
Role of Diacylglycerol Kinases in Acute Myeloid Leukemia

 , ,
, ,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cancer Tissue Database Exploration
2.2. Coexpression
2.3. Cell Lines
2.4. Chemicals and Inhibitors
2.5. DGKA Protein Purification
2.6. DGKA Activity Assay
2.7. Reverse Transcription Polymerase Chain Reaction Assay
2.8. AlamarBlue and Trypan Blue Viability Assays
2.9. Real-Time Viability Assay
2.10. Cell Cycle
3. Results
3.1. Overexpression of Selected DGK Isoforms in AML
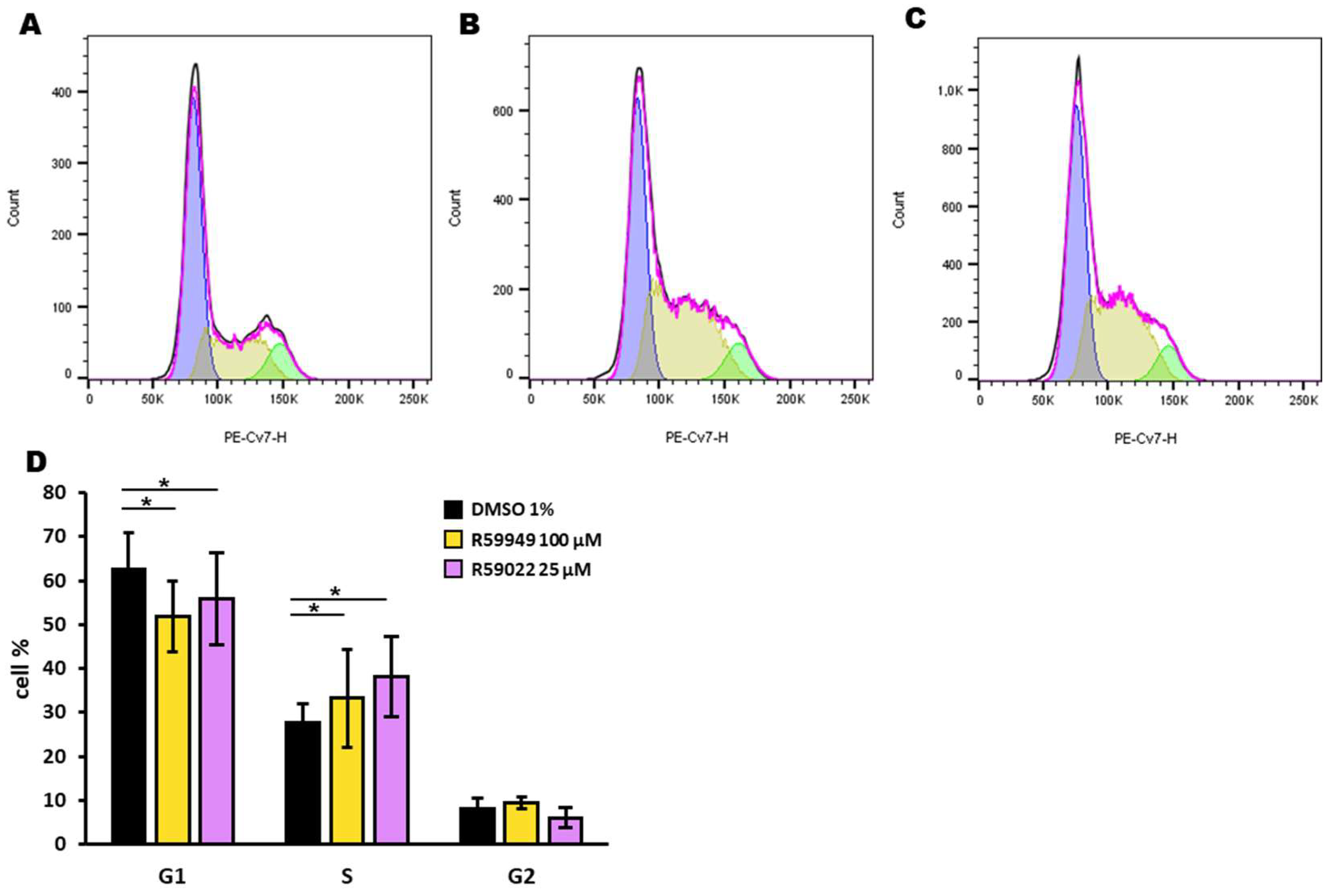
3.2. Effect of DGK Inhibitors on AML Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zjablovskaja, P.; Florian, M.C. Acute Myeloid Leukemia: Aging and Epigenetics. Cancers 2019, 12, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 ELN Recommendations from an International Expert Panel. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Vergez, F.; Largeaud, L.; Bertoli, S.; Nicolau, M.L.; Rieu, J.B.; Vergnolle, I.; Saland, E.; Sarry, A.; Tavitian, S.; Huguet, F.; et al. Phenotypically-defined stages of leukemia arrest predict main driver mutations subgroups, and outcome in acute myeloid leukemia. Blood Cancer J. 2022, 12, 117. [Google Scholar] [CrossRef]
- Hwang, S.M. Classification of acute myeloid leukemia. Blood Res. 2020, 55, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Baldanzi, G.; Malerba, M. DGKα in Neutrophil Biology and Its Implications for Respiratory Diseases. Int. J. Mol. Sci. 2019, 20, 5673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldanzi, G.; Ragnoli, B.; Malerba, M. Potential role of diacylglycerol kinases in immune-mediated diseases. Clin. Sci. 2020, 134, 1637–1658. [Google Scholar] [CrossRef]
- Dominguez, C.L.; Floyd, D.H.; Xiao, A.; Mullins, G.R.; Kefas, B.A.; Xin, W.; Yacur, M.N.; Abounader, R.; Lee, J.K.; Wilson, G.M.; et al. Diacylglycerol kinase α is a critical signaling node and novel therapeutic target in glioblastoma and other cancers. Cancer Discov. 2013, 3, 782–797. [Google Scholar] [CrossRef] [Green Version]
- Olmez, I.; Love, S.; Xiao, A.; Manigat, L.; Randolph, P.; McKenna, B.D.; Neal, B.P.; Boroda, S.; Li, M.; Brenneman, B.; et al. Targeting the mesenchymal subtype in glioblastoma and other cancers via inhibition of diacylglycerol kinase alpha. Neuro-Oncology 2018, 20, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, K.; Yasuda, S.; Kai, M.; Imai, S.; Yamada, K.; Yamashita, T.; Jimbow, K.; Kanoh, H.; Sakane, F. Diacylglycerol kinase alpha suppresses tumor necrosis factor-alpha-induced apoptosis of human melanoma cells through NF-kappaB activation. Biochim. Biophys. Acta 2007, 1771, 462–474. [Google Scholar] [CrossRef]
