

Role of the Renin-Angiotensin System in Long COVID’s Cardiovascular Injuries



Abstract
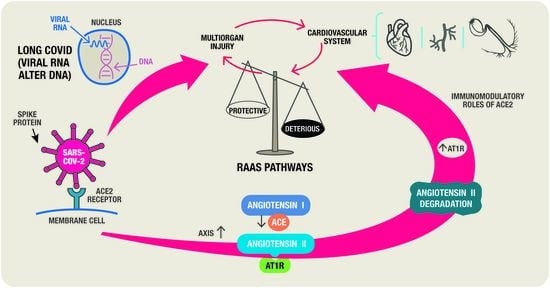
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
3. The Alteration of RAS Components—A Key Role in SARS-CoV-2 Infection
- Harmful arm-ACE/Ang II/angiotensin II receptor type 1 (AT1R) pathway; because it separates Ang II from Ang I and interacts with the AT1R to cause vasoconstriction, inflammation, hypertrophy, blood coagulation, cell growth and proliferation, extracellular matrix remodelling, and stimulation of oxidative stress and fibrosis.
- Protective arm-ACE2-Ang (1–7)-MasR receptor (MasR); Ang (1–7) interacts with MasR to promote vasodilation, anti-inflammatory effects, antifibrotic effects, anti-proliferative effects, and vascular protection and mediates endothelial nitric oxide synthase activation and suppress apoptosis, negatively regulating RAS. The Ang (1–7) is produced either by the cleavage of Ang II by ACE2 or through the metabolism of inactive Ang (1–9) (cleaved from Ang I by ACE2). The cardiac antihypertrophic actions of Ang II and Ang (1–7) are mediated through the activation of Ang type 2 receptors (AT2R) and MasR, respectively [19]. Since only AT1R is blocked, using an ARB (angiotensin receptor blockers) for the therapy of COVID-19 patients may have favorable outcomes because any formed Ang II may have anti-inflammatory effects through its interaction with AT2R or through conversion by residual ACE2 to Ang (1–7) acting through AT2R, MasR, and Mas-related G protein-coupled receptor D) [20]. Thus, by preventing or reversing Ang II-induced cardiac hypertrophy, AT1R inhibition or activation of AT2R and Mas receptors as well as decreased oxidative stress may have a cardioprotective role.
3.1. SARS-CoV-2 Cell Entrance
3.2. ACE2—A Key Pathogenic Role in COVID-19
3.2.1. ACE2 Downregulation
3.2.2. Imbalance in the RAS
3.2.3. Inflammatory Response and Lung Injury
3.2.4. The Kallikrein-Kinin System (KKS)
4. RAS Involvement in COVID Cardiovascular Injuries
4.1. Inflammatory Response
4.2. Endothelial Dysfunction
4.3. Microvascular Abnormalities: Hypercoagulability and Thrombus Formation
4.4. Fibrosis
5. Conclusions
6. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Coronavirus Disease (COVID-19) Pandemic. Available online: https://www.who.int/europe/emergencies/situations/covid-19 (accessed on 6 June 2023).
- World Health Organization. Weekly Epidemiological Update on COVID-19—8 June 2023. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---8-june-2023 (accessed on 9 June 2023).
- World Health Organization. Post COVID-19 Condition (Long COVID). Available online: https://www.who.int/europe/news-room/fact-sheets/item/post-covid-19-condition (accessed on 10 June 2023).
- Thaweethai, T.; Jolley, S.E.; Karlson, E.W.; Levitan, E.B.; Levy, B.; McComsey, G.A.; McCorkell, L.; Nadkarni, G.N.; Parthasarathy, S.; Singhe, U.; et al. Development of a Definition of Postacute Sequelae of SARS-CoV-2 Infection. JAMA 2023, 329, 1934–1946. [Google Scholar] [CrossRef]
- Rubin, R. As Their Numbers Grow, COVID-19 “Long Haulers” Stump Experts. JAMA 2020, 324, 1381–1383. [Google Scholar] [CrossRef] [PubMed]
- COVID.gov—What Is Long COVID. Available online: https://www.covid.gov/longcovid/definitions (accessed on 10 June 2023).
- Ballering, A.V.; van Zon, S.K.R.; Olde Hartman, T.C.; Rosmalen, J.G.M. Persistence of somatic symptoms after COVID-19 in the Netherlands: An observational cohort study. Lancet 2022, 400, 452–461. [Google Scholar] [CrossRef]
- Raveendran, A.V. Long COVID-19, Challenges in the diagnosis and proposed diagnostic criteria. Diabetes Metab. Syndr. 2021, 15, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Raveendran, A.V.; Jayadevan, R.; Sashidharan, S. Long COVID: An overview. Diabetes Metab. Syndr. 2021, 15, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Sudre, C.H.; Murray, B.; Varsavsky, T.; Graham, M.S.; Penfold, R.S.; Bowyer, R.C.; Pujol, J.C.; Klaser, K.; Antonelli, M.; Canas, L.S.; et al. Attributes and predictors of long COVID. Nat. Med. 2021, 27, 626–631. [Google Scholar] [CrossRef]
- O’Mahoney, L.L.; Routen, A.; Gillies, C.; Ekezie, W.; Welford, A.; Zhang, A.; Karamchandani, U.; Simms-Williams, N.; Cassambai, S.; Ardavani, A.; et al. The prevalence and long-term health effects of Long COVID among hospitalised and non-hospitalised populations: A systematic review and meta-analysis. EClinicalMedicine 2023, 55, 101762. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, X.; Wei, X.; Li, J.; Yang, J.; Tan, H.; Zhu, J.; Zhang, Q.; Wu, J.; Liu, L.; et al. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. 2020, 92, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Driggin, E.; Madhavan, M.V.; Bikdeli, B.; Chuich, T.; Laracy, J.; Biondi-Zoccai, G.; Brown, T.S.; Der Nigoghossian, C.; Zidar, D.A.; Haythe, J.; et al. Cardiovascular Considerations for Patients, Health Care Workers, and Health Systems during the COVID-19 Pandemic. J. Am. Coll. Cardiol. 2020, 75, 2352–2371. [Google Scholar] [CrossRef]
- Bavishi, C.; Bonow, R.O.; Trivedi, V.; Abbott, J.D.; Messerli, F.H.; Bhatt, D.L. Special Article—Acute myocardial injury in patients hospitalized with COVID-19 infection: A review. Prog. Cardiovasc. Dis. 2020, 63, 682–689. [Google Scholar] [CrossRef]
- Campbell, D.J. Clinical relevance of local Renin Angiotensin systems. Front. Endocrinol. 2014, 14, 113. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.A.; Turner, L.; Marentes Ruiz, C.J.; Tanaka, M.L.; Congrave-Wilson, Z.; Lee, Y.; Jumarang, J.; Perez, S.; Peralta, A.; Pannaraj, P.S. Clinical manifestations of COVID-19 differ by age and obesity status. Influenza Other Respir. Viruses 2022, 16, 255–264. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin–Angiotensin–Aldosterone System Inhibitors in Patients with COVID-19. N. Eng. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Costa, L.B.; Perez, L.G.; Palmeira, V.A.; Macedo e Cordeiro, T.; Teatini Ribeiro, V.; Lanza, K.; Simões e Silva, A.C. Insights on SARS-CoV-2 Molecular Interactions With the Renin-Angiotensin System. Front. Cell Dev. Biol. 2020, 8, 559841. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, S.K.; Dhalla, N.S. Angiotensin II-Induced Signal Transduction Mechanisms for Cardiac Hypertrophy. Cells 2022, 11, 3336. [Google Scholar] [CrossRef] [PubMed]
- Lumbers, E.R.; Head, R.; Smith, G.R.; Delforce, S.J.; Jarrott, B.; Martin, J.H.; Pringle, K.G. The interacting physiology of COVID-19 and the renin- angiotensin-aldosterone system: Key agents for treatment. Pharmacol. Res. Perspect. 2022, 10, e00917. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Hrenak, J.; Adamcova, M.; Paulis, L. Renin-Angiotensin-Aldosterone System: Friend or Foe-The Matter of Balance. Insight on History, Therapeutic Implications and COVID-19 Interactions. Int. J. Mol. Sci. 2021, 22, 3217. [Google Scholar] [CrossRef]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care. 2020, 24, 422. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabassum, A.; Iqbal, M.S.; Sultan, S.; Alhuthali, R.A.; Alshubaili, D.I.; Sayyam, R.S.; Abyad, L.M.; Qasem, A.H.; Arbaeen, A. Dysregulated Bradykinin: Mystery in the Pathogenesis of COVID-19. Mediat. Inflamm. 2022, 2022, 7423537. [Google Scholar] [CrossRef] [PubMed]
- Raman, B.; Bluemke, D.A.; Lüscher, T.F.; Neubauer, S. Long COVID: Post-acute sequelae of COVID-19 with a cardiovascular focus. Eur. Heart J. 2022, 43, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Maas, C.; Renne, T. Coagulation factor XII in thrombosis and inflammation. Blood 2018, 131, 1903–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieffer, E.; Schieffer, B. The rationale for the treatment of long-COVID symptoms—A cardiologist’s view. Front. Cardiovasc. Med. 2022, 9, 992686. [Google Scholar] [CrossRef]
- Schmidt, B.; Schieffer, B. Angiotensin II AT1 receptor antagonists. Clinical implications of active metabolites. J. Med. Chem. 2003, 46, 2261–2270. [Google Scholar] [CrossRef]
- Writing Committee for the REMAP-CAP Investigators. Effect of Angiotensin-Converting Enzyme Inhibitor and Angiotensin Receptor Blocker Initiation on Organ Support–Free Days in Patients Hospitalized with COVID-19, A Randomized Clinical Trial. JAMA 2023, 329, 1183–1196. [Google Scholar] [CrossRef]
- Self, W.H.; Shotwell, M.S.; Gibbs, K.W.; de Wit, M.; Files, C.; Harkins, M.; Hudock, K.M.; Merck, L.H.; Moskowitz, A.; Apodaca, K.D.; et al. Renin-Angiotensin System Modulation with Synthetic Angiotensin (1–7) and Angiotensin II Type 1 Receptor–Biased Ligand in Adults with COVID-19, Two Randomized Clinical Trials. JAMA 2023, 329, 1170–1182. [Google Scholar] [CrossRef]
- Lee, M.M.Y.; McMurray, J.J.V. Lack of Benefit of Renin-Angiotensin System Inhibitors in COVID-19. JAMA 2023, 329, 1155–1156. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.D.; Macedo, A.V.S.; de Barros E Silva, P.G.M.; Moll-Bernardes, R.J.; Feldman, A.; D’Andréa Saba Arruda, G.; de Souza, A.S.; de Albuquerque, D.C.; Mazza, L.; Santos, M.F.; et al. Continuing versus suspending angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: Impact on adverse outcomes in hospitalized patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)–The BRACE CORONA Trial. Am. Heart J. 2020, 226, 49–59. [Google Scholar] [CrossRef]
- Cohen, J.B.; Hanff, T.C.; William, P.; Sweitzer, N.; Rosado-Santander, N.R.; Medina, C.; Rodriguez-Mori, J.R.; Renna, N.; Chang, T.I.; Corrales-Medina, V.; et al. Continuation versus discontinuation of renin–angiotensin system inhibitors in patients admitted to hospital with COVID-19, A prospective, randomised, open-label trial. Lancet Respir. Med. 2021, 9, 275–284. [Google Scholar] [CrossRef]
- Chan Sui Ko, A.; Candellier, A.; Mercier, M.; Joseph, C.; Carette, H.; Basille, D.; Lion-Daolio, S.; Devaux, S.; Schmit, J.L.; Lanoix, J.P.; et al. No Impact of Corticosteroid Use During the Acute Phase on Persistent Symptoms Post-COVID-19. Int. J. Gen. Med. 2022, 18, 6645–6651. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liu, D.X. Coronavirus infection, ER stress, apoptosis and innate immunity. Front. Microbiol. 2014, 5, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inde, Z.; Croker, B.A.; Yapp, C.; Joshi, G.N.; Spetz, J.; Fraser, C.; Qin, X.; Xu, L.; Deskin, B.; Ghelf, E.; et al. Age-dependent regulation of SARS-CoV-2 cell entry genes and cell death programs correlates with COVID-19 severity. Sci. Adv. 2021, 7, 8609–8627. [Google Scholar] [CrossRef]
- Aleksova, A.; Ferro, F.; Gagno, G.; Cappelletto, C.; Santon, D.; Rossi, M.; Ippolito, G.; Zumla, A.; Beltrami, A.P.; Sinagra, G. COVID-19 and renin-angiotensin system inhibition: Role of angiotensin converting enzyme 2 (ACE2)—Is there any scientific evidence for controversy? J. Intern. Med. 2020, 288, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, U.M.; Abokor, A.A.; Edwards, J.M.; Edwards, J.M.; Waigi, E.W.; Royfman, R.S.; Hasan, S.A.M.; Smedlund, K.B.; Gregio Hardy, A.M.; Chakravarti, R.; et al. SARS-CoV-2, ACE2 expression, and systemic organ invasion. Physiol. Genom. 2021, 53, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Polidoro, R.B.; Hagan, R.S.; de Santis Santiago, R.; Schmidt, N.W. Overview: Systemic Inflammatory Response Derived From Lung Injury Caused by SARS-CoV-2 Infection Explains Severe Outcomes in COVID-19. Front. Immunol. 2020, 11, 1626. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Recinos, A.; LeJeune, W.S.; Sun, H.; Lee, C.Y.; Tieu, B.C.; Lu, M.; Hou, T.; Boldogh, I.; Tilton, R.G.; Brasier, A.R. Angiotensin II induces IL-6 expression and the Jak-STAT3 pathway in aortic adventitia of LDL receptor-deficient mice. Atherosclerosis 2007, 194, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Zhao, T.; Zhou, X.; Xiang, Y.; Gutierrez-Castrellon, P.; Ma, X. Inflammatory pathways in COVID-19, Mechanism and therapeutic interventions. MedComm 2022, 3, e154. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimes, J.M.; Grimes, K.V. p38 MAPK inhibition: A promising therapeutic approach for COVID-19. J. Mol. Cell. Cardiol. 2020, 144, 63–65. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. A hypothesis for pathobiology and treatment of COVID-19, The centrality of ACE1/ACE2 imbalance. Br. J. Pharmacol. 2020, 177, 4825–4844. [Google Scholar] [CrossRef] [PubMed]
- Vollbracht, C.; Kraft, K. Oxidative Stress and HyperInflammation as Major Drivers of Severe COVID-19 and Long COVID: Implications for the Benefit of HighDose Intravenous Vitamin C. Front. Pharmacol. 2022, 13, 899198. [Google Scholar] [CrossRef]
- de las Heras, N.; Martín Giménez, V.M.; Ferder, L.; Manucha, W.; Lahera, V. Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19, Therapeutic Effects of Vitamin D. Antioxidants 2020, 9, 897. [Google Scholar] [CrossRef]
- Zhu, Z.; Zheng, Z.; Liu, J. Comparison of COVID-19 and Lung Cancer via Reactive Oxygen Species Signaling. Front. Oncol. 2021, 11, 708263. [Google Scholar] [CrossRef]
- Alobaidy, A.S.H.; Elhelaly, M.; Amer, M.E.; Shemies, R.A.; Othman, A.I.; El-Missiry, M.A. Angiotensin converting enzyme 2 gene expression and markers of oxidative stress are correlated with disease severity in patients with COVID-19. Mol. Biol. Rep. 2023, 50, 5827–5836. [Google Scholar] [CrossRef]
- Khazaal, S.; Harb, J.; Rima, M.; Annweiler, C.; Wu, Y.; Cao, Z.; Khattar, Z.A.; Legros, C.; Kovacic, H.; Fajloun, Z.; et al. The Pathophysiology of Long COVID throughout the Renin-Angiotensin System. Molecules 2022, 27, 2903. [Google Scholar] [CrossRef]
- Jakovac, H.; Ferenčić, A.; Stemberger, C.; Mohar Vitezić, B.; Cuculić, D. Detection of SARS-CoV-2 Antigens in the AV-Node of a Cardiac Conduction System—A Case Report. Trop. Med. Infect. Dis. 2022, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Tavazzi, G.; Pellegrini, C.; Maurelli, M.; Belliato, M.; Sciutti, F.; Bottazzi, A.; Sepe, P.A.; Resasco, T.; Camporotondo, R.; Bruno, R.; et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur. J. Heart Fail. 2020, 22, 911–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrofanova, L.B.; Makarov, I.A.; Gorshkov, A.N.; Runov, A.L.; Vonsky, M.S.; Pisareva, M.M.; Komissarov, A.B.; Makarova, T.A.; Li, Q.; Karonova, T.L.; et al. Comparative Study of the Myocardium of Patients from Four COVID-19 Waves. Diagnostics 2023, 13, 1645. [Google Scholar] [CrossRef] [PubMed]
- Bojkova, D.; Wagner, J.U.G.; Shumliakivska, M.; Aslan, G.S.; Saleem, U.; Hansen, A.; Luxán, G.; Günther, S.; Duc Pham, M.; Krishnan, J.; et al. SARS-CoV-2 infects and induces cytotoxic effects in human cardiomyocytes. Cardiovasc. Res. 2020, 116, 2207–2215. [Google Scholar] [CrossRef]
- Sakamoto, A.; Kawakami, R.; Kawai, K.; Gianatti, A.; Pellegrini, D.; Kutys, R.; Guo, L.; Mori, M.; Cornelissen, A.; Sato, Y.; et al. ACE2 (Angiotensin-Converting Enzyme 2) and TMPRSS2 (Transmembrane Serine Protease 2) Expression and Localization of SARS-CoV-2 Infection in the Human Heart. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 542–544. [Google Scholar] [CrossRef]
- Yang, J.; Chen, T.; Zhou, Y. Mediators of SARS-CoV-2 entry are preferentially enriched in cardiomyocytes. Hereditas 2021, 158, 4. [Google Scholar] [CrossRef]
- Liu, T.; Luo, S.; Libby, P.; Shi, G.P. Cathepsin L-selective inhibitors: A potentially promising treatment for COVID-19 patients. Pharmacol. Ther. 2020, 213, 107587. [Google Scholar] [CrossRef]
- Nappi, F.; Avtaar Singh, S.S. Endothelial Dysfunction in SARS-CoV-2 Infection. Biomedicines 2022, 10, 654. [Google Scholar] [CrossRef]
- Tucker, N.R.; Chaffin, M.; Bedi, K.C., Jr.; Papangeli, I.; Akkad, A.D.; Arduini, A.; Hayat, S.; Eraslan, G.; Muus, C.; Bhattacharyya, R.P.; et al. Myocyte-Specific Upregulation of ACE2 in Cardiovascular Disease. Circulation 2020, 142, 708–710. [Google Scholar]
- Di Toro, A.; Bozzani, A.; Tavazzi, G.; Urtis, M.; Giuliani, L.; Pizzoccheri, R.; Aliberti, F.; Fergnani, V.; Arbustini, E. Long COVID: Long-term effects? Eur. Hear J. Suppl. 2021, 23, E1–E5. [Google Scholar] [CrossRef]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Lüscher, T.F.; Shechter, M.; Taddei, S.; et al. The assessment of endothelial function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef] [Green Version]
- Del Turco, S.; Vianello, A.; Ragusa, R.; Caselli, C.; Basta, G. COVID-19 and cardiovascular consequences: Is the endothelial dysfunction the hardest challenge? Thromb. Res. 2020, 196, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef] [PubMed]
- Proal, A.D.; VanElzakker, M.B. Long COVID or Post-acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front. Microbiol. 2021, 12, 698169. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef]
- Arthur, J.M.; Forrest, J.C.; Boehme, K.W.; Kennedy, J.L.; Owens, S.; Herzog, C.; Liu, J.; Harville, T.O. Development of ACE2 autoantibodies after SARS-CoV-2 infection. PLoS ONE 2021, 16, e0257016. [Google Scholar] [CrossRef]
- Shi, H.; Zuo, Y.; Navaz, S.; Harbaugh, A.; Hoy, C.K.; Gandhi, A.A.; Sule, G.; Yalavarthi, S.; Gockman, K.; Madisonet, J.A.; et al. Endothelial Cell–Activating Antibodies in COVID-19. Arthritis Rheumatol. 2022, 74, 1132–1138. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Team, R.A. The microvascular hypothesis underlying neurologic manifestations of long COVID-19 and possible therapeutic strategies. Cardiovasc. Endocrinol. Metab. 2021, 10, 193–203. [Google Scholar] [CrossRef]
- Vallejo Camazón, N.; Teis, A.; Martínez Membrive, M.J.; Llibre, C.; Bayés-Genís, A.; Mateu, L. Long COVID-19 and microvascular disease-related angina. Rev. Esp. Cardiol. 2022, 75, 444–446. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M.; Mehrad, B. New mechanisms of pulmonary fibrosis. Chest 2009, 136, 1364–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zheng, X.; Tong, Q.; Li, W.; Wang, B.; Sutter, K.; Trilling, M.; Dittmer, M.U.; Yang, D. Overlapping and discrete aspects of the pathology and pathogenesis of the emerging human pathogenic coronaviruses SARS-CoV, MERS-CoV, and 2019-nCoV. J. Med. Virol. 2020, 92, 491–494. [Google Scholar] [CrossRef] [Green Version]
- Méndez-García, L.A.; Escobedo, G.; Minguer-Uribe, A.G.; Viurcos-Sanabria, R.; Aguayo-Guerrero, J.A.; Carrillo-Ruiz, J.D.; Solleiro-Villavicencio, H. Role of the renin-angiotensin system in the development of COVID-19-associated neurological manifestations. Front. Cell Neurosci. 2022, 16, 977039. [Google Scholar] [CrossRef]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-β, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef]
- Castro-Malaspina, H.; Rabellino, E.M.; Yen, A.; Nachman, R.L.; Moore, M.A. Human megakaryocyte stimulation of proliferation of bone marrow fibroblasts. Blood 1981, 57, 781–787. [Google Scholar] [CrossRef] [Green Version]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19, The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Bitto, N.; Liguori, E.; La Mura, V. Coagulation, Microenvironment and Liver Fibrosis. Cells 2018, 7, 85. [Google Scholar] [CrossRef] [Green Version]
- Thille, A.W.; Esteban, A.; Fernández-Segoviano, P.; Rodriguez, J.M.; Aramburu, J.A.; Vargas-Errazuriz, P.; Martin-Pellicer, A.; Lorente, J.A.; Frutos-Vivar, F. Chronology of histological lesions in acute respiratory distress syndrome with diffuse alveolar damage: A prospective cohort study of clinical autopsies. Lancet Respir. Med. 2013, 1, 395–401. [Google Scholar] [CrossRef]
- Huang, W.T.; Akhter, H.; Jiang, C.; MacEwen, M.; Ding, Q.; Antony, V.; Thannickal, V.J.; Liu, R.M. Plasminogen activator inhibitor 1, fibroblast apoptosis resistance, and aging-related susceptibility to lung fibrosis. Exp. Gerontol. 2015, 61, 62–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkataraman, T.; Frieman, M.B. The role of epidermal growth factor receptor (EGFR) signaling in SARS coronavirus-induced pulmonary fibrosis. Antivir. Res. 2017, 143, 142. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.W.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.A.; Zaki, A.; Fouchier, R.A.M.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Tanaka, K.; Fujita, T.; Umezawa, H.; Amano, H.; Yoshioka, K.; Naito, Y.; Hatano, M.; Kimura, S.; Tatsumi, K.; et al. Bidirectional role of IL-6 signal in pathogenesis of lung fibrosis. Respir. Res. 2015, 16, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cojocaru, E.; Cojocaru, C.; Vlad, C.-E.; Eva, L. Role of the Renin-Angiotensin System in Long COVID’s Cardiovascular Injuries. Biomedicines 2023, 11, 2004. https://doi.org/10.3390/biomedicines11072004
Cojocaru E, Cojocaru C, Vlad C-E, Eva L. Role of the Renin-Angiotensin System in Long COVID’s Cardiovascular Injuries. Biomedicines. 2023; 11(7):2004. https://doi.org/10.3390/biomedicines11072004
Chicago/Turabian StyleCojocaru, Elena, Cristian Cojocaru, Cristiana-Elena Vlad, and Lucian Eva. 2023. "Role of the Renin-Angiotensin System in Long COVID’s Cardiovascular Injuries" Biomedicines 11, no. 7: 2004. https://doi.org/10.3390/biomedicines11072004
APA StyleCojocaru, E., Cojocaru, C., Vlad, C. -E., & Eva, L. (2023). Role of the Renin-Angiotensin System in Long COVID’s Cardiovascular Injuries. Biomedicines, 11(7), 2004. https://doi.org/10.3390/biomedicines11072004