
Switch-Independent 3A: An Epigenetic Regulator in Cancer with New Implications for Pulmonary Arterial Hypertension



Abstract
: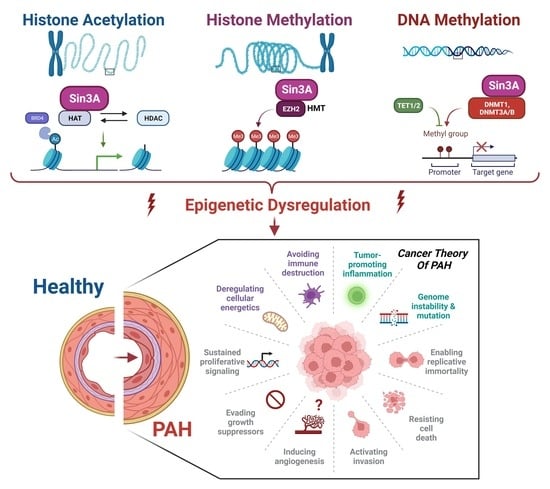
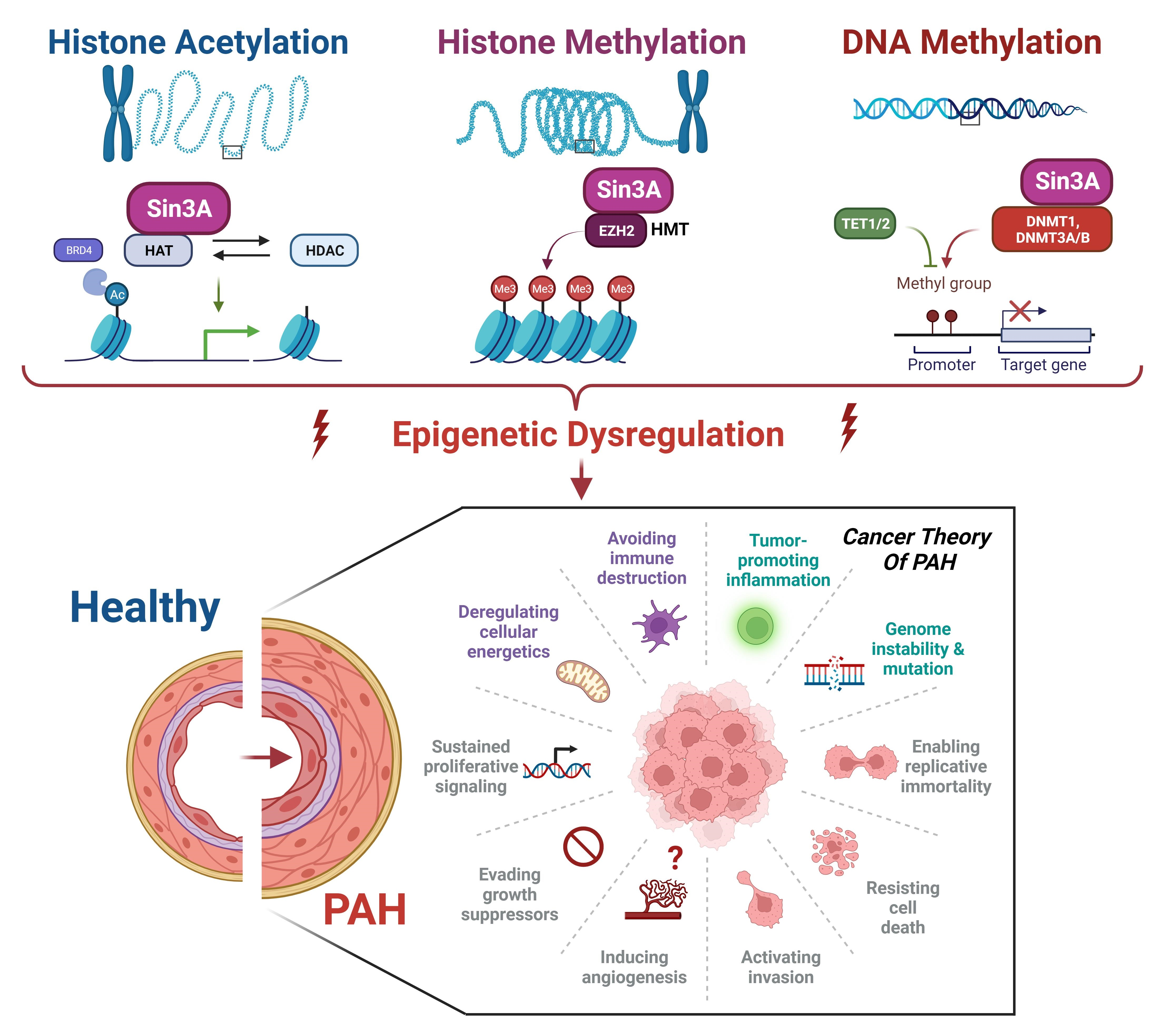{kind=link}
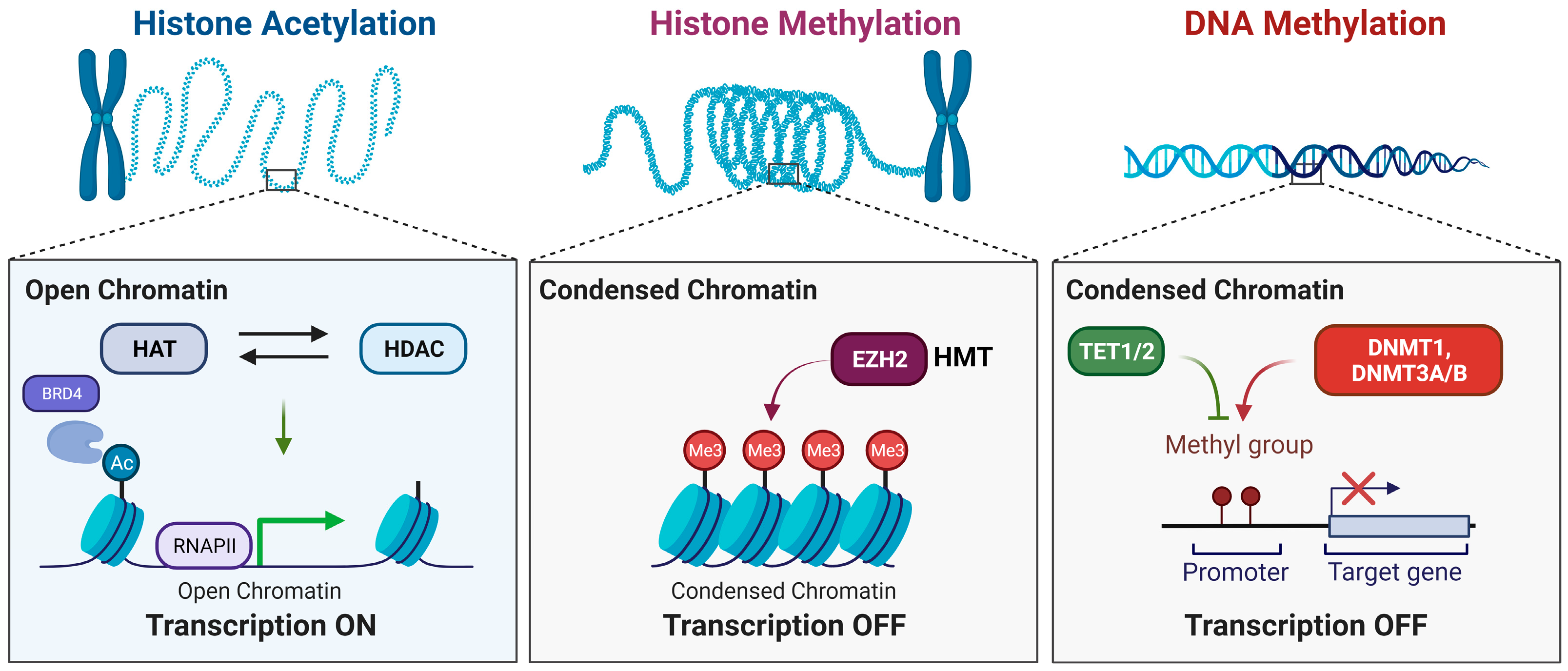{kind=link}
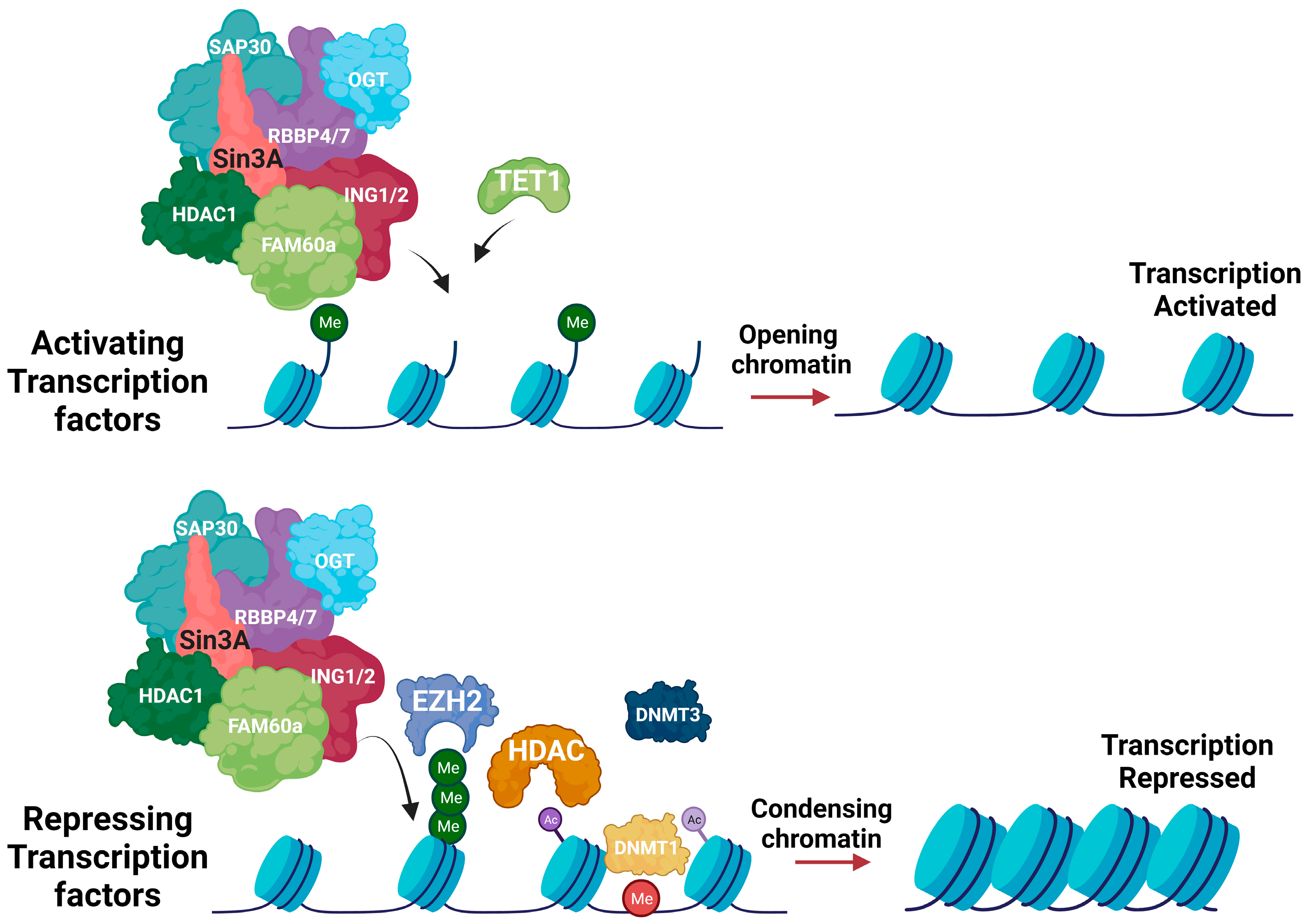{kind=link}
{kind=link}
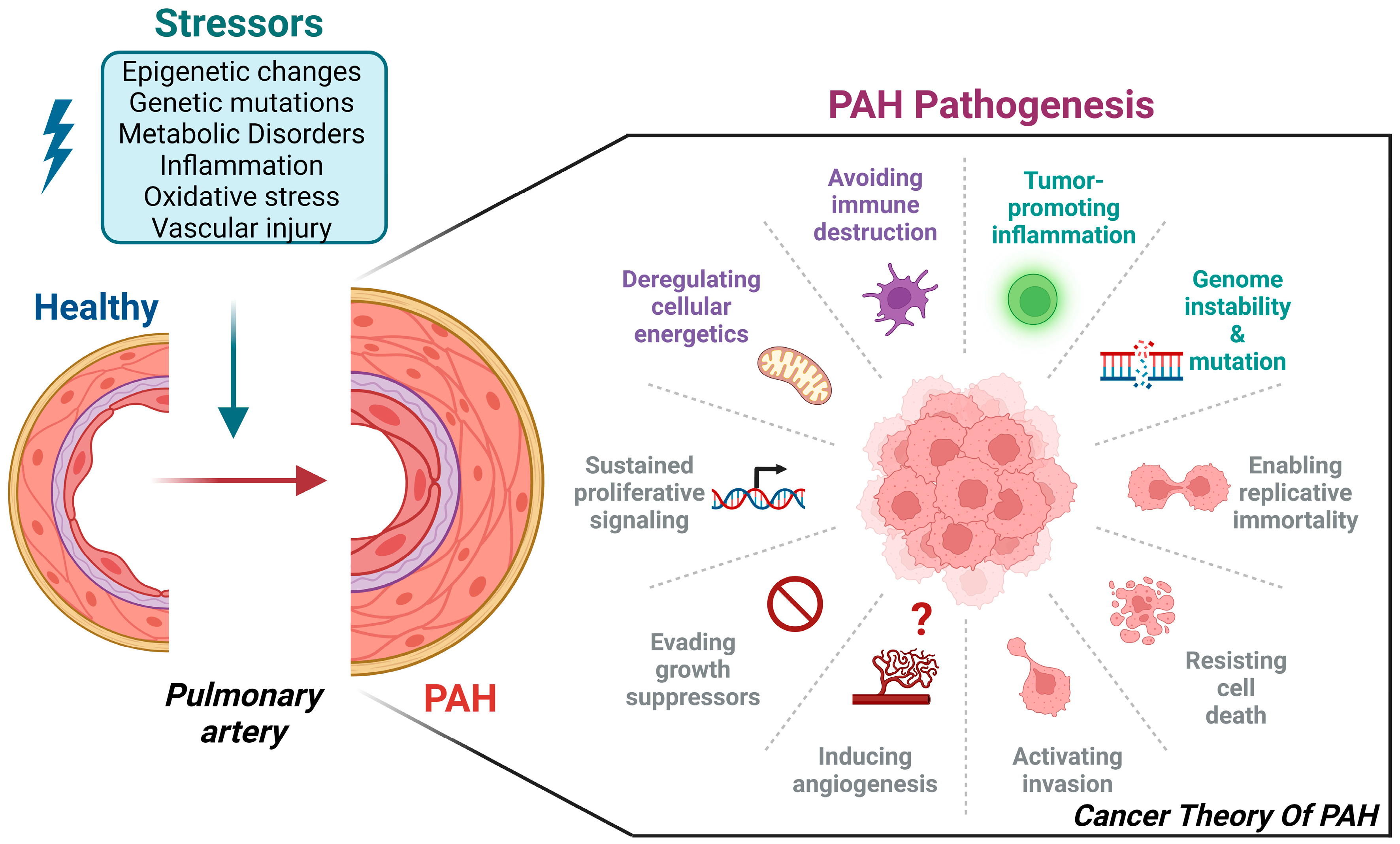
1. Introduction
2. Key Epigenetic Regulators
3. Epigenetic Regulator SIN3a
3.1. Molecular Function of SIN3a
3.2. Interactions with Histone Deacetylases (HDACs) and Other Partners
3.3. Role of SIN3a in Transcriptional Regulation and Chromatin Remodeling: Co-Repressor and Activator
4. Emerging Roles of Epigenetic Regulator SIN3a in Cancer
4.1. SIN3a’s Involvement in Cancer Development and Progression
4.2. SIN3a’s Impact on Oncogenes and Tumor Suppressor Genes
4.3. Mechanisms of SIN3a-Mediated Epigenetic Alterations: Examples in Different Types of Cancer
4.4. Targeting SIN3a as a Potential Therapeutic Approach in Cancer
5. The Cancer-like Theory of PAH
6. New Insights into the Roles of SIN3a in PAH
6.1. SIN3a Molecular Mechanisms in PAH
6.2. Comparison of SIN3a’s Effects on Cancer and Its Implications for PAH
6.3. Exploring SIN3a as a Novel Therapeutic Target for PAH
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5mC | 5-methylcytosine |
| BET | Bromodomain and extra-terminal family proteins |
| BMPR2 | Bone morphogenetic protein receptor type 2 |
| BRD4 | Bromodomain-containing protein 4 |
| DNMT | DNA methyltransferases |
| EZH2 | Enhancer of zeste homolog 2 |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylase |
| PASMCs | Pulmonary arterial smooth muscle cells |
| lncRNA | Long non-coding RNA |
| miRNA | Micro RNA |
| PAECs | Human pulmonary arterial endothelial cells |
| PAH | Pulmonary arterial hypertension |
