
Regulation of Soluble E-Cadherin Signaling in Non-Small-Cell Lung Cancer Cells by Nicotine, BDNF, and β-Adrenergic Receptor Ligands

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. MMP9 Level Measurement
2.4. Activated EGFR Assay
2.5. Activated IGF-1R Assay
2.6. Phosphoinositide 3-Kinase (PI3K) Assay
2.7. Extracellular Signal-Regulated Kinase (ERK) Assay
2.8. p53 Transcription Factor Activity Assay
2.9. MTT Assay
2.10. Apoptosis Assay
2.11. Measurement of sE-Cad Levels
2.12. Immunodepletion
2.13. Western Blotting
2.14. SiRNA Transfection
2.15. Statistical Analysis
3. Results
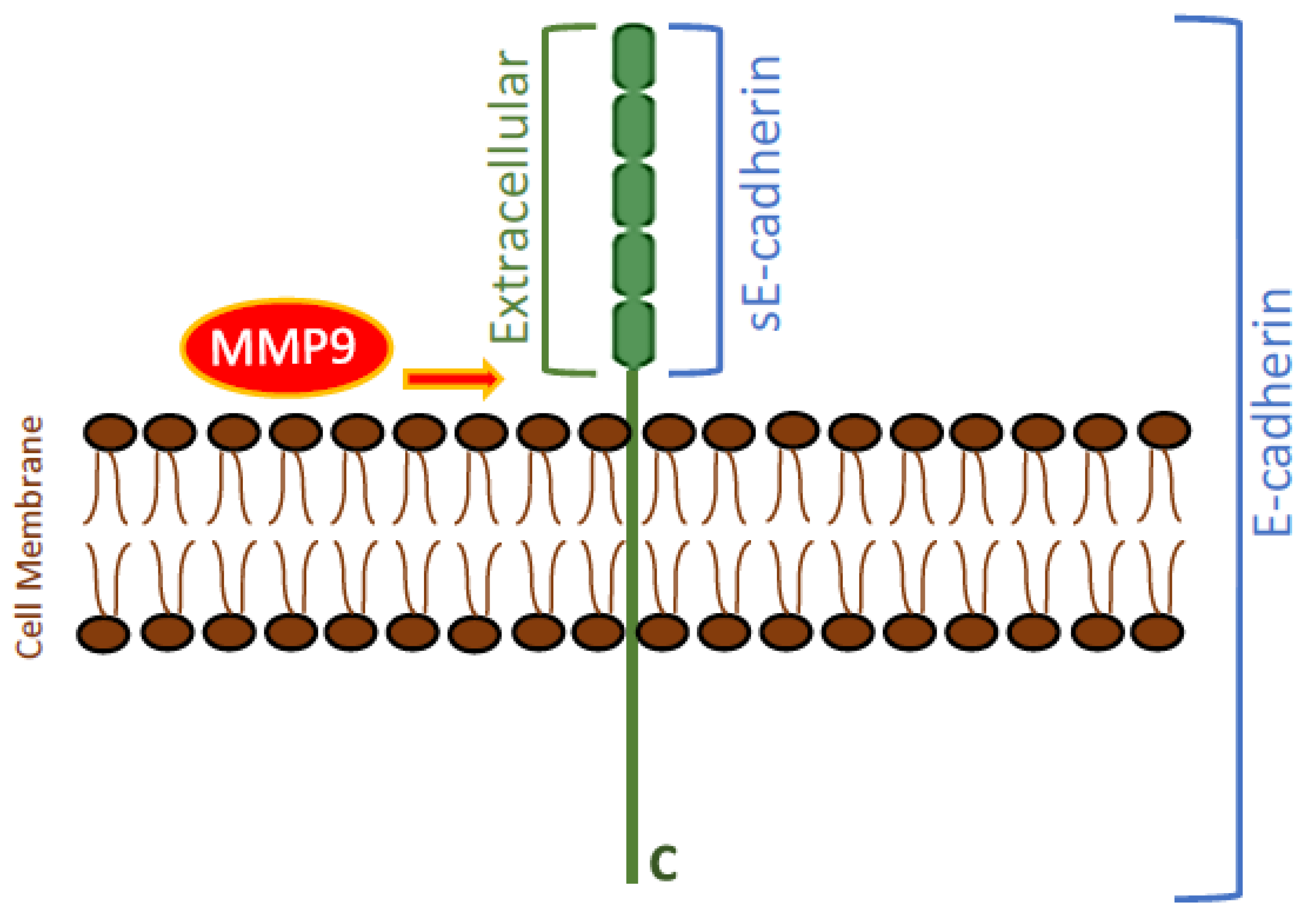
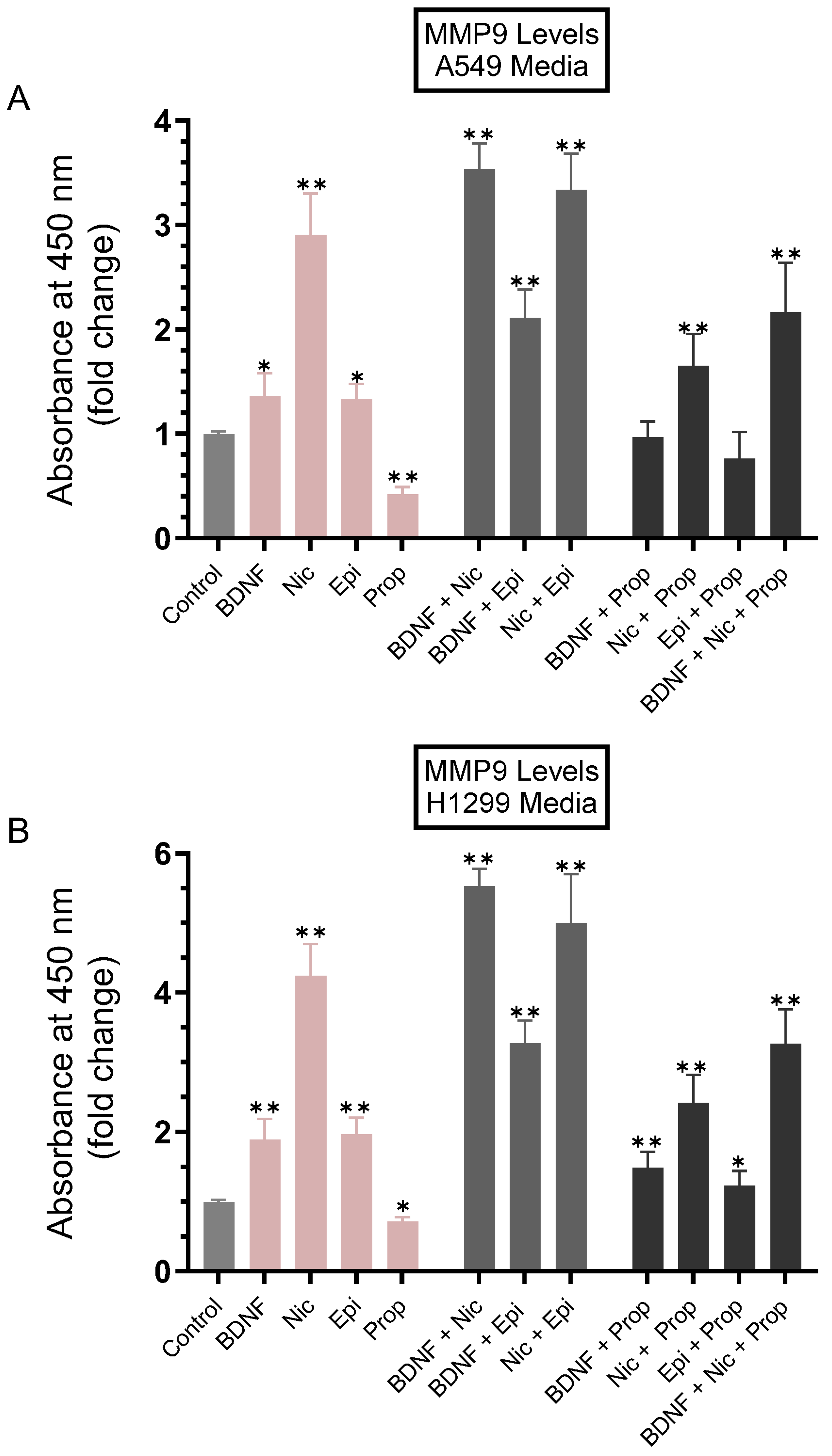
3.1. MMP9 Levels Increased in the Conditioned Media of A549 and H1299 Cells Treated with Either BDNF, Nicotine, or Epinephrine but Decreased upon Cell Treatment with Non-Selective β-Adrenergic Receptors Blocker, Propranolol
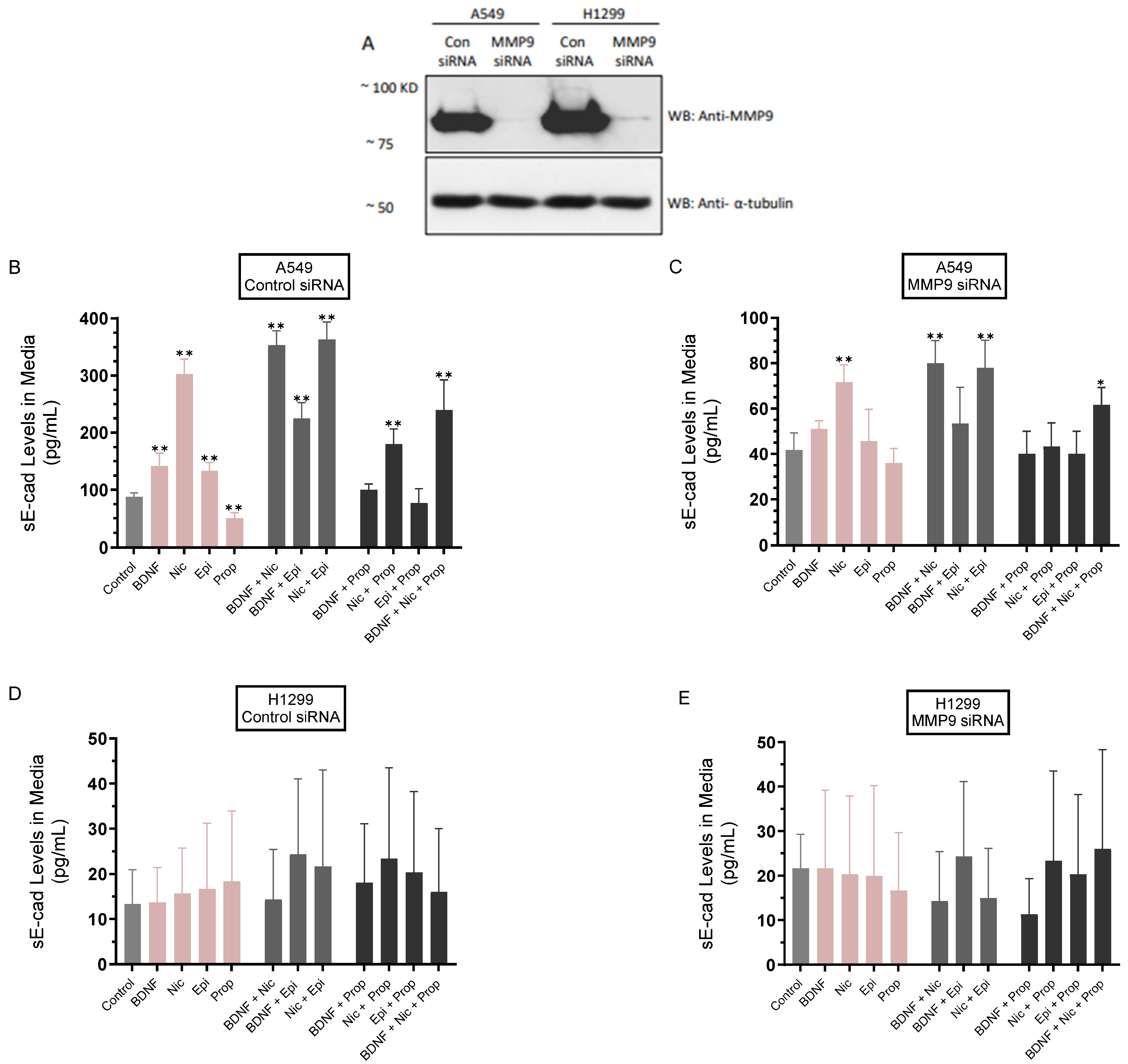
3.2. The Levels of sE-Cad Were Downregulated in the Media of A549 Cells Transfected with MMP9 siRNA
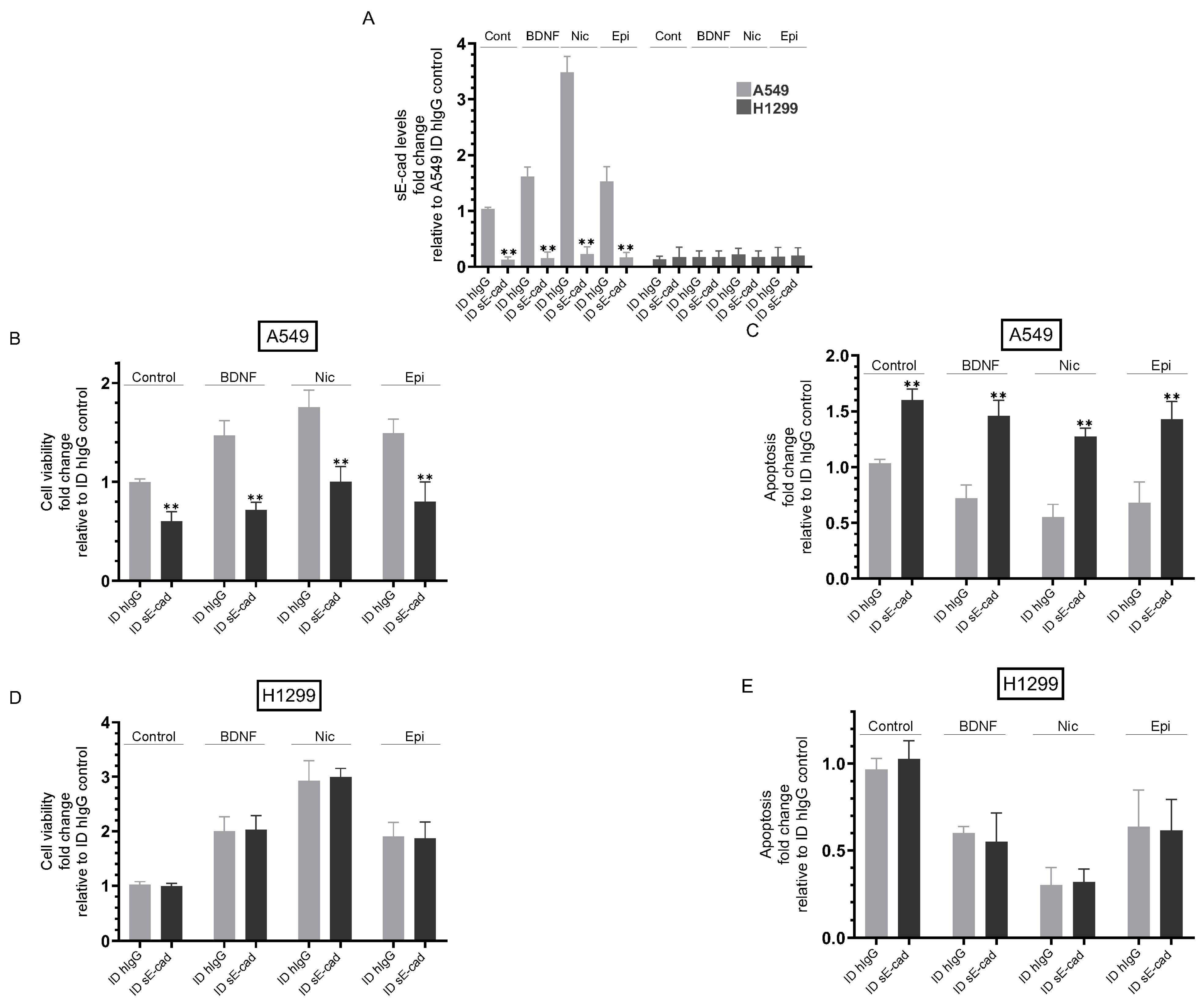
3.3. Immunodepletion of sE-Cad Decreased Viability and Increased Apoptosis of A549 Cells
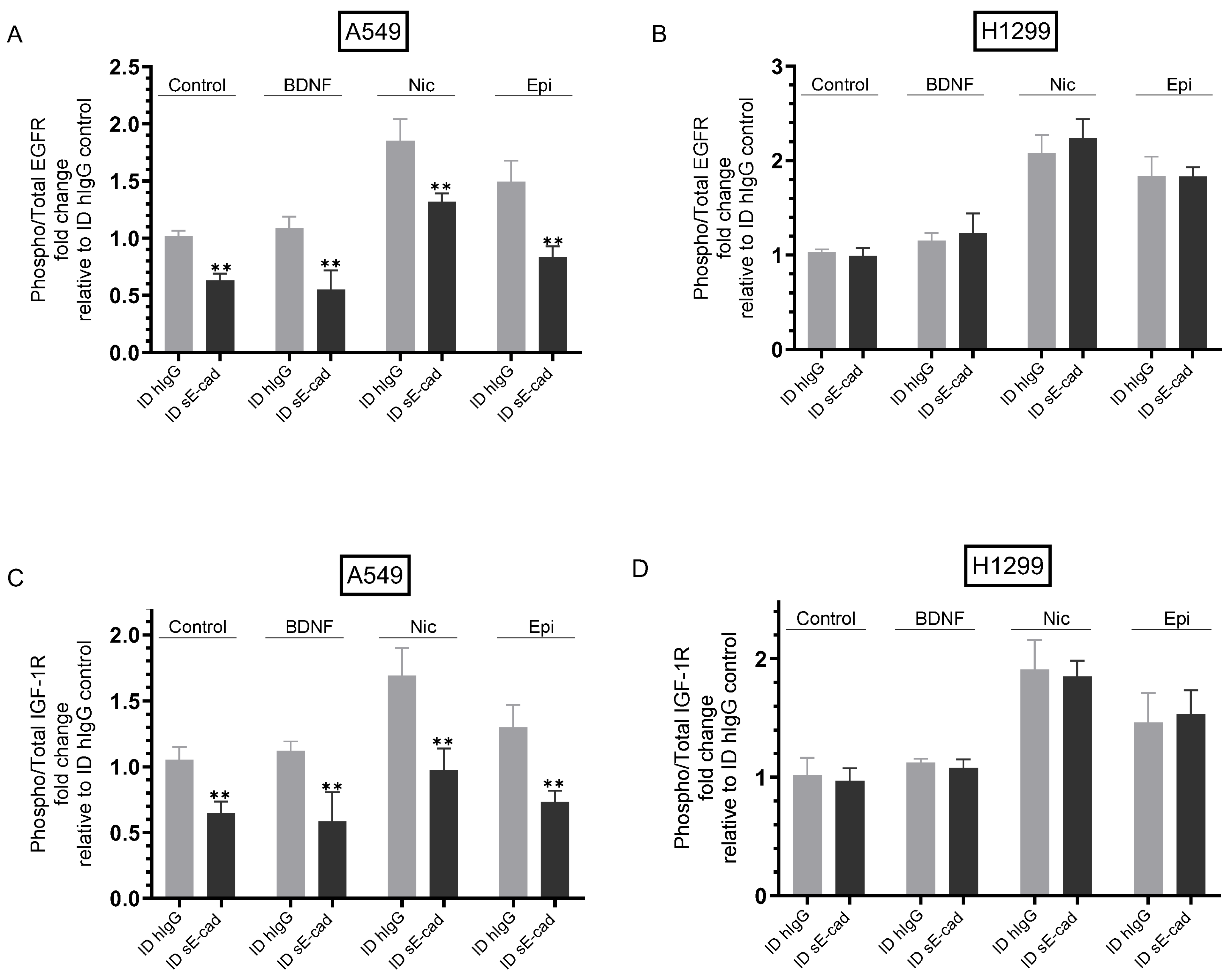
3.4. The Activities of EGFR and IGF-1R Were Blocked by Immunodepletion of sE-Cad from the Media of A549 Cells
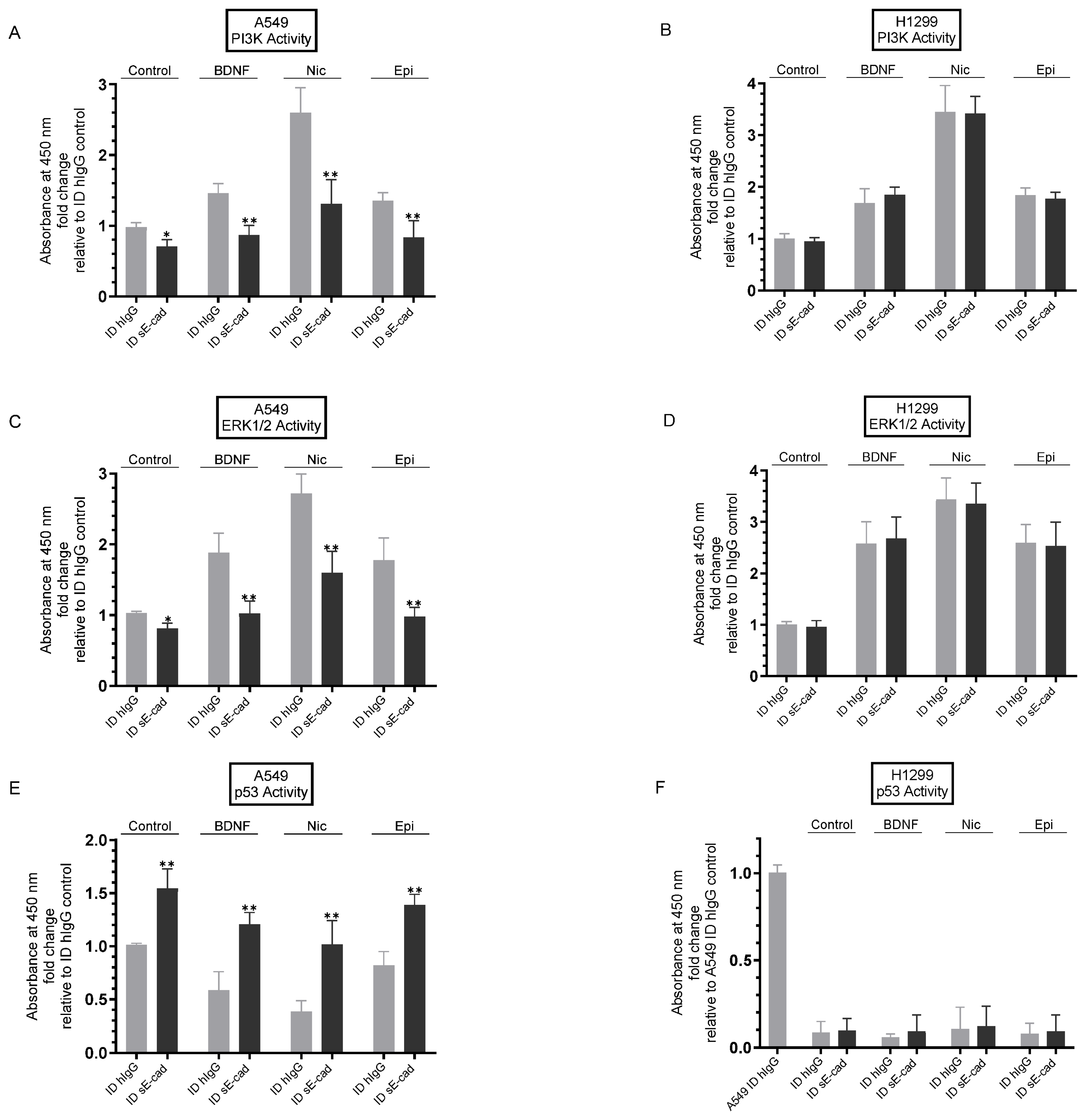
3.5. Immunodepletion of sE-Cad Led to Decreased PI3K and ERK1/2 Activities and Enhanced p53 Activation in A549 Cells
4. Discussion
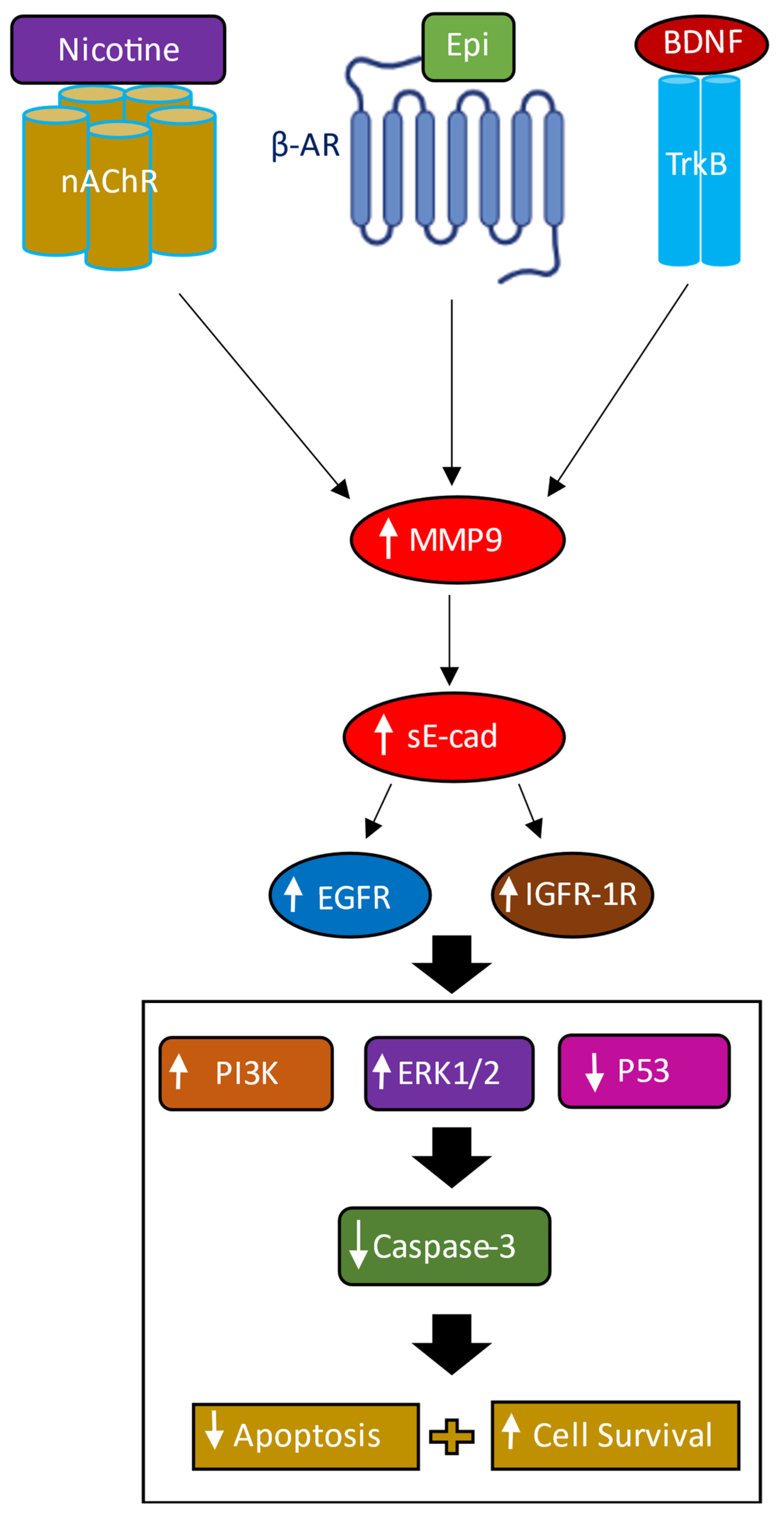
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mithoowani, H.; Febbraro, M. Non-Small-Cell Lung Cancer in 2022: A Review for General Practitioners in Oncology. Curr. Oncol. 2022, 29, 1828–1839. [Google Scholar] [CrossRef]
- Custodio, A.B.; González-Larriba, J.L.; Bobokova, J.; Calles, A.; Alvarez, R.; Cuadrado, E.; Manzano, A.; Díaz-Rubio, E. Prognostic and Predictive Markers of Benefit from Adjuvant Chemotherapy in Early-Stage Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2009, 4, 891–910. [Google Scholar] [CrossRef]
- Rotow, J.; Bivona, T.G. Understanding and Targeting Resistance Mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Siddiqui, F.; Siddiqui, A.H. Lung Cancer. In StatPearls; StatPearls Publishing: Treasure Island, FL USA, 2021. [Google Scholar]
- Reddy, P.; Liu, L.; Ren, C.; Lindgren, P.; Boman, K.; Shen, Y.; Lundin, E.; Ottander, U.; Rytinki, M.; Liu, K. Formation of E-Cadherin-Mediated Cell-Cell Adhesion Activates AKT and Mitogen Activated Protein Kinase via Phosphatidylinositol 3 Kinase and Ligand-Independent Activation of Epidermal Growth Factor Receptor in Ovarian Cancer Cells. Mol. Endocrinol. 2005, 19, 2564–2578. [Google Scholar] [CrossRef]
- Fonseca, I.C.C.F.E.; da Luz, F.A.C.; Uehara, I.A.; Silva, M.J.B. Cell-Adhesion Molecules and Their Soluble Forms: Promising Predictors of “Tumor Progression” and Relapse in Leukemia. Tumor Biol. 2018, 40, 1010428318811525. [Google Scholar] [CrossRef]
- David, J.M.; Rajasekaran, A.K. Dishonorable Discharge: The Oncogenic Roles of Cleaved E-Cadherin Fragments. Cancer Res. 2012, 72, 2917–2923. [Google Scholar] [CrossRef]
- Hu, Q.; Kuang, J.; Yang, Q.-K.; Bian, X.; Yu, S.-C. Beyond a Tumor Suppressor: Soluble E-cadherin Promotes the Progression of Cancer. Int. J. Cancer 2016, 138, 2804–2812. [Google Scholar] [CrossRef]
- Miller, M.A.; Sullivan, R.J.; Lauffenburger, D.A. Molecular Pathways: Receptor Ectodomain Shedding in Treatment, Resistance, and Monitoring of Cancer. Clin. Cancer Res. 2017, 23, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Symowicz, J.; Adley, B.P.; Gleason, K.J.; Johnson, J.J.; Ghosh, S.; Fishman, D.A.; Hudson, L.G.; Stack, M.S. Engagement of Collagen-Binding Integrins Promotes Matrix Metalloproteinase-9-Dependent E-Cadherin Ectodomain Shedding in Ovarian Carcinoma Cells. Cancer Res. 2007, 67, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, M.M.; Day, M.L. Soluble E-Cadherin: More than a Symptom of Disease. Front. Biosci. (Landmark Ed.) 2012, 17, 1948–1964. [Google Scholar] [CrossRef]
- Brouxhon, S.M.; Kyrkanides, S.; Teng, X.; Athar, M.; Ghazizadeh, S.; Simon, M.; O’Banion, M.K.; Ma, L. Soluble E-Cadherin: A Critical Oncogene Modulating Receptor Tyrosine Kinases, MAPK and PI3K/Akt/mTOR Signaling. Oncogene 2014, 33, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Brouxhon, S.M.; Kyrkanides, S.; Teng, X.; O’Banion, M.K.; Clarke, R.; Byers, S.; Ma, L. Soluble-E-Cadherin Activates HER and IAP Family Members in HER2+ and TNBC Human Breast Cancers. Mol. Carcinog. 2014, 53, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Yang, L.; Li, T.; Zhang, Y. Cadherin Signaling in Cancer: Its Functions and Role as a Therapeutic Target. Front. Oncol. 2019, 9, 989. [Google Scholar] [CrossRef]
- Friedman, J.R.; Richbart, S.D.; Merritt, J.C.; Brown, K.C.; Nolan, N.A.; Akers, A.T.; Lau, J.K.; Robateau, Z.R.; Miles, S.L.; Dasgupta, P. Acetylcholine Signaling System in Progression of Lung Cancers. Pharmacol. Ther. 2019, 194, 222–254. [Google Scholar] [CrossRef]
- Gahring, L.C.; Myers, E.J.; Dunn, D.M.; Weiss, R.B.; Rogers, S.W. Nicotinic Alpha 7 Receptor Expression and Modulation of the Lung Epithelial Response to Lipopolysaccharide. PLoS ONE 2017, 12, e0175367. [Google Scholar] [CrossRef]
- Jiang, S.-H.; Hu, L.-P.; Wang, X.; Li, J.; Zhang, Z.-G. Neurotransmitters: Emerging Targets in Cancer. Oncogene 2020, 39, 503–515. [Google Scholar] [CrossRef]
- Lazarevic-Pasti, T.; Leskovac, A.; Momic, T.; Petrovic, S.; Vasic, V. Modulators of Acetylcholinesterase Activity: From Alzheimer’s Disease to Anti-Cancer Drugs. Curr. Med. Chem. 2017, 24, 3283–3309. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian Nicotinic Acetylcholine Receptors: From Structure to Function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef]
- Martínez-López de Castro, A.; Nieto-Cerón, S.; Aurelio, P.-C.; Galbis-Martínez, L.; Latour-Pérez, J.; Torres-Lanzas, J.; Tovar-Zapata, I.; Martínez-Hernández, P.; Rodríguez-López, J.N.; Cabezas-Herrera, J. Cancer-Associated Differences in Acetylcholinesterase Activity in Bronchial Aspirates from Patients with Lung Cancer. Clin. Sci. 2008, 115, 245–253. [Google Scholar] [CrossRef]
- Small, D.H.; Michaelson, S.; Sberna, G. Non-Classical Actions of Cholinesterases: Role in Cellular Differentiation, Tumorigenesis and Alzheimer’s Disease. Neurochem. Int. 1996, 28, 453–483. [Google Scholar] [CrossRef]
- Russo, P.; Cardinale, A.; Margaritora, S.; Cesario, A. Nicotinic Receptor and Tobacco-Related Cancer. Life Sci. 2012, 91, 1087–1092. [Google Scholar] [CrossRef]
- Xi, H.-J.; Wu, R.-P.; Liu, J.-J.; Zhang, L.-J.; Li, Z.-S. Role of Acetylcholinesterase in Lung Cancer. Thorac. Cancer 2015, 6, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Schaal, C.; Chellappan, S.P. Nicotine-Mediated Cell Proliferation and Tumor Progression in Smoking-Related Cancers. Mol. Cancer Res. 2014, 12, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Egleton, R.D.; Brown, K.C.; Dasgupta, P. Nicotinic Acetylcholine Receptors in Cancer: Multiple Roles in Proliferation and Inhibition of Apoptosis. Trends Pharmacol. Sci. 2008, 29, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Spindel, E.R. Cholinergic Targets in Lung Cancer. Curr. Pharm. Des. 2016, 22, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.B.; Le, X.; Heymach, J.V. β-Adrenergic Signaling in Lung Cancer: A Potential Role for Beta-Blockers. J. Neuroimmune Pharmacol. 2020, 15, 27–36. [Google Scholar] [CrossRef]
- Al-Wadei, H.A.N.; Al-Wadei, M.H.; Schuller, H.M. Cooperative Regulation of Non-Small Cell Lung Carcinoma by Nicotinic and Beta-Adrenergic Receptors: A Novel Target for Intervention. PLoS ONE 2012, 7, e29915. [Google Scholar] [CrossRef]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Frisch, S.M.; Schaller, M.; Cieply, B. Mechanisms That Link the Oncogenic Epithelial-Mesenchymal Transition to Suppression of Anoikis. J. Cell Sci. 2013, 126, 21–29. [Google Scholar] [CrossRef]
- De la Cruz-Morcillo, M.A.; Berger, J.; Sánchez-Prieto, R.; Saada, S.; Naves, T.; Guillaudeau, A.; Perraud, A.; Sindou, P.; Lacroix, A.; Descazeaud, A.; et al. P75 Neurotrophin Receptor and Pro-BDNF Promote Cell Survival and Migration in Clear Cell Renal Cell Carcinoma. Oncotarget 2016, 7, 34480–34497. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Hui, L.-P.; Li, C.-Y.; Gao, J.; Cui, Z.-S.; Qiu, X.-S. More Expression of BDNF Associates with Lung Squamous Cell Carcinoma and Is Critical to the Proliferation and Invasion of Lung Cancer Cells. BMC Cancer 2016, 16, 171. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Liu, B.; Ji, R.; Jiang, X.; Yan, X.; Xin, Y. Targeting the BDNF/TrkB Pathway for the Treatment of Tumors. Oncol. Lett. 2019, 17, 2031–2039. [Google Scholar] [CrossRef]
- Radin, D.P.; Patel, P. BDNF: An Oncogene or Tumor Suppressor? Anticancer Res. 2017, 37, 3983–3990. [Google Scholar] [CrossRef] [PubMed]
- Dorandish, S.; Atali, S.; Ray, R.; Al Khashali, H.; Coleman, K.-L.; Guthrie, J.; Heyl, D.; Evans, H.G. Differences in the Relative Abundance of ProBDNF and Mature BDNF in A549 and H1299 Human Lung Cancer Cell Media. Int. J. Mol. Sci. 2021, 22, 7059. [Google Scholar] [CrossRef] [PubMed]
- Al Khashali, H.; Ray, R.; Coleman, K.-L.; Atali, S.; Haddad, B.; Wareham, J.; Guthrie, J.; Heyl, D.; Evans, H.G. Regulation of the Soluble Amyloid Precursor Protein α (sAPPα) Levels by Acetylcholinesterase and Brain-Derived Neurotrophic Factor in Lung Cancer Cell Media. Int. J. Mol. Sci. 2022, 23, 10746. [Google Scholar] [CrossRef]
- Ray, R.; Al Khashali, H.; Haddad, B.; Wareham, J.; Coleman, K.-L.; Alomari, D.; Ranzenberger, R.; Guthrie, J.; Heyl, D.; Evans, H.G. Regulation of Cisplatin Resistance in Lung Cancer Cells by Nicotine, BDNF, and a β-Adrenergic Receptor Blocker. Int. J. Mol. Sci. 2022, 23, 12829. [Google Scholar] [CrossRef] [PubMed]
- Muterspaugh, R.; Price, D.; Esckilsen, D.; McEachern, S.; Guthrie, J.; Heyl, D.; Evans, H.G. Interaction of Insulin-Like Growth Factor-Binding Protein 3 With Hyaluronan and Its Regulation by Humanin and CD44. Biochemistry 2018, 57, 5726–5737. [Google Scholar] [CrossRef]
- Dorandish, S.; Devos, J.; Clegg, B.; Price, D.; Muterspaugh, R.; Guthrie, J.; Heyl, D.L.; Evans, H.G. Biochemical Determinants of the IGFBP-3-Hyaluronan Interaction. FEBS Open Bio 2020, 10, 1668–1684. [Google Scholar] [CrossRef]
- Price, D.; Dorandish, S.; Williams, A.; Iwaniec, B.; Stephens, A.; Marshall, K.; Guthrie, J.; Heyl, D.; Evans, H.G. Humanin Blocks the Aggregation of Amyloid-β Induced by Acetylcholinesterase, an Effect Abolished in the Presence of IGFBP-3. Biochemistry 2020, 59, 1981–2002. [Google Scholar] [CrossRef]
- Price, D.; Muterspaugh, R.; Clegg, B.; Williams, A.; Stephens, A.; Guthrie, J.; Heyl, D.; Guy Evans, H. IGFBP-3 Blocks Hyaluronan-CD44 Signaling, Leading to Increased Acetylcholinesterase Levels in A549 Cell Media and Apoptosis in a P53-Dependent Manner. Sci. Rep. 2020, 10, 5083–5099. [Google Scholar] [CrossRef]
- Al Khashali, H.; Wareham, J.; Ray, R.; Haddad, B.; Coleman, K.-L.; Ranzenberger, R.; McCombs, P.; Guthrie, J.; Heyl, D.; Evans, H.G. Opposing Roles of IGFBP-3 and Heparanase in Regulating A549 Lung Cancer Cell Survival. Cells 2022, 11, 3533. [Google Scholar] [CrossRef]
- Coleman, K.-L.; Chiaramonti, M.; Haddad, B.; Ranzenberger, R.; Henning, H.; Al Khashali, H.; Ray, R.; Darweesh, B.; Guthrie, J.; Heyl, D.; et al. Phosphorylation of IGFBP-3 by Casein Kinase 2 Blocks Its Interaction with Hyaluronan, Enabling HA-CD44 Signaling Leading to Increased NSCLC Cell Survival and Cisplatin Resistance. Cells 2023, 12, 405. [Google Scholar] [CrossRef] [PubMed]
- Atali, S.; Dorandish, S.; Devos, J.; Williams, A.; Price, D.; Taylor, J.; Guthrie, J.; Heyl, D.; Evans, H.G. Interaction of Amyloid Beta with Humanin and Acetylcholinesterase Is Modulated by ATP. FEBS Open Bio 2020, 10, 2805–2823. [Google Scholar] [CrossRef] [PubMed]
- Dorandish, S.; Williams, A.; Atali, S.; Sendo, S.; Price, D.; Thompson, C.; Guthrie, J.; Heyl, D.; Evans, H.G. Regulation of Amyloid-β Levels by Matrix Metalloproteinase-2/9 (MMP2/9) in the Media of Lung Cancer Cells. Sci. Rep. 2021, 11, 9708. [Google Scholar] [CrossRef]
- Evans, H.G.; Guthrie, J.W.; Jujjavarapu, M.; Hendrickson, N.; Eitel, A.; Park, Y.; Garvey, J.; Newman, R.; Esckilsen, D.; Heyl, D.L. D-Amino Acid Analogues of the Antimicrobial Peptide CDT Exhibit Anti- Cancer Properties in A549, a Human Lung Adenocarcinoma Cell Line. Protein Pept. Lett. 2017, 24, 590–598. [Google Scholar] [CrossRef]
- Njomen, E.; Evans, H.G.; Gedara, S.H.; Heyl, D.L. Humanin Peptide Binds to Insulin-Like Growth Factor-Binding Protein 3 (IGFBP3) and Regulates Its Interaction with Importin-β. Protein Pept. Lett. 2015, 22, 869–876. [Google Scholar] [CrossRef]
- Patel, B.B.; Barrero, C.A.; Braverman, A.; Kim, P.D.; Jones, K.A.; Chen, D.E.; Bowler, R.P.; Merali, S.; Kelsen, S.G.; Yeung, A.T. Assessment of Two Immunodepletion Methods: Off-Target Effects and Variations in Immunodepletion Efficiency May Confound Plasma Proteomics. J. Proteome Res. 2012, 11, 5947–5958. [Google Scholar] [CrossRef]
- Brouxhon, S.M.; Kyrkanides, S.; Teng, X.; Raja, V.; O’Banion, M.K.; Clarke, R.; Byers, S.; Silberfeld, A.; Tornos, C.; Ma, L. Monoclonal Antibody against the Ectodomain of E-Cadherin (DECMA-1) Suppresses Breast Carcinogenesis: Involvement of the HER/PI3K/Akt/mTOR and IAP Pathways. Clin. Cancer Res. 2013, 19, 3234–3246. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zhang, C.; Chen, Y.; Zhou, T. Upregulation of Acetylcholinesterase Mediated by P53 Contributes to Cisplatin-Induced Apoptosis in Human Breast Cancer Cell. J. Cancer 2015, 6, 48–53. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Nakade, J.; Tachibana, M.; Ibi, D.; Someya, E.; Koike, H.; Kamei, H.; Nabeshima, T.; Itohara, S.; Takuma, K.; et al. Matrix Metalloproteinase-9 Contributes to Kindled Seizure Development in Pentylenetetrazole-Treated Mice by Converting pro-BDNF to Mature BDNF in the Hippocampus. J. Neurosci. 2011, 31, 12963–12971. [Google Scholar] [CrossRef]
- Greenberg, M.E.; Xu, B.; Lu, B.; Hempstead, B.L. New Insights in the Biology of BDNF Synthesis and Release: Implications in CNS Function. J. Neurosci. 2009, 29, 12764–12767. [Google Scholar] [CrossRef] [PubMed]
- Dagnell, C.; Kemi, C.; Klominek, J.; Eriksson, P.; Sköld, C.M.; Eklund, A.; Grunewald, J.; Olgart Höglund, C. Effects of Neurotrophins on Human Bronchial Smooth Muscle Cell Migration and Matrix Metalloproteinase-9 Secretion. Transl. Res. 2007, 150, 303–310. [Google Scholar] [CrossRef]
- Kuzniewska, B.; Rejmak, E.; Malik, A.R.; Jaworski, J.; Kaczmarek, L.; Kalita, K. Brain-Derived Neurotrophic Factor Induces Matrix Metalloproteinase 9 Expression in Neurons via the Serum Response Factor/c-Fos Pathway. Mol. Cell. Biol. 2013, 33, 2149–2162. [Google Scholar] [CrossRef] [PubMed]
- Merchant, N.; Nagaraju, G.P.; Rajitha, B.; Lammata, S.; Jella, K.K.; Buchwald, Z.S.; Lakka, S.S.; Ali, A.N. Matrix Metalloproteinases: Their Functional Role in Lung Cancer. Carcinogenesis 2017, 38, 766–780. [Google Scholar] [CrossRef]
- Gong, L.; Wu, D.; Zou, J.; Chen, J.; Chen, L.; Chen, Y.; Ni, C.; Yuan, H. Prognostic Impact of Serum and Tissue MMP-9 in Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Oncotarget 2016, 7, 18458–18468. [Google Scholar] [CrossRef]
- Alsharairi, N.A. Scutellaria Baicalensis and Their Natural Flavone Compounds as Potential Medicinal Drugs for the Treatment of Nicotine-Induced Non-Small-Cell Lung Cancer and Asthma. Int. J. Environ. Res. Public Health 2021, 18, 5243. [Google Scholar] [CrossRef]
- Huang, Q.; Tan, Q.; Mao, K.; Yang, G.; Ma, G.; Luo, P.; Wang, S.; Mei, P.; Wu, F.; Xu, J.; et al. The Role of Adrenergic Receptors in Lung Cancer. Am. J. Cancer Res. 2018, 8, 2227–2237. [Google Scholar]
- Chaudhary, K.R.; Yan, S.X.; Heilbroner, S.P.; Sonett, J.R.; Stoopler, M.B.; Shu, C.; Halmos, B.; Wang, T.J.C.; Hei, T.K.; Cheng, S.K. Effects of β-Adrenergic Antagonists on Chemoradiation Therapy for Locally Advanced Non-Small Cell Lung Cancer. J. Clin. Med. 2019, 8, 575. [Google Scholar] [CrossRef]
- Min, H.-Y.; Boo, H.-J.; Lee, H.J.; Jang, H.-J.; Yun, H.J.; Hwang, S.J.; Smith, J.K.; Lee, H.-J.; Lee, H.-Y. Smoking-Associated Lung Cancer Prevention by Blockade of the Beta-Adrenergic Receptor-Mediated Insulin-like Growth Factor Receptor Activation. Oncotarget 2016, 7, 70936–70947. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Chen, C.; Wu, F.; Qiu, L.; Ke, Q.; Sun, R.; Duan, Q.; Luo, M.; Luo, Z. Curcumin Inhibits the Migration and Invasion of Non-Small-Cell Lung Cancer Cells Through Radiation-Induced Suppression of Epithelial-Mesenchymal Transition and Soluble E-Cadherin Expression. Technol. Cancer Res. Treat. 2020, 19, 1533033820947485. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-H.; Liu, C.-W.; Tsai, C.-H.; Peng, Y.-J.; Yang, Y.-H.; Liao, P.-L.; Lee, C.-C.; Cheng, Y.-W.; Kang, J.-J. Cytoplasmic Aryl Hydrocarbon Receptor Regulates Glycogen Synthase Kinase 3 Beta, Accelerates Vimentin Degradation, and Suppresses Epithelial-Mesenchymal Transition in Non-Small Cell Lung Cancer Cells. Arch. Toxicol. 2017, 91, 2165–2178. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Hu, X.; Lu, C.; Su, C.; Luo, S.; Luo, Z.W. Promoter-Hypermethylation Associated Defective Expression of E-Cadherin in Primary Non-Small Cell Lung Cancer. Lung Cancer 2008, 62, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Girard, L.; Hollestelle, A.; Minna, J.D.; Gazdar, A.F.; Soussi, T. Analysis of TP53 Mutation Status in Human Cancer Cell Lines: A Reassessment. Hum. Mutat. 2014, 35, 756–765. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, C.-X.; Fang, E.-H.; Wang, G.-B.; Tong, Q. Role of Epithelial-Mesenchymal Transition in Gastric Cancer Initiation and Progression. World J. Gastroenterol. 2014, 20, 5403–5410. [Google Scholar] [CrossRef]
- Dasgupta, P.; Rizwani, W.; Pillai, S.; Kinkade, R.; Kovacs, M.; Rastogi, S.; Banerjee, S.; Carless, M.; Kim, E.; Coppola, D.; et al. Nicotine Induces Cell Proliferation, Invasion and Epithelial-Mesenchymal Transition in a Variety of Human Cancer Cell Lines. Int. J. Cancer 2009, 124, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.-J.; Chang, L.W.; Lin, P.; Wang, Y.-J. Epigenetic Effects and Molecular Mechanisms of Tumorigenesis Induced by Cigarette Smoke: An Overview. J. Oncol. 2011, 2011, 654931. [Google Scholar] [CrossRef]
- Sousa, B.; Pereira, J.; Paredes, J. The Crosstalk between Cell Adhesion and Cancer Metabolism. Int. J. Mol. Sci. 2019, 20, 1933. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ray, R.; Goel, S.; Al Khashali, H.; Darweesh, B.; Haddad, B.; Wozniak, C.; Ranzenberger, R.; Khalil, J.; Guthrie, J.; Heyl, D.; et al. Regulation of Soluble E-Cadherin Signaling in Non-Small-Cell Lung Cancer Cells by Nicotine, BDNF, and β-Adrenergic Receptor Ligands. Biomedicines 2023, 11, 2555. https://doi.org/10.3390/biomedicines11092555
Ray R, Goel S, Al Khashali H, Darweesh B, Haddad B, Wozniak C, Ranzenberger R, Khalil J, Guthrie J, Heyl D, et al. Regulation of Soluble E-Cadherin Signaling in Non-Small-Cell Lung Cancer Cells by Nicotine, BDNF, and β-Adrenergic Receptor Ligands. Biomedicines. 2023; 11(9):2555. https://doi.org/10.3390/biomedicines11092555
Chicago/Turabian StyleRay, Ravel, Stuti Goel, Hind Al Khashali, Ban Darweesh, Ben Haddad, Caroline Wozniak, Robert Ranzenberger, Jeneen Khalil, Jeffrey Guthrie, Deborah Heyl, and et al. 2023. "Regulation of Soluble E-Cadherin Signaling in Non-Small-Cell Lung Cancer Cells by Nicotine, BDNF, and β-Adrenergic Receptor Ligands" Biomedicines 11, no. 9: 2555. https://doi.org/10.3390/biomedicines11092555
APA StyleRay, R., Goel, S., Al Khashali, H., Darweesh, B., Haddad, B., Wozniak, C., Ranzenberger, R., Khalil, J., Guthrie, J., Heyl, D., & Evans, H. G. (2023). Regulation of Soluble E-Cadherin Signaling in Non-Small-Cell Lung Cancer Cells by Nicotine, BDNF, and β-Adrenergic Receptor Ligands. Biomedicines, 11(9), 2555. https://doi.org/10.3390/biomedicines11092555