- Yamaki, A.; Akiyama, R.; Murakami, C.; Takao, S.; Murakami, Y.; Mizuno, S.; Takahashi, D.; Kado, S.; Taketomi, A.; Shirai, Y.; et al. Diacylglycerol kinase α-selective inhibitors induce apoptosis and reduce viability of melanoma and several other cancer cell lines. J. Cell. Biochem. 2019, 120, 10043–10056. [Google Scholar] [CrossRef] [PubMed]
- Takeishi, K.; Taketomi, A.; Shirabe, K.; Toshima, T.; Motomura, T.; Ikegami, T.; Yoshizumi, T.; Sakane, F.; Maehara, Y. Diacylglycerol kinase alpha enhances hepatocellular carcinoma progression by activation of Ras-Raf-MEK-ERK pathway. J. Hepatol. 2012, 57, 77–83. [Google Scholar] [CrossRef]
- Baldanzi, G.; Mitola, S.; Cutrupi, S.; Filigheddu, N.; van Blitterswijk, W.J.; Sinigaglia, F.; Bussolino, F.; Graziani, A. Activation of diacylglycerol kinase alpha is required for VEGF-induced angiogenic signaling in vitro. Oncogene 2004, 23, 4828–4838. [Google Scholar] [CrossRef] [Green Version]
- Flores, I.; Casaseca, T.; Martinez-A, C.; Kanoh, H.; Merida, I. Phosphatidic acid generation through interleukin 2 (IL-2)-induced alpha-diacylglycerol kinase activation is an essential step in IL-2-mediated lymphocyte proliferation. J. Biol. Chem. 1996, 271, 10334–10340. [Google Scholar] [CrossRef] [Green Version]
- Bacchiocchi, R.; Baldanzi, G.; Carbonari, D.; Capomagi, C.; Colombo, E.; van Blitterswijk, W.J.; Graziani, A.; Fazioli, F. Activation of alpha-diacylglycerol kinase is critical for the mitogenic properties of anaplastic lymphoma kinase. Blood 2005, 106, 2175–2182. [Google Scholar] [CrossRef] [Green Version]
- Fazio, A.; Owusu Obeng, E.; Rusciano, I.; Marvi, M.V.; Zoli, M.; Mongiorgi, S.; Ramazzotti, G.; Follo, M.Y.; McCubrey, J.A.; Cocco, L.; et al. Subcellular Localization Relevance and Cancer-Associated Mechanisms of Diacylglycerol Kinases. Int. J. Mol. Sci. 2020, 21, 5297. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.Y.; Kim, Y.Y.; Yu, H.S.; Lee, M.; Kim, S.; Lee, J. CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res. 2018, 78, 4692–4703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, E.; Singh, B.K.; Paustian, A.M.; Kambayashi, T. Diacylglycerol Kinase ζ Is a Target to Enhance NK Cell Function. J. Immunol. 2016, 197, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Baldanzi, G. Immune Checkpoint Receptors Signaling in T Cells. Int. J. Mol. Sci. 2022, 23, 3529. [Google Scholar] [CrossRef] [PubMed]
- Ruffo, E.; Malacarne, V.; Larsen, S.E.; Das, R.; Patrussi, L.; Wülfing, C.; Biskup, C.; Kapnick, S.M.; Verbist, K.; Tedrick, P.; et al. Inhibition of diacylglycerol kinase α restores restimulation-induced cell death and reduces immunopathology in XLP-1. Sci. Transl. Med. 2016, 8, 321ra327. [Google Scholar] [CrossRef] [Green Version]
- Velnati, S.; Centonze, S.; Rossino, G.; Purghè, B.; Antona, A.; Racca, L.; Mula, S.; Ruffo, E.; Malacarne, V.; Malerba, M.; et al. Wiskott-Aldrich syndrome protein interacts and inhibits diacylglycerol kinase alpha promoting IL-2 induction. Front. Immunol. 2023, 14, 1043603. [Google Scholar] [CrossRef]
- Velnati, S.; Massarotti, A.; Antona, A.; Talmon, M.; Fresu, L.G.; Galetto, A.S.; Capello, D.; Bertoni, A.; Mercalli, V.; Graziani, A.; et al. Structure activity relationship studies on Amb639752: Toward the identification of a common pharmacophoric structure for DGKα inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Velnati, S.; Ruffo, E.; Massarotti, A.; Talmon, M.; Varma, K.S.S.; Gesu, A.; Fresu, L.G.; Snow, A.L.; Bertoni, A.; Capello, D.; et al. Identification of a novel DGKα inhibitor for XLP-1 therapy by virtual screening. Eur. J. Med. Chem. 2019, 164, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dong, C.; Tian, Y.; Li, X.; Wang, B.; Zhai, D.; Bai, Y.; Chao, X. Knockdown of diacylglycerol kinase zeta (DGKZ) induces apoptosis and G2/M phase arrest in human acute myeloid leukemia HL-60 cells through MAPK/survivin/caspase pathway. Pharmazie 2019, 74, 418–422. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldanzi, G.; Cutrupi, S.; Chianale, F.; Gnocchi, V.; Rainero, E.; Porporato, P.; Filigheddu, N.; van Blitterswijk, W.J.; Parolini, O.; Bussolino, F.; et al. Diacylglycerol kinase-alpha phosphorylation by Src on Y335 is required for activation, membrane recruitment and Hgf-induced cell motility. Oncogene 2008, 27, 942–956. [Google Scholar] [CrossRef] [Green Version]
- Stefanowicz-Hajduk, J.; Ochocka, J.R. Real-time cell analysis system in cytotoxicity applications: Usefulness and comparison with tetrazolium salt assays. Toxicol. Rep. 2020, 7, 335–344. [Google Scholar] [CrossRef]
- Kramer, M.H.; Zhang, Q.; Sprung, R.; Day, R.B.; Erdmann-Gilmore, P.; Li, Y.; Xu, Z.; Helton, N.M.; George, D.R.; Mi, Y.; et al. Proteomic and Phosphoproteomic Landscapes of Acute Myeloid Leukemia. Blood 2022, 140, 1533–1548. [Google Scholar] [CrossRef]
- Mou, T.; Pawitan, Y.; Stahl, M.; Vesterlund, M.; Deng, W.; Jafari, R.; Bohlin, A.; Österroos, A.; Siavelis, L.; Bäckvall, H.; et al. The transcriptome-wide landscape of molecular subtype-specific mRNA expression profiles in acute myeloid leukemia. Am. J. Hematol. 2021, 96, 580–588. [Google Scholar] [CrossRef]
