

Exploring the Therapeutic Potential of Phosphorylated Cis-Tau Antibody in a Pig Model of Traumatic Brain Injury
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Surgery
2.2. Biomarker Study
2.3. Diffusion Tensor Imaging
2.4. Statistical Analysis
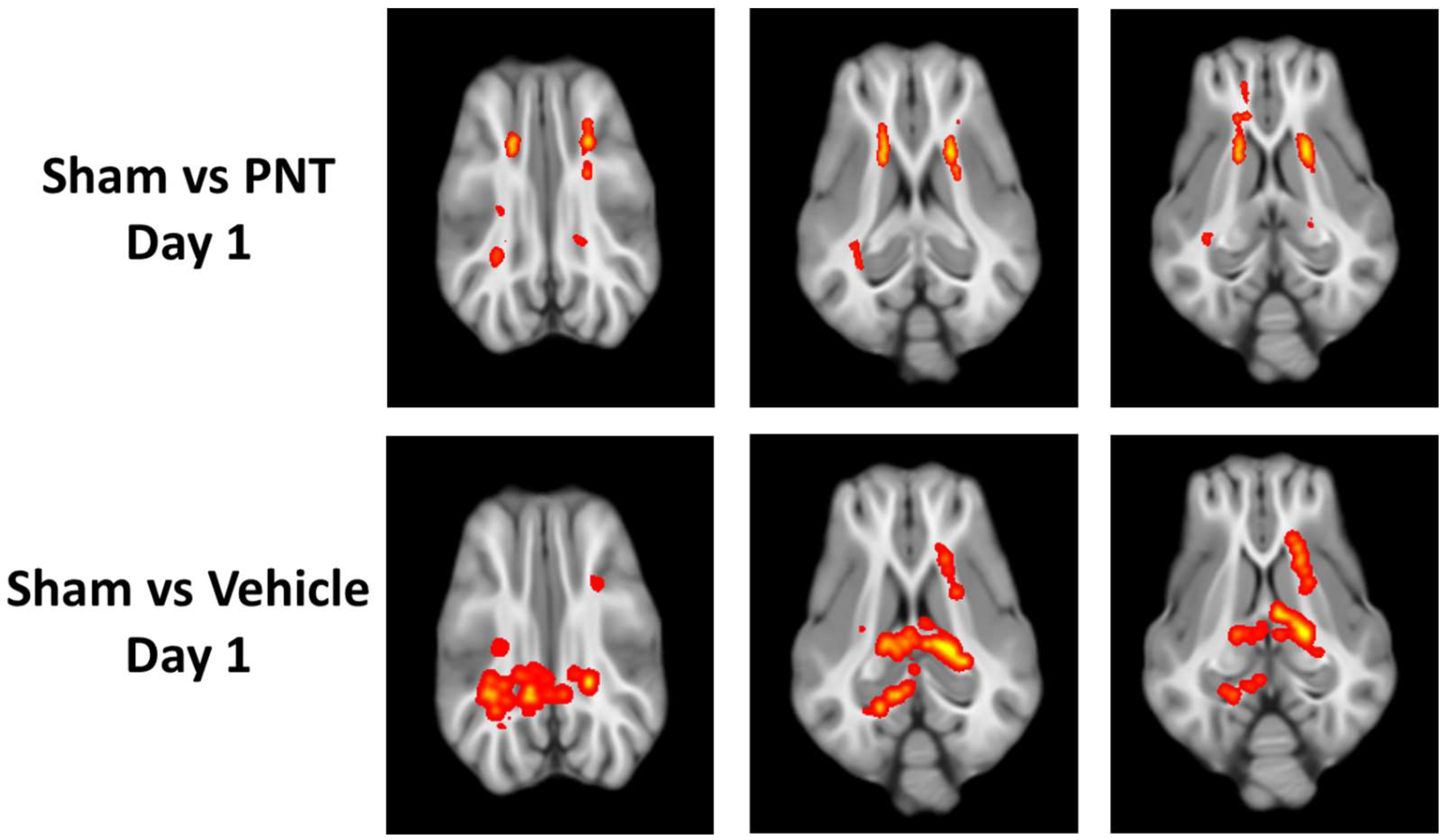
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shin, R.W.; Iwaki, T.; Kitamoto, T.; Tateishi, J. Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer’s disease brain tissues. Lab. Investig. 1991, 64, 693–702. [Google Scholar] [PubMed]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Tagge, C.A.; Fisher, A.M.; Minaeva, O.V.; Gaudreau-Balderrama, A.; Moncaster, J.A.; Zhang, X.L.; Wojnarowicz, M.W.; Casey, N.; Lu, H.; Kokiko-Cochran, O.N.; et al. Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain 2018, 141, 422–458. [Google Scholar] [CrossRef]
- Kondo, A.; Shahpasand, K.; Mannix, R.; Qiu, J.; Moncaster, J.; Chen, C.H.; Yao, Y.; Lin, Y.M.; Driver, J.A.; Sun, Y.; et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 2015, 523, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Mez, J.; Daneshvar, D.H.; Kiernan, P.T.; Abdolmohammadi, B.; Alvarez, V.E.; Huber, B.R.; Alosco, M.L.; Solomon, T.M.; Nowinski, C.J.; McHale, L.; et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. JAMA 2017, 318, 360–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, A.C.; Stern, R.A.; Nowinski, C.J.; Stein, T.D.; Alvarez, V.E.; Daneshvar, D.H.; Lee, H.S.; Wojtowicz, S.M.; Hall, G.; Baugh, C.M.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, A.; Takeuchi, H.; Tanaka, F. Tau Pathology in Chronic Traumatic Encephalopathy and Alzheimer’s Disease: Similarities and Differences. Front. Neurol. 2019, 10, 980. [Google Scholar] [CrossRef]
- Albayram, O.; Kondo, A.; Mannix, R.; Smith, C.; Tsai, C.Y.; Li, C.; Herbert, M.K.; Qiu, J.; Monuteaux, M.; Driver, J.; et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat. Commun. 2017, 8, 1000. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Albayram, O.; Kondo, A.; Wang, B.; Kim, N.; Arai, K.; Tsai, C.Y.; Bassal, M.A.; Herbert, M.K.; Washida, K.; et al. Cis P-tau underlies vascular contribution to cognitive impairment and dementia and can be effectively targeted by immunotherapy in mice. Sci. Transl. Med. 2021, 13, eaaz7615. [Google Scholar] [CrossRef]
- Foster, K.; Manca, M.; McClure, K.; Koivula, P.; Trojanowski, J.Q.; Havas, D.; Chancellor, S.; Goldstein, L.; Brunden, K.R.; Kraus, A.; et al. Preclinical characterization and IND-enabling safety studies for PNT001, an antibody that recognizes cis-pT231 tau. Alzheimers Dement 2023, in press. [Google Scholar] [CrossRef]
- Margulies, S.S.; Kilbaugh, T.; Sullivan, S.; Smith, C.; Propert, K.; Byro, M.; Saliga, K.; Costine, B.A.; Duhaime, A.C. Establishing a Clinically Relevant Large Animal Model Platform for TBI Therapy Development: Using Cyclosporin A as a Case Study. Brain Pathol. 2015, 25, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.H.; Johnson, V.E.; Stewart, W. Chronic neuropathologies of single and repetitive TBI: Substrates of dementia? Nat. Rev. Neurol. 2013, 9, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeKosky, S.T.; Blennow, K.; Ikonomovic, M.D.; Gandy, S. Acute and chronic traumatic encephalopathies: Pathogenesis and biomarkers. Nat. Rev. Neurol. 2013, 9, 192–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omalu, B.I.; DeKosky, S.T.; Minster, R.L.; Kamboh, M.I.; Hamilton, R.L.; Wecht, C.H. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 2005, 57, 128–134. [Google Scholar] [CrossRef]
- Goldstein, L.E.; Fisher, A.M.; Tagge, C.A.; Zhang, X.L.; Velisek, L.; Sullivan, J.A.; Upreti, C.; Kracht, J.M.; Ericsson, M.; Wojnarowicz, M.W.; et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 2012, 4, 134ra160. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Hefti, M.M.; Mazandi, V.M.; Issadore, D.A.; Meaney, D.; Christman Schneider, A.; Diaz-Arrastia, R.; Kilbaugh, T.J. Plasma Neurofilament Light and Glial Fibrillary Acidic Protein Levels over thirty days in a Porcine Model of Traumatic Brain Injury. J. Neurotrauma 2022, 39, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.S.N.; Zimmerman, K.A.; Moro, F.; Heslegrave, A.; Maillard, S.A.; Bernini, A.; Miroz, J.P.; Donat, C.K.; Lopez, M.Y.; Bourke, N.; et al. Axonal marker neurofilament light predicts long-term outcomes and progressive neurodegeneration after traumatic brain injury. Sci. Transl. Med. 2021, 13, eabg9922. [Google Scholar] [CrossRef]
- Shahim, P.; Zetterberg, H.; Tegner, Y.; Blennow, K. Serum neurofilament light as a biomarker for mild traumatic brain injury in contact sports. Neurology 2017, 88, 1788–1794. [Google Scholar] [CrossRef] [Green Version]
- Shahim, P.; Tegner, Y.; Marklund, N.; Blennow, K.; Zetterberg, H. Neurofilament light and tau as blood biomarkers for sports-related concussion. Neurology 2018, 90, e1780–e1788. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Rodriguez, A.; Arevalo, M.A. The Contribution of Astrocyte Autophagy to Systemic Metabolism. Int. J. Mol. Sci. 2020, 21, 2479. [Google Scholar] [CrossRef] [Green Version]
- Middeldorp, J.; Hol, E.M. GFAP in health and disease. Prog. Neurobiol. 2011, 93, 421–443. [Google Scholar] [CrossRef]
- Villapol, S.; Byrnes, K.R.; Symes, A.J. Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Front. Neurol. 2014, 5, 82. [Google Scholar] [CrossRef] [Green Version]