| RBP1 | Retinol-binding protein 1 |
| RV | Right ventricle |
| RVSP | Right ventricular systolic pressure |
| SIN3 | SWI-independent-3 complex |
| SuHx | Sugen-chronic hypoxia |
| TET | Ten-eleven translocation factor |
References
- Li, Y. Modern epigenetics methods in biological research. Methods 2021, 187, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. Epigenetics: A historical overview. Epigenetics 2006, 1, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Ohm, J.E. Epigenetic gene silencing in cancer—A mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 2006, 6, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef]
- Ratti, M.; Lampis, A.; Ghidini, M.; Salati, M.; Mirchev, M.B.; Valeri, N.; Hahne, J.C. MicroRNAs (miRNAs) and Long Non-Coding RNAs (lncRNAs) as New Tools for Cancer Therapy: First Steps from Bench to Bedside. Target. Oncol. 2020, 15, 261–278. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Lewis, M.J.; Liu, J.; Libby, E.F.; Lee, M.; Crawford, N.P.; Hurst, D.R. SIN3A and SIN3B differentially regulate breast cancer metastasis. Oncotarget 2016, 7, 78713–78725. [Google Scholar] [CrossRef]
- Ren, J.; Li, X.; Dong, H.; Suo, L.; Zhang, J.; Zhang, L.; Zhang, J. miR-210-3p regulates the proliferation and apoptosis of non-small cell lung cancer cells by targeting SIN3A. Exp. Ther. Med. 2019, 18, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Toolabi, N.; Daliri, F.S.; Mokhlesi, A.; Talkhabi, M. Identification of key regulators associated with colon cancer prognosis and pathogenesis. J. Cell Commun. Signal. 2022, 16, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Sangodkar, J.; Negre, N.; Narla, G.; Cagan, R.L. Sin3a acts through a multi-gene module to regulate invasion in Drosophila and human tumors. Oncogene 2013, 32, 3184–3197. [Google Scholar] [CrossRef] [PubMed]
- Solaimani, P.; Wang, F.; Hankinson, O. SIN3A, generally regarded as a transcriptional repressor, is required for induction of gene transcription by the aryl hydrocarbon receptor. J. Biol. Chemtranscriptional. 2014, 289, 33655–33662. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, J.H.; David, G.; Zhong, S.; van der Torre, J.; Wong, W.H.; Depinho, R.A. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005, 19, 1581–1595. [Google Scholar] [CrossRef]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef]
- Spiekerkoetter, E.; Goncharova, E.A.; Guignabert, C.; Stenmark, K.; Kwapiszewska, G.; Rabinovitch, M.; Voelkel, N.; Bogaard, H.J.; Graham, B.; Pullamsetti, S.S.; et al. Hot topics in the mechanisms of pulmonary arterial hypertension disease: Cancer-like pathobiology, the role of the adventitia, systemic involvement, and right ventricular failure. Pulm. Circ. 2019, 9, 2045894019889775. [Google Scholar] [CrossRef]
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef]
- Dave, J.; Jagana, V.; Janostiak, R.; Bisserier, M. Unraveling the epigenetic landscape of pulmonary arterial hypertension: Implications for personalized medicine development. J. Transl. Med. 2023, 21, 477. [Google Scholar] [CrossRef]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thebaud, B.; Husain, A.N.; et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef]
- Bisserier, M.; Mathiyalagan, P.; Zhang, S.; Elmastour, F.; Dorfmuller, P.; Humbert, M.; David, G.; Tarzami, S.; Weber, T.; Perros, F.; et al. Regulation of the Methylation and Expression Levels of the BMPR2 Gene by SIN3a as a Novel Therapeutic Mechanism in Pulmonary Arterial Hypertension. Circulation 2021, 144, 52–73. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.L.; Huang, Z.N.; Zhu, Z.; Cui, Y.Y.; Li, M.Q.; Huang, R.M.; Yan, J.; Shen, B. Downregulation of TET1 Promotes Bladder Cancer Cell Proliferation and Invasion by Reducing DNA Hydroxymethylation of AJAP1. Front. Oncol. 2020, 10, 667. [Google Scholar] [CrossRef] [PubMed]
- Potus, F.; Pauciulo, M.W.; Cook, E.K.; Zhu, N.; Hsieh, A.; Welch, C.L.; Shen, Y.; Tian, L.; Lima, P.; Mewburn, J.; et al. Novel Mutations and Decreased Expression of the Epigenetic Regulator TET2 in Pulmonary Arterial Hypertension. Circulation 2020, 141, 1986–2000. [Google Scholar] [CrossRef] [PubMed]
- Chelladurai, P.; Dabral, S.; Basineni, S.R.; Chen, C.N.; Schmoranzer, M.; Bender, N.; Feld, C.; Notzold, R.R.; Dobreva, G.; Wilhelm, J.; et al. Isoform-specific characterization of class I histone deacetylases and their therapeutic modulation in pulmonary hypertension. Sci. Rep. 2020, 10, 12864. [Google Scholar] [CrossRef] [PubMed]
- Chelladurai, P.; Kuenne, C.; Bourgeois, A.; Gunther, S.; Valasarajan, C.; Cherian, A.V.; Rottier, R.J.; Romanet, C.; Weigert, A.; Boucherat, O.; et al. Epigenetic reactivation of transcriptional programs orchestrating fetal lung development in human pulmonary hypertension. Sci. Transl. Med. 2022, 14, eabe5407. [Google Scholar] [CrossRef]
- Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef]
- Wang, R.; Li, Q.; Helfer, C.M.; Jiao, J.; You, J. Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J. Biol. Chem. 2012, 287, 10738–10752. [Google Scholar] [CrossRef]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- Gan, L.; Yang, Y.; Li, Q.; Feng, Y.; Liu, T.; Guo, W. Epigenetic regulation of cancer progression by EZH2: From biological insights to therapeutic potential. Biomark. Res. 2018, 6, 10. [Google Scholar] [CrossRef]
- Silverstein, R.A.; Ekwall, K. Sin3: A flexible regulator of global gene expression and genome stability. Curr. Genet. 2005, 47, 1–17. [Google Scholar] [CrossRef]
- Guo, Z.; Chu, C.; Lu, Y.; Zhang, X.; Xiao, Y.; Wu, M.; Gao, S.; Wong, C.C.L.; Zhan, X.; Wang, C. Structure of a SIN3-HDAC complex from budding yeast. Nat. Struct. Mol. Biol. 2023, 30, 753–760. [Google Scholar] [CrossRef] [PubMed]
- McDonel, P.; Demmers, J.; Tan, D.W.; Watt, F.; Hendrich, B.D. Sin3a is essential for the genome integrity and viability of pluripotent cells. Dev. Biol. 2012, 363, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, P.; Pellegrino, J.; David, G. A novel mammalian complex containing Sin3B mitigates histone acetylation and RNA polymerase II progression within transcribed loci. Mol. Cell. Biol. 2011, 31, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Laherty, C.D.; Yang, W.M.; Sun, J.M.; Davie, J.R.; Seto, E.; Eisenman, R.N. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 1997, 89, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Kadamb, R.; Mittal, S.; Bansal, N.; Batra, H.; Saluja, D. Sin3: Insight into its transcription regulatory functions. Eur. J. Cell Biol. 2013, 92, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; David, G.; Farias, E.; Waxman, S. Emerging Roles of Epigenetic Regulator Sin3 in Cancer. Adv. Cancer Res. 2016, 130, 113–135. [Google Scholar] [CrossRef] [PubMed]
- van Oevelen, C.; Bowman, C.; Pellegrino, J.; Asp, P.; Cheng, J.; Parisi, F.; Micsinai, M.; Kluger, Y.; Chu, A.; Blais, A.; et al. The mammalian Sin3 proteins are required for muscle development and sarcomere specification. Mol. Cell. Biol. 2010, 30, 5686–5697. [Google Scholar] [CrossRef] [PubMed]
- Icardi, L.; Mori, R.; Gesellchen, V.; Eyckerman, S.; De Cauwer, L.; Verhelst, J.; Vercauteren, K.; Saelens, X.; Meuleman, P.; Leroux-Roels, G.; et al. The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc. Natl. Acad. Sci. USA 2012, 109, 12058–12063. [Google Scholar] [CrossRef]
- Kuzmichev, A.; Zhang, Y.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Role of the Sin3-histone deacetylase complex in growth regulation by the candidate tumor suppressor p33(ING1). Mol. Cell. Biol. 2002, 22, 835–848. [Google Scholar] [CrossRef]
- Zhang, Y.; Reinberg, D. Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes Dev. 2001, 15, 2343–2360. [Google Scholar] [CrossRef]
- Sif, S.; Saurin, A.J.; Imbalzano, A.N.; Kingston, R.E. Purification and characterization of mSin3A-containing Brg1 and hBrm chromatin remodeling complexes. Genes Dev. 2001, 15, 603–618. [Google Scholar] [CrossRef] [PubMed]
- Nagl, N.G., Jr.; Wang, X.; Patsialou, A.; Van Scoy, M.; Moran, E. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J. 2007, 26, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chen, C.N.; Hajji, N.; Oliver, E.; Cotroneo, E.; Wharton, J.; Wang, D.; Li, M.; McKinsey, T.A.; Stenmark, K.R.; et al. Histone deacetylation inhibition in pulmonary hypertension: Therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 2012, 126, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Johnson, H.; Jing, X.; Michalkiewicz, T.; Huang, Y.W.; Lane, R.H.; Konduri, G.G. Persistent pulmonary hypertension alters the epigenetic characteristics of endothelial nitric oxide synthase gene in pulmonary artery endothelial cells in a fetal lamb model. Physiol. Genom. 2018, 50, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Cavasin, M.A.; Demos-Davies, K.; Horn, T.R.; Walker, L.A.; Lemon, D.D.; Birdsey, N.; Weiser-Evans, M.C.; Harral, J.; Irwin, D.C.; Anwar, A.; et al. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ. Res. 2012, 110, 739–748. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.A.; Ramadoss, M.; Mahadevan, V. Histone Deacetylase (HDAC) Inhibitors—Emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr. Neuropharmacol. 2016, 14, 55–71. [Google Scholar] [CrossRef]
- Yang, S.S.; Zhang, R.; Wang, G.; Zhang, Y.F. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl. Neurodegener. 2017, 6, 19. [Google Scholar] [CrossRef]
- Bansal, N.; Kadamb, R.; Mittal, S.; Vig, L.; Sharma, R.; Dwarakanath, B.S.; Saluja, D. Tumor Suppressor Protein p53 Recruits Human Sin3B/HDAC1 Complex for Down-Regulation of Its Target Promoters in Response to Genotoxic Stress. PLoS ONE 2011, 6, e26156. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Tong, J.K.; Schreiber, S.L. Genomewide studies of histone deacetylase function in yeast. Proc. Natl. Acad. Sci. USA 2000, 97, 13708–13713. [Google Scholar] [CrossRef]
- Chaubal, A.; Pile, L.A. Same agent, different messages: Insight into transcriptional regulation by SIN3 isoforms. Epigenetics Chromatin 2018, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Ellison-Zelski, S.J.; Alarid, E.T. Maximum growth and survival of estrogen receptor-alpha positive breast cancer cells requires the Sin3A transcriptional repressor. Mol. Cancer 2010, 9, 263. [Google Scholar] [CrossRef]
- Cowley, S.M.; Iritani, B.M.; Mendrysa, S.M.; Xu, T.; Cheng, P.F.; Yada, J.; Liggitt, H.D.; Eisenman, R.N. The mSin3A chromatin-modifying complex is essential for embryogenesis and T-cell development. Mol. Cell. Biol. 2005, 25, 6990–7004. [Google Scholar] [CrossRef]
- Prall, O.W.; Rogan, E.M.; Sutherland, R.L. Estrogen regulation of cell cycle progression in breast cancer cells. J. Steroid Biochem. Mol. Biol. 1998, 65, 169–174. [Google Scholar] [CrossRef]
- Gompel, A.; Somai, S.; Chaouat, M.; Kazem, A.; Kloosterboer, H.J.; Beusman, I.; Forgez, P.; Mimoun, M.; Rostene, W. Hormonal regulation of apoptosis in breast cells and tissues. Steroids 2000, 65, 593–598. [Google Scholar] [CