjkic, T., Schito, P., Russo, T., Nimac, J., Chami, A. A., Mele, A., Vidatic, L., Kriz, J., Julien, J. -P., Apic, G., Russell, R. B., Rogelj, B., Cannon, J. R., Baralle, M., Agosta, F., Hecimovic, S., Mazzini, L., Buratti, E., & Munitic, I. (2023). Emerging Trends in the Field of Inflammation and Proteinopathy in ALS/FTD Spectrum Disorder. Biomedicines, 11(6), 1599. https://doi.org/10.3390/biomedicines11061599