- Castellani, R.J.; Perry, G. Tau Biology, Tauopathy, Traumatic Brain Injury, and Diagnostic Challenges. J. Alzheimers Dis. 2019, 67, 447–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafiei, S.S.; Guerrero-Munoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Yoshiyama, Y.; Uryu, K.; Higuchi, M.; Longhi, L.; Hoover, R.; Fujimoto, S.; McIntosh, T.; Lee, V.M.; Trojanowski, J.Q. Enhanced neurofibrillary tangle formation, cerebral atrophy, and cognitive deficits induced by repetitive mild brain injury in a transgenic tauopathy mouse model. J. Neurotrauma 2005, 22, 1134–1141. [Google Scholar] [CrossRef] [Green Version]
- Bittar, A.; Bhatt, N.; Hasan, T.F.; Montalbano, M.; Puangmalai, N.; McAllen, S.; Ellsworth, A.; Carretero Murillo, M.; Taglialatela, G.; Lucke-Wold, B.; et al. Neurotoxic tau oligomers after single versus repetitive mild traumatic brain injury. Brain Commun. 2019, 1, fcz004. [Google Scholar] [CrossRef]
- Ihara, Y. PHF and PHF-like fibrils--cause or consequence? Neurobiol. Aging 2001, 22, 123–126. [Google Scholar] [CrossRef]
- Ren, Z.; Iliff, J.J.; Yang, L.; Yang, J.; Chen, X.; Chen, M.J.; Giese, R.N.; Wang, B.; Shi, X.; Nedergaard, M. ‘Hit & Run’ model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J. Cereb. Blood Flow Metab. 2013, 33, 834–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, S.S.; Mazandi, V.M.; Schneider, A.L.C.; Morton, S.; Starr, J.P.; Weeks, M.K.; Widmann, N.J.; Jang, D.H.; Kao, S.-H.; Ahlijanian, M.K.; et al. Exploring the Therapeutic Potential of Phosphorylated Cis-Tau Antibody in a Pig Model of Traumatic Brain Injury. Biomedicines 2023, 11, 1807. https://doi.org/10.3390/biomedicines11071807
Shin SS, Mazandi VM, Schneider ALC, Morton S, Starr JP, Weeks MK, Widmann NJ, Jang DH, Kao S-H, Ahlijanian MK, et al. Exploring the Therapeutic Potential of Phosphorylated Cis-Tau Antibody in a Pig Model of Traumatic Brain Injury. Biomedicines. 2023; 11(7):1807. https://doi.org/10.3390/biomedicines11071807
Chicago/Turabian StyleShin, Samuel S., Vanessa M. Mazandi, Andrea L. C. Schneider, Sarah Morton, Jonathan P. Starr, M. Katie Weeks, Nicholas J. Widmann, David H. Jang, Shih-Han Kao, Michael K. Ahlijanian, and et al. 2023. "Exploring the Therapeutic Potential of Phosphorylated Cis-Tau Antibody in a Pig Model of Traumatic Brain Injury" Biomedicines 11, no. 7: 1807. https://doi.org/10.3390/biomedicines11071807
APA StyleShin, S. S., Mazandi, V. M., Schneider, A. L. C., Morton, S., Starr, J. P., Weeks, M. K., Widmann, N. J., Jang, D. H., Kao, S. -H., Ahlijanian, M. K., & Kilbaugh, T. J. (2023). Exploring the Therapeutic Potential of Phosphorylated Cis-Tau Antibody in a Pig Model of Traumatic Brain Injury. Biomedicines, 11(7), 1807. https://doi.org/10.3390/biomedicines11071807