

Desmosomal Arrhythmogenic Cardiomyopathy: The Story Telling of a Genetically Determined Heart Muscle Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Discovery of Arrhythmogenic Cardiomyopathy
2. Contemporary History of Arrhythmogenic Cardiomyopathy
3. AC Is a Heart Muscle Disease with Peculiar Electrical Instability
4. Pathological Substrates
5. Nomenclature and Classification
6. Arrhythmogenic Cardiomyopathy as a Cause of Sudden Cardiac Death
7. Arrhythmogenic Cardiomyopathy: A Genetically Determined Heart Muscle Disease
8. Non-Desmosomal Arrhythmogenic Cardiomyopathy
9. Advances in Clinical Diagnosis
10. Prevention of SCD
11. Research Globalization
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Marrone, D.; Zampieri, F.; Basso, C.; Zanatta, A.; Thiene, G. History of the discovery of Arrhythmogenic Cardiomyopathy. Eur. Heart J. 2019, 40, 1100–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancisi, G.M. De Motu Cordis et Aneurysmatibus; Book II, Ch. VI, Par. XLVII; G.M. Salvioni: Roma, Italy, 1728. [Google Scholar]
- Lancisi, G.M. De Motu Cordis et Aneurysmatibus; Caput V: Naples, Italy, 1736. [Google Scholar]
- Hippocrates. On Diseases; Book I; Sacred Disease; Book V.
- Laennec, R. De L’Auscultation Médiate ou Traité du Diagnostic des Maladies des Poumons et du Coeur, Fondé Principalment sur ce Nouveau Moyen d’Exploration; Book II, Ch. XV; Brosson & Chaudé: Paris, France, 1819. [Google Scholar]
- Thiene, G. The research venture in arrhythmogenic right ventricular cardiomyopathy: A paradigm of translational medicine. Eur. Heart J. 2015, 36, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef] [Green Version]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef]
- Eliot, G. Middlemarch. A Study of Provincial Life; Book IV, Ch. XLII; W. Blackwood and Sons: Edinburgh-London, Scotland, 1871; p. 307. [Google Scholar]
- Dickens, C. The Posthumous Papers of the Pickwick Club; Chapman and Hall: London, UK, 2014; pp. 1836–1837. [Google Scholar]
- Kaufmann, E. Lehrbuch der Speziellen Pathologischen Anatomie für Studirende und Ärzte, 1st ed.; Reimer: Berlin, Germany, 1896; p. 28, Trattato di Patologia Speciale, 5th ed.; Vallardi: Milano, Italy, 1902; 1959; p. 125. [Google Scholar]
- Basso, C.; Thiene, G. Adipositas cordis, fatty infiltration of the right ventricle, and Arrhythmogenic Right Ventricular Cardiomyopathy. Just a matter of fat? Cardiovasc. Pathol. 2005, 14, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Osler, W. The principles and Practice of Medicine, 6th ed.; D. Appleton & Co.: New York, NY, USA, 1905; p. 820. [Google Scholar]
- MacDermot, H.E. Maude Abbott: A Memoir; Macmillan: Toronto, ON, Canada, 1941; p. 90. [Google Scholar]
- Segall, H.N. Parchment heart (Osler). Am. Heart J. 1950, 40, 948–950. [Google Scholar] [CrossRef]
- Uhl, H.S.M. A previously undescribed congenital malformation of the heart: Almost total absence of the myocardium of the right ventricle. Bull. Johns. Hopkins Hosp. 1952, 91, 197–209. [Google Scholar]
- Froment, R.; Perrin, A.; Loire, R.; Dalloz, C.L. Ventricule droit papyracé du jeune adulte par dystrophie congénitale. Arch. Mal. Coeur 1968, 61, 477–503. [Google Scholar]