- Mérida, I.; Andrada, E.; Gharbi, S.I.; Ávila-Flores, A. Redundant and specialized roles for diacylglycerol kinases α and ζ in the control of T cell functions. Sci. Signal. 2015, 8, re6. [Google Scholar] [CrossRef] [PubMed]
- Boroda, S.; Niccum, M.; Raje, V.; Purow, B.W.; Harris, T.E. Dual activities of ritanserin and R59022 as DGKα inhibitors and serotonin receptor antagonists. Biochem. Pharmacol. 2017, 123, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Kunii, N.; Sakuma, M.; Yamaki, A.; Mizuno, S.; Sato, M.; Sakai, H.; Kado, S.; Kumagai, K.; Kojima, H.; et al. A novel diacylglycerol kinase α-selective inhibitor, CU-3, induces cancer cell apoptosis and enhances immune response. J. Lipid Res. 2016, 57, 368–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liang, J.; Yang, W.; Guo, W.; Song, W.; Zhang, W.; Wu, X.; He, B. A distinct lipid metabolism signature of acute myeloid leukemia with prognostic value. Front. Oncol. 2022, 12, 876981. [Google Scholar] [CrossRef]
- Wang, L.; Sun, Y.; Meng, L.; Xu, X. First case of AML with rare chromosome translocations: A case report of twins. BMC Cancer 2018, 18, 458. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [Green Version]
- Poli, A.; Fiume, R.; Baldanzi, G.; Capello, D.; Ratti, S.; Gesi, M.; Manzoli, L.; Graziani, A.; Suh, P.G.; Cocco, L.; et al. Nuclear Localization of Diacylglycerol Kinase Alpha in K562 Cells Is Involved in Cell Cycle Progression. J. Cell. Physiol. 2016, 232, 2550–2557. [Google Scholar] [CrossRef] [PubMed]
- Batista, E.L.; Warbington, M.; Badwey, J.A.; Van Dyke, T.E. Differentiation of HL-60 cells to granulocytes involves regulation of select diacylglycerol kinases (DGKs). J. Cell. Biochem. 2005, 94, 774–793. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Sakane, F.; Imai, S.; Tsushima, S.; Murakami, T.; Kanoh, H. Regulatory role of diacylglycerol kinase gamma in macrophage differentiation of leukemia cells. Biochem. Biophys. Res. Commun. 2003, 305, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Flores, I.; Jones, D.R.; Ciprés, A.; Díaz-Flores, E.; Sanjuan, M.A.; Mérida, I. Diacylglycerol kinase inhibition prevents IL-2-induced G1 to S transition through a phosphatidylinositol-3 kinase-independent mechanism. J. Immunol. 1999, 163, 708–714. [Google Scholar] [CrossRef]
- Audia, A.; Bhat, K.P. Ritanserin, a novel agent targeting the mesenchymal subtype of glioblastomas. Neuro-Oncology 2018, 20, 151–152. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.; Menezes, A.C.; Azevedo, A.; Leckenby, A.; Davies, S.; Seedhouse, C.; Gilkes, A.; Knapper, S.; Tonks, A.; Darley, R.L. Protein Kinase C Epsilon Overexpression Is Associated with Poor Patient Outcomes in AML and Promotes Daunorubicin Resistance Through p-Glycoprotein-Mediated Drug Efflux. Front. Oncol. 2022, 12, 840046. [Google Scholar] [CrossRef] [PubMed]
- Di Marcantonio, D.; Martinez, E.; Sidoli, S.; Vadaketh, J.; Nieborowska-Skorska, M.; Gupta, A.; Meadows, J.M.; Ferraro, F.; Masselli, E.; Challen, G.A.; et al. Protein Kinase C Epsilon Is a Key Regulator of Mitochondrial Redox Homeostasis in Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 608–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Jia, J.; Yao, M.; Kang, J.; Wang, Y.; Yan, X.; Zhang, L.; Lv, Q.; Chen, X.; Lu, F. Diacylglycerol kinase γ predicts prognosis and functions as a tumor suppressor by negatively regulating glucose transporter 1 in hepatocellular carcinoma. Exp. Cell Res. 2018, 373, 211–220. [Google Scholar] [CrossRef]
- Velnati, S.; Centonze, S.; Girivetto, F.; Capello, D.; Biondi, R.M.; Bertoni, A.; Cantello, R.; Ragnoli, B.; Malerba, M.; Graziani, A.; et al. Identification of Key Phospholipids That Bind and Activate Atypical PKCs. Biomedicines 2021, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Lincoln, H.C.; Kim, C.R.; Frey, J.W.; Goodman, C.A.; Zhong, X.P.; Hornberger, T.A. The role of diacylglycerol kinase ζ and phosphatidic acid in the mechanical activation of mammalian target of rapamycin (mTOR) signaling and skeletal muscle hypertrophy. J. Biol. Chem. 2014, 289, 1551–1563. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gravina, T.; Boggio, C.M.T.; Gorla, E.; Racca, L.; Polidoro, S.; Centonze, S.; Ferrante, D.; Lunghi, M.; Graziani, A.; Corà, D.; et al. Role of Diacylglycerol Kinases in Acute Myeloid Leukemia. Biomedicines 2023, 11, 1877. https://doi.org/10.3390/biomedicines11071877
Gravina T, Boggio CMT, Gorla E, Racca L, Polidoro S, Centonze S, Ferrante D, Lunghi M, Graziani A, Corà D, et al. Role of Diacylglycerol Kinases in Acute Myeloid Leukemia. Biomedicines. 2023; 11(7):1877. https://doi.org/10.3390/biomedicines11071877
Chicago/Turabian StyleGravina, Teresa, Chiara Maria Teresa Boggio, Elisa Gorla, Luisa Racca, Silvia Polidoro, Sara Centonze, Daniela Ferrante, Monia Lunghi, Andrea Graziani, Davide Corà, and et al. 2023. "Role of Diacylglycerol Kinases in Acute Myeloid Leukemia" Biomedicines 11, no. 7: 1877. https://doi.org/10.3390/biomedicines11071877
APA StyleGravina, T., Boggio, C. M. T., Gorla, E., Racca, L., Polidoro, S., Centonze, S., Ferrante, D., Lunghi, M., Graziani, A., Corà, D., & Baldanzi, G. (2023). Role of Diacylglycerol Kinases in Acute Myeloid Leukemia. Biomedicines, 11(7), 1877. https://doi.org/10.3390/biomedicines11071877