- Shirani, J.; Berezowski, K.; Roberts, W.C. Quantitative measurement of normal and excessive (cor adiposum) subepicardial adipose tissue, its clinical significance, and its effect on electrocardiographic QRS voltage. Am. J. Cardiol. 1995, 76, 414–418. [Google Scholar] [CrossRef]
- Dalla Volta, S.; Battaglia, G.; Zerbini, E. “Auricularization” of the right ventricular pressure. Am. Heart J. 1961, 61, 25–33. [Google Scholar] [CrossRef]
- Dalla Volta, S.; Fameli, O.; Maschio, G. Le syndrome clinique et hémodynamique de l’auricularisation du ventricule droit. Arch. Mal. Coeur 1965, 58, 1129–1143. [Google Scholar] [PubMed]
- Fontaine, G.; Frank, R.; Vedel, J.; Grosgogeat, Y.; Cabrol, C.; Facquet, J. Stimulation studies and epicardial mapping in ventricular tachycardia: Study of mechanisms and selection for surgery. In Reentrant Arrhythmias; Kulbertus, H.E., Ed.; MTP Publishing: Dalton, PA, USA, 1977; pp. 334–350. [Google Scholar]
- Fontaine, G.; Frank, R.; Gallais-Hamonno, F.; Allali, I.; Phan-Thuc, H.; Grosgogeat, Y. Electrocardiographie des potentiels tardifs du syndrome de post-excitation [Electrocardiography of delayed potentials in post-excitation syndrome]. Arch. Mal. Coeur Vaiss. 1978, 71, 854–864. [Google Scholar] [PubMed]
- Beffagna, G.; Zorzi, A.; Pilichou, K.; Marra, M.P.; Rigato, I.; Corrado, D.; Migliore, F.; Rampazzo, A.; Bauce, B.; Basso, C.; et al. Arrhythmogenic Cardiomyopathy. Eur. Heart J. 2020, 41, 4457–4462. [Google Scholar] [CrossRef] [PubMed]
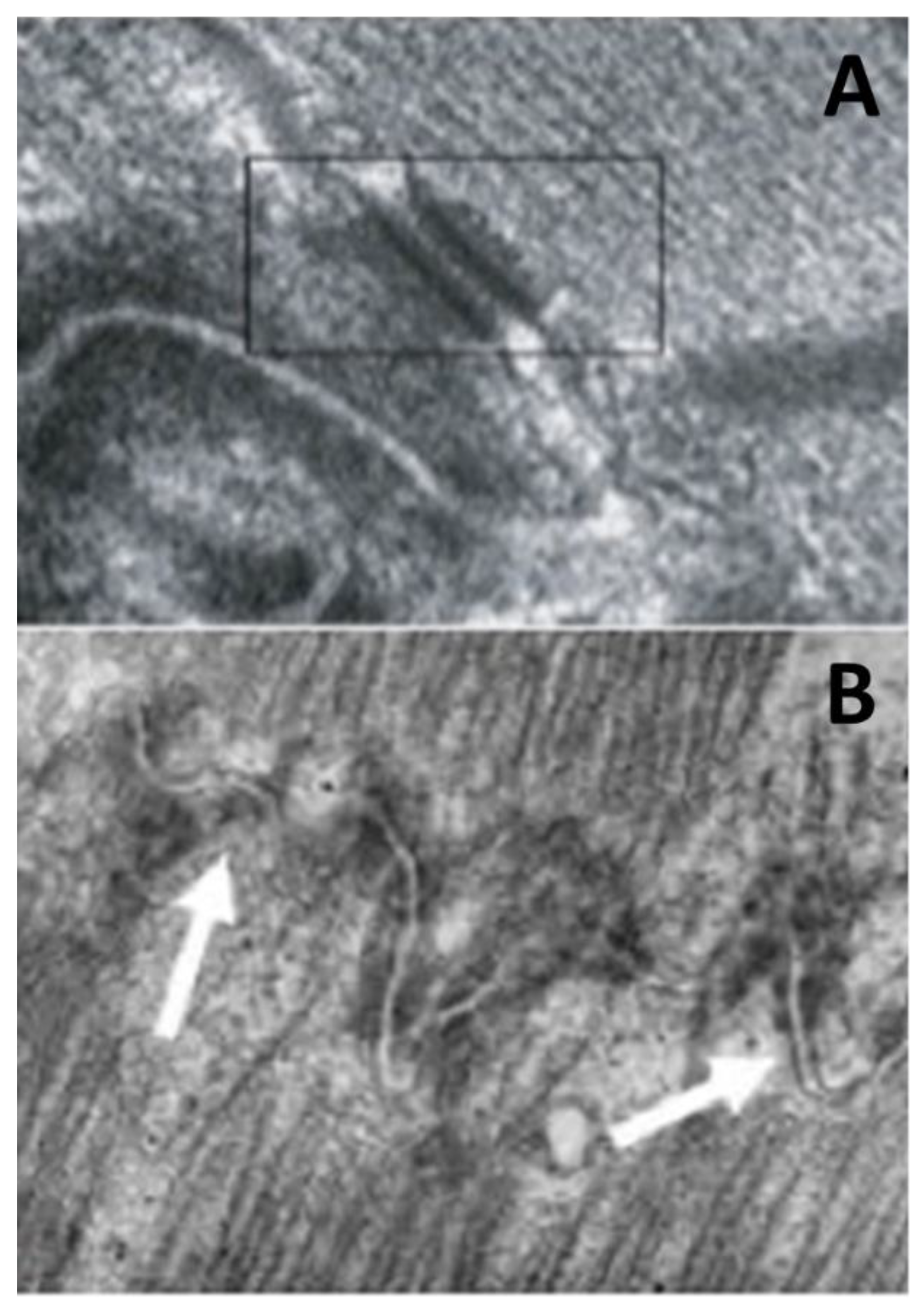
- Basso, C.; Czarnowska, E.; Della Barbera, M.; Bauce, B.; Beffagna, G.; Wlodarska, E.K.; Pilichou, K.; Ramondo, A.; Lorenzon, A.; Wozniek, O.; et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: An electron microscopy investigation on endomyocardial biopsies. Eur. Heart J. 2006, 27, 1847–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, M.; Calabrese, F.; Thiene, G.; Angelini, A.; Basso, C.; Nava, A.; Rossi, L. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am. J. Pathol. 1998, 152, 479–484. [Google Scholar]
- Thiene, G.; Corrado, D.; Nava, A.; Rossi, L.; Poletti, A.; Boffa, G.M.; Daliento, L.; Pennelli, N. Right ventricular cardiomyopathy: Is there evidence of an inflammatory aetiology? Eur. Heart J. 1991, 12 (Suppl. D), 22–25. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef]
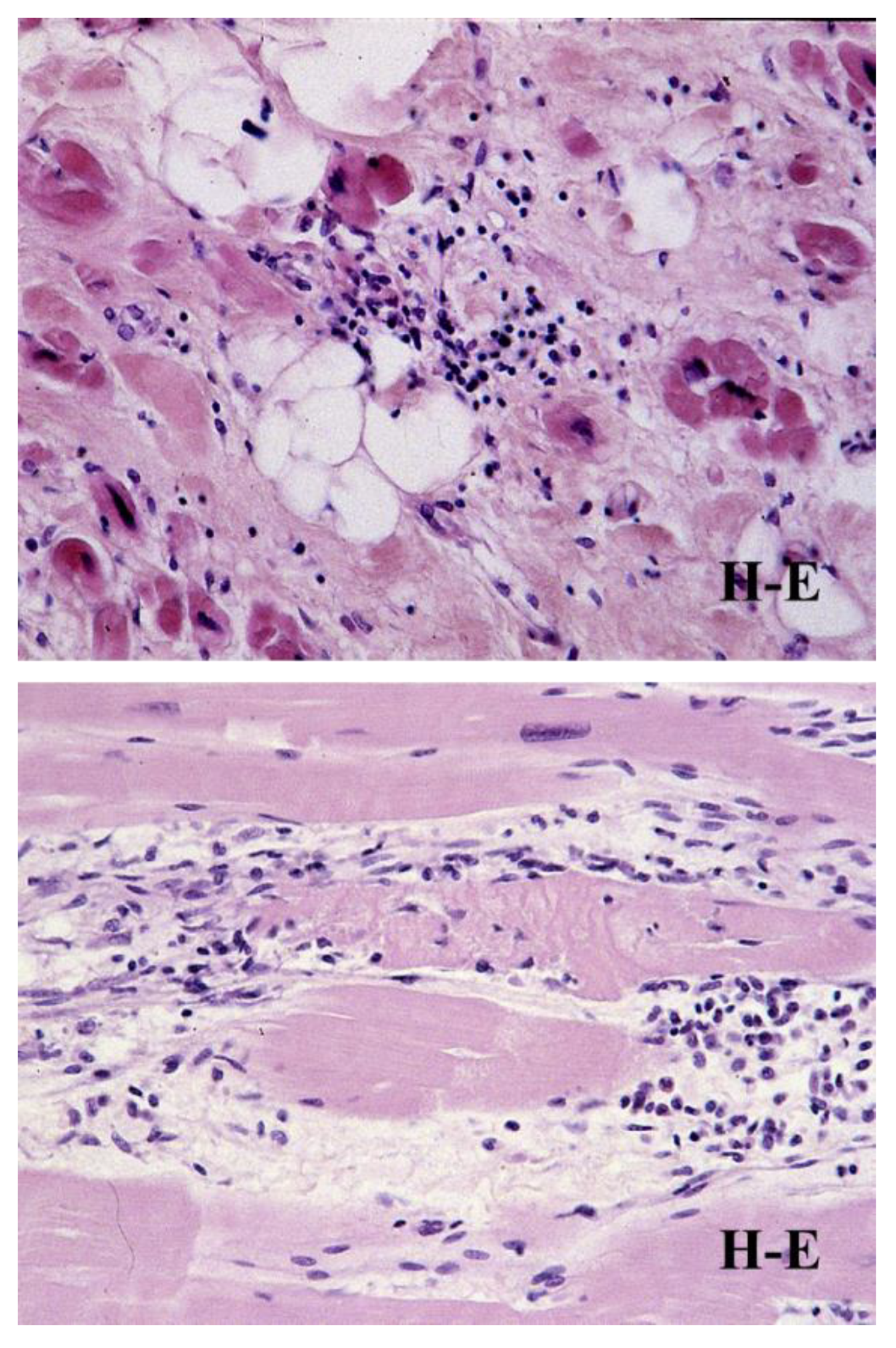
- D’Amati, G.; Leone, O.; di Gioia, C.R.; Magelli, C.; Arpesella, G.; Grillo, P.; Marino, B.; Fiore, F.; Gallo, P. Arrhythmogenic right ventricular cardiomyopathy: Clinicopathologic correlation based on a revised definition of pathologic patterns. Hum. Pathol. 2001, 32, 1078–1086. [Google Scholar] [CrossRef]
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Dello Russo, A.; Farina, F.M.; Casella, M.; Catto, V.; Pontone, G.; et al. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Heart J. 2016, 37, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef]
- Nava, A.; Rossi, L.; Thiene, G. Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia; International Congress Series (Book 1122); Excerpta Medica: Amstelveen, The Netherlands, 1997. [Google Scholar]
- Richardson, P.; McKenna, W.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar] [PubMed]
- Basso, C.; Nava, A.; Thiene, G. Cardiomiopatia Aritmogena; Arti Grafiche Color Black: Milano, Italy, 2001. [Google Scholar]
- Corrado, D.; Basso, C.; Rizzoli, G.; Schiavon, M.; Thiene, G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J. Am. Coll. Cardiol. 2003, 42, 1959–1963. [Google Scholar] [CrossRef] [PubMed]
- Winkel, B.G.; Holst, A.G.; Theilade, J.; Kristensen, I.B.; Thomsen, J.L.; Ottesen, G.L.; Bundgaard, H.; Svendsen, J.H.; Haunsø, S.; Tfelt-Hansen, J. Nationwide study of sudden cardiac death in persons aged 1–35 years. Eur. Heart J. 2011, 32, 983–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finocchiaro, G.; Papadakis, M.; Robertus, J.L.; Dhutia, H.; Steriotis, A.K.; Tome, M.; Mellor, G.; Merghani, A.; Malhotra, A.; Behr, E.; et al. Etiology of Sudden Death in Sports: Insights From a United Kingdom Regional Registry. J. Am. Coll. Cardiol. 2016, 67, 2108–2115. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connell, V.; Wallace, J.; et al. A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N. Engl. J. Med. 2016, 374, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Pavei, A.; Michieli, P.; Schiavon, M.; Thiene, G. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA 2006, 296, 1593–1601. [Google Scholar] [CrossRef] [Green Version]
- Nava, A.; Thiene, G.; Canciani, B.; Scognamiglio, R.; Daliento, L.; Buja, G.; Martini, B.; Stritoni, P.; Fasoli, G. Familial occurrence of right ventricular dysplasia: A study involving nine families. J. Am. Coll. Cardiol. 1988, 12, 1222–1228. [Google Scholar] [CrossRef] [Green Version]
- Nava, A.; Bauce, B.; Basso, C.; Muriago, M.; Rampazzo, A.; Villanova, C.; Daliento, L.; Buja, G.; Corrado, D.; Danieli, G.A.; et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2000, 36, 2226–2233. [Google Scholar] [CrossRef] [Green Version]
- Protonotarios, N.; Tsatsopoulou, A.; Patsourakos, P.; Alexopoulos, D.; Gezerlis, P.; Simitsis, S.; Scampardonis, G. Cardiac abnormalities in familial palmoplantar keratosis. Br. Heart J. 1986, 56, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, P.; Brinkmann, V.; Ledermann, B.; Behrend, M.; Grund, C.; Thalhammer, C.; Vogel, F.; Birchmeier, C.; Günthert, U.; Franke, W.W.; et al. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J. Cell Biol. 1996, 135, 215–225. [Google Scholar] [CrossRef]
- Coonar, A.S.; Protonotarios, N.; Tsatsopoulou, A.; Needham, E.W.; Houlston, R.S.; Cliff, S.; Otter, M.I.; Murday, V.A.; Mattu, R.K.; McKenna, W.J. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation 1998, 97, 2049–2058. [Google Scholar] [CrossRef] [Green Version]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Huerta, L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J. Am. Acad. Dermatol. 1998, 39, 418–421. [Google Scholar] [CrossRef]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, S.R.; Gard, J.J.; Carvajal-Huerta, L.; Ruiz-Cabezas, J.C.; Thiene, G.; Saffitz, J.E. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc. Pathol. 2004, 13, 26–32. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauce, B.; Basso, C.; Rampazzo, A.; Beffagna, G.; Daliento, L.; Frigo, G.; Malacrida, S.; Settimo, L.; Danieli, G.; Thiene, G.; et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur. Heart J. 2005, 26, 1666–1675. [Google Scholar] [CrossRef] [Green Version]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Nava, A.; Basso, C.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Frigo, G.; Vettori, A.; Valente, M.; Towbin, J.; et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 2006, 113, 1171–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syrris, P.; Ward, D.; Evans, A.; Asimaki, A.; Gandjbakhch, E.; Sen-Chowdhry, S.; McKenna, W.J. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am. J. Hum. Genet. 2006, 79, 978–984. [Google Scholar] [CrossRef] [Green Version]
- Beffagna, G.; De Bortoli, M.; Nava, A.; Salamon, M.; Lorenzon, A.; Zaccolo, M.; Mancuso, L.; Sigalotti, L.; Bauce, B.; Occhi, G.; et al. Missense mutations in desmocollin-2 N-terminus, associated with arrhythmogenic right ventricular cardiomyopathy, affect intracellular localization of desmocollin-2 in vitro. BMC Med. Genet. 2007, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigato, I.; Bauce, B.; Rampazzo, A.; Zorzi, A.; Pilichou, K.; Mazzotti, E.; Migliore, F.; Marra, M.P.; Lorenzon, A.; De Bortoli, M.; et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; van den Hoff, M.J.; de Bakker, J.M.; et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef] [Green Version]
- Giuliodori, A.; Beffagna, G.; Marchetto, G.; Fornetto, C.; Vanzi, F.; Toppo, S.; Facchinello, N.; Santimaria, M.; Vettori, A.; Rizzo, S.; et al. Loss of cardiac Wnt/β-catenin signalling in desmoplakin-deficient AC8 zebrafish models is rescuable by genetic and pharmacological intervention. Cardiovasc. Res. 2018, 114, 1082–1097. [Google Scholar] [CrossRef]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Karin Arndt, A.; Adams, E.; Liu, Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J.; et al. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2014, 6, 240ra74. [Google Scholar] [CrossRef] [Green Version]
- Risato, G.; Celeghin, R.; Brañas Casas, R.; Dinarello, A.; Zuppardo, A.; Vettori, A.; Pilichou, K.; Thiene, G.; Basso, C.; Argenton, F.; et al. Hyperactivation of Wnt/β-catenin and Jak/Stat3 pathways in human and zebrafish foetal growth restriction models: Implications for pharmacological rescue. Front. Cell Dev. Biol. 2022, 10, 943127. [Google Scholar] [CrossRef]
- Rizzo, S.; Lodder, E.M.; Verkerk, A.O.; Wolswinkel, R.; Beekman, L.; Pilichou, K.; Basso, C.; Remme, C.A.; Thiene, G.; Bezzina, C.R. Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc. Res. 2012, 95, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Christensen, A.H.; Andersen, C.B.; Tybjaerg-Hansen, A.; Haunso, S.; Svendsen, J.H. Mutation analysis and evaluation of the cardiac localization of TMEM43 in arrhythmogenic right ventricular cardiomyopathy. Clin. Genet. 2011, 80, 256–264. [Google Scholar] [CrossRef]
- Haywood, A.F.; Merner, N.D.; Hodgkinson, K.A.; Houston, J.; Syrris, P.; Booth, V.; Connors, S.; Pantazis, A.; Quarta, G.; Elliott, P.; et al. Recurrent missense mutations in TMEM43 (ARVD5) due to founder effects cause arrhythmogenic cardiomyopathies in the UK and Canada. Eur. Heart J. 2013, 34, 1002–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkapic, D.; Neumann, T.; Schmitt, J.; Sperzel, J.; Berkowitsch, A.; Kuniss, M.; Hamm, C.W.; Pitschner, H.F. Electrical storm in a patient with arrhythmogenic right ventricular cardiomyopathy and SCN5A mutation. Europace 2008, 10, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, J.; Dai, X.; Cao, Q.; Xiong, Q.; Liu, X.; Liu, X.; Shen, Y.; Chen, Q.; Hua, W.; et al. SCN5A mutation in Chinese patients with arrhythmogenic right ventricular dysplasia. Herz 2014, 39, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, A.S.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Sobreira, N.L.; Amat-Alarcon, N.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Tintelen, J.P.; Van Gelder, I.C.; Asimaki, A.; Suurmeijer, A.J.; Wiesfeld, A.C.; Jongbloed, J.D.; van den Wijngaard, A.; Kuks, J.B.; van Spaendonck-Zwarts, K.Y.; Notermans, N.; et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 2009, 6, 1574–1583. [Google Scholar] [CrossRef]
- Otten, E.; Asimaki, A.; Maass, A.; van Langen, I.M.; van der Wal, A.; de Jonge, N.; van den Berg, M.P.; Saffitz, J.E.; Wilde, A.A.; Jongbloed, J.D.; et al. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm. 2010, 7, 1058–1064. [Google Scholar] [CrossRef]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [Green Version]
- Hedberg, C.; Melberg, A.; Kuhl, A.; Jenne, D.; Oldfors, A. Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy 7 is caused by a DES mutation. Eur. J. Hum. Genet. 2012, 20, 984–985. [Google Scholar] [CrossRef]
- Lorenzon, A.; Beffagna, G.; Bauce, B.; De Bortoli, M.; Li Mura, I.E.; Calore, M.; Dazzo, E.; Basso, C.; Nava, A.; Thiene, G.; et al. Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am. J. Cardiol. 2013, 111, 400–405. [Google Scholar] [CrossRef]
- Brodehl, A.; Dieding, M.; Klauke, B.; Dec, E.; Madaan, S.; Huang, T.; Gargus, J.; Fatima, A.; Saric, T.; Cakar, H.; et al. The novel desmin mutant p.A120D impairs filament formation, prevents intercalated disk localization, and causes sudden cardiac death. Circ. Cardiovasc. Genet. 2013, 6, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripoll-Vera, T.; Zorio, E.; Gámez, J.M.; Molina, P.; Govea, N.; Crémer, D. Phenotypic Patterns of Cardiomyopathy Caused by Mutations in the Desmin Gene. A Clinical and Genetic Study in Two Inherited Heart Disease Units. Rev. Esp. Cardiol. 2015, 68, 1027–1029. [Google Scholar] [CrossRef]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.Á.; Álvarez, M.; López-Fernández, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Zwaag, P.A.; van Rijsingen, I.A.; de Ruiter, R.; Nannenberg, E.A.; Groeneweg, J.A.; Post, J.G.; Hauer, R.N.; van Gelder, I.C.; van den Berg, M.P.; van der Harst, P.; et al. Recurrent and founder mutations in the Netherlands-Phospholamban p.Arg14del mutation causes arrhythmogenic cardiomyopathy. Neth. Heart J. 2013, 21, 286–293. [Google Scholar] [CrossRef] [Green Version]
- van Rijsingen, I.A.; van der Zwaag, P.A.; Groeneweg, J.A.; Nannenberg, E.A.; Jongbloed, J.D.; Zwinderman, A.H.; Pinto, Y.M.; Dit Deprez, R.H.; Post, J.G.; Tan, H.L.; et al. Outcome in phospholamban R14del carriers: Results of a large multicentre cohort study. Circ. Cardiovasc. Genet. 2014, 7, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Gho, J.M.; van Es, R.; Stathonikos, N.; Harakalova, M.; te Rijdt, W.P.; Suurmeijer, A.J.; van der Heijden, J.F.; de Jonge, N.; Chamuleau, S.A.; de Weger, R.A.; et al. High resolution systematic digital histological quantification of cardiac fibrosis and adipose tissue in phospholamban p.Arg14del mutation associated cardiomyopathy. PLoS ONE 2014, 9, e94820. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; van der Zwaag, P.A.; Olde Nordkamp, L.R.; Bikker, H.; Jongbloed, J.D.; Jongbloed, R.; Wiesfeld, A.C.; Cox, M.G.; van der Heijden, J.F.; Atsma, D.E.; et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy according to revised 2010 task force criteria with inclusion of non-desmosomal phospholamban mutation carriers. Am. J. Cardiol. 2013, 112, 1197–1206. [Google Scholar] [CrossRef]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Valtuille, L.; Paterson, I.; Kim, D.H.; Mullen, J.; Sergi, C.; Oudit, G.Y. A case of lamin A/C mutation cardiomyopathy with overlap features of ARVC: A critical role of genetic testing. Int. J. Cardiol. 2013, 168, 4325–4327. [Google Scholar] [CrossRef]
- Alastalo, T.P.; West, G.; Li, S.P.; Keinänen, A.; Helenius, M.; Tyni, T.; Lapatto, R.; Turanlahti, M.; Heikkilä, P.; Kääriäinen, H.; et al. LMNA Mutation c.917T>G (p.L306R) Leads to Deleterious Hyper-Assembly of Lamin A/C and Associates with Severe Right Ventricular Cardiomyopathy and Premature Aging. Hum. Mutat. 2015, 36, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Takahashi, N.; Fujii, Y.; Umehara, A.; Nishiuchi, S.; Makiyama, T.; Ohno, S.; Horie, M. LMNA cardiomyopathy detected in Japanese arrhythmogenic right ventricular cardiomyopathy cohort. J. Cardiol. 2016, 68, 346–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Brun, F.; Gigli, M.; Graw, S.L.; Judge, D.P.; Merlo, M.; Murray, B.; Calkins, H.; Sinagra, G.; Taylor, M.R.; Mestroni, L.; et al. FLNC truncations cause arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2020, 57, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Oz, S.; Yonath, H.; Visochyk, L.; Ofek, E.; Landa, N.; Reznik-Wolf, H.; Ortiz-Genga, M.; Monserrat, L.; Ben-Gal, T.; Goitein, O.; et al. Reduction in Filamin C transcript is associated with arrhythmogenic cardiomyopathy in Ashkenazi Jews. Int. J. Cardiol. 2020, 317, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.L.; Akhtar, M.M.; Sabater-Molina, M.; Futema, M.; Asimaki, A.; Protonotarios, A.; Dalageorgou, C.; Pittman, A.M.; Suarez, M.P.; Aguilera, B.; et al. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int. J. Cardiol. 2020, 307, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Celeghin, R.; Cipriani, A.; Bariani, R.; Bueno Marinas, M.; Cason, M.; Bevilacqua, M.; De Gaspari, M.; Rizzo, S.; Rigato, I.; Da Pozzo, S.; et al. Filamin-C variant-associated cardiomyopathy: A pooled analysis of individual patient data to evaluate the clinical profile and risk of sudden cardiac death. Heart Rhythm. 2022, 19, 235–243. [Google Scholar] [CrossRef]
- Turkowski, K.L.; Tester, D.J.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.J. Whole exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit. Heart Dis. 2017, 12, 226–235. [Google Scholar] [CrossRef]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001605. [Google Scholar] [CrossRef] [Green Version]
- Ghidoni, A.; Elliott, P.M.; Syrris, P.; Calkins, H.; James, C.A.; Judge, D.P.; Murray, B.; Barc, J.; Probst, V.; Schott, J.J.; et al. Cadherin 2-Related Arrhythmogenic Cardiomyopathy: Prevalence and Clinical Features. Circ. Genom. Precis. Med. 2021, 14, e003097. [Google Scholar] [CrossRef]
- De Bortoli, M.; Postma, A.V.; Poloni, G.; Calore, M.; Minervini, G.; Mazzotti, E.; Rigato, I.; Ebert, M.; Lorenzon, A.; Vazza, G.; et al. Whole-Exome Sequencing Identifies Pathogenic Variants in TJP1 Gene Associated with Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e002123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quang, K.L.; Maguy, A.; Qi, X.Y.; Naud, P.; Xiong, F.; Tadevosyan, A.; Shi, Y.F.; Chartier, D.; Tardif, J.C.; Dobrev, D.; et al. Loss of cardiomyocyte integrin-linked kinase produces an arrhythmogenic cardiomyopathy in mice. Circ. Arrhythm. Electrophysiol. 2015, 8, 921–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodehl, A.; Rezazadeh, S.; Williams, T.; Munsie, N.M.; Liedtke, D.; Oh, T.; Ferrier, R.; Shen, Y.; Jones, S.J.M.; Stiegler, A.L.; et al. Mutations in ILK, encoding integrin-linked kinase, are associated with arrhythmogenic cardiomyopathy. Transl. Res. 2019, 208, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Abdelfatah, N.; Chen, R.; Duff, H.J.; Seifer, C.M.; Buffo, I.; Huculak, C.; Clarke, S.; Clegg, R.; Jassal, D.S.; Gordon, P.M.K.; et al. Characterization of a Unique Form of Arrhythmic Cardiomyopathy Caused by Recessive Mutation in LEMD2. JACC Basic Transl. Sci. 2019, 4, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.L.; Wallace, M.J.; Refaey, M.E.; Roberts, J.D.; Koenig, S.N.; Mohler, P.J. Arrhythmogenic Cardiomyopathy: Molecular Insights for Improved Therapeutic Design. J. Cardiovasc. Dev. Dis. 2020, 7, 21. [Google Scholar] [CrossRef]
- Celeghin, R.; Pilichou, K. The complex molecular genetics of arrhythmogenic cardiomyopathy. Int. J. Cardiol. 2019, 284, 59–60. [Google Scholar] [CrossRef]
- James, C.A.; Jongbloed, J.D.H.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International Evidence Based Reappraisal of Genes Associated with Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ. Genom. Precis. Med. 2021, 14, e003273. [Google Scholar] [CrossRef]
- McKenna, W.J.; Thiene, G.; Nava, A.; Fontaliran, F.; Blomstrom-Lundqvist, C.; Fontaine, G.; Camerini, F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br. Heart J. 1994, 71, 215–218. [Google Scholar]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Daliento, L.; Rizzoli, G.; Thiene, G.; Nava, A.; Rinuncini, M.; Chioin, R.; Dalla Volta, S. Diagnostic accuracy of right ventriculography in arrhythmogenic right ventricular cardiomyopathy. Am. J. Cardiol. 1990, 66, 741–745. [Google Scholar] [CrossRef]
- Basso, C.; Ronco, F.; Marcus, F.; Abudureheman, A.; Rizzo, S.; Frigo, A.C.; Bauce, B.; Maddalena, F.; Nava, A.; Corrado, D.; et al. Quantitative assessment of endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy/dysplasia: An in vitro validation of diagnostic criteria. Eur. Heart J. 2008, 29, 2760–2771. [Google Scholar] [CrossRef] [Green Version]
- Zorzi, A.; Perazzolo Marra, M.; Rigato, I.; De Lazzari, M.; Susana, A.; Niero, A.; Pilichou, K.; Migliore, F.; Rizzo, S.; Giorgi, B.; et al. Nonischemic Left Ventricular Scar as a Substrate of Life-Threatening Ventricular Arrhythmias and Sudden Cardiac Death in Competitive Athletes. Circ. Arrhythm. Electrophysiol. 2016, 9, e004229. [Google Scholar] [CrossRef] [PubMed]
- Norman, M.; Simpson, M.; Mogensen, J.; Shaw, A.; Hughes, S.; Syrris, P.; Sen-Chowdhry, S.; Rowland, E.; Crosby, A.; McKenna, W.J. Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation 2005, 112, 636–642. [Google Scholar] [CrossRef] [PubMed]
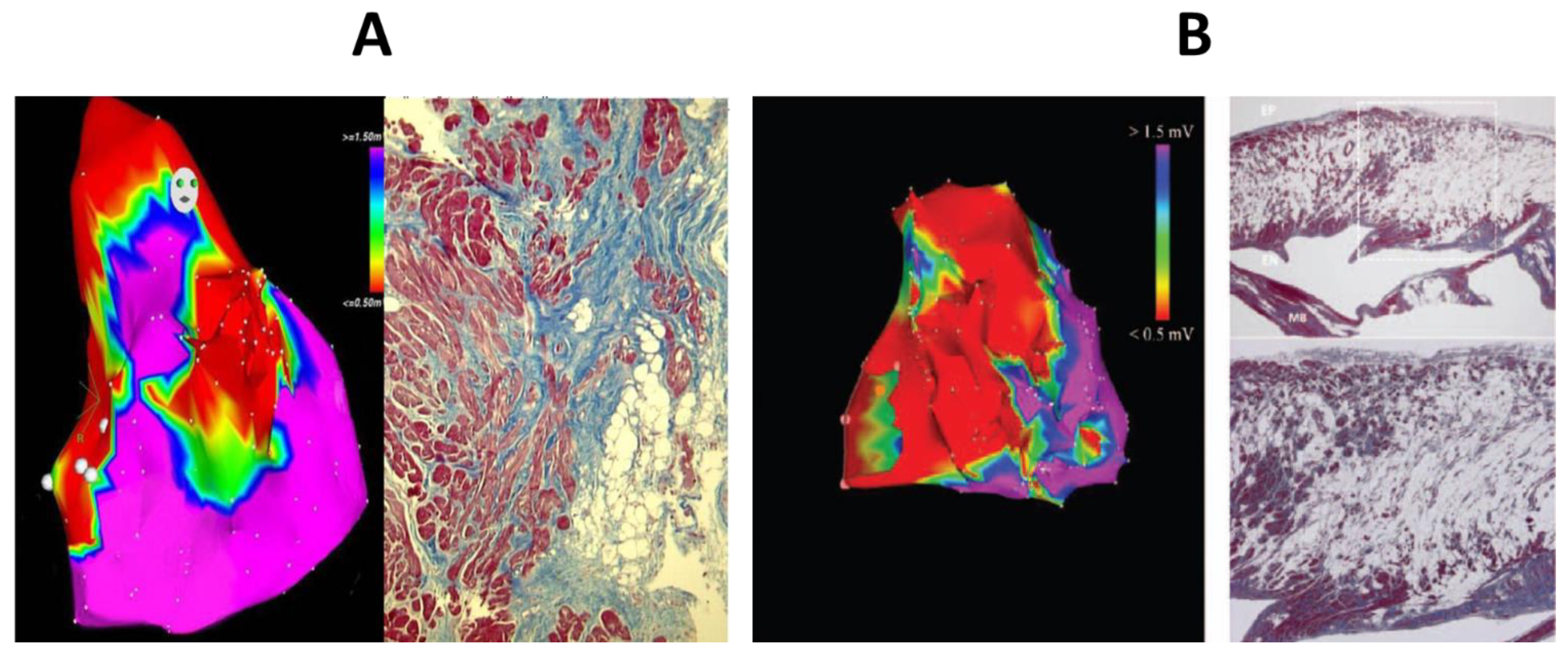
- Corrado, D.; Basso, C.; Leoni, L.; Tokajuk, B.; Turrini, P.; Bauce, B.; Migliore, F.; Pavei, A.; Tarantini, G.; Napodano, M.; et al. Three-dimensional electroanatomical voltage mapping and histologic evaluation of myocardial substrate in right ventricular outflow tract tachycardia. J. Am. Coll. Cardiol. 2008, 51, 731–739. [Google Scholar] [CrossRef]
- Migliore, F.; Zorzi, A.; Michieli, P.; Perazzolo Marra, M.; Siciliano, M.; Rigato, I.; Bauce, B.; Basso, C.; Toazza, D.; Schiavon, M.; et al. Prevalence of cardiomyopathy in Italian asymptomatic children with electrocardiographic T-wave inversion at preparticipation screening. Circulation 2012, 125, 529–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Leoni, L.; Link, M.S.; Della Bella, P.; Gaita, F.; Curnis, A.; Salerno, J.U.; Igidbashian, D.; Raviele, A.; Disertori, M.; et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2003, 108, 3084–3091. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Basso, C.; Pilichou, K.; Thiene, G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart 2011, 97, 530–539. [Google Scholar] [CrossRef]
- Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic RV Cardiomyopathy/Dysplasia; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]






























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thiene, G.; Basso, C.; Pilichou, K.; Bueno Marinas, M. Desmosomal Arrhythmogenic Cardiomyopathy: The Story Telling of a Genetically Determined Heart Muscle Disease. Biomedicines 2023, 11, 2018. https://doi.org/10.3390/biomedicines11072018
Thiene G, Basso C, Pilichou K, Bueno Marinas M. Desmosomal Arrhythmogenic Cardiomyopathy: The Story Telling of a Genetically Determined Heart Muscle Disease. Biomedicines. 2023; 11(7):2018. https://doi.org/10.3390/biomedicines11072018
Chicago/Turabian StyleThiene, Gaetano, Cristina Basso, Kalliopi Pilichou, and Maria Bueno Marinas. 2023. "Desmosomal Arrhythmogenic Cardiomyopathy: The Story Telling of a Genetically Determined Heart Muscle Disease" Biomedicines 11, no. 7: 2018. https://doi.org/10.3390/biomedicines11072018
APA StyleThiene, G., Basso, C., Pilichou, K., & Bueno Marinas, M. (2023). Desmosomal Arrhythmogenic Cardiomyopathy: The Story Telling of a Genetically Determined Heart Muscle Disease. Biomedicines, 11(7), 2018. https://doi.org/10.3390/biomedicines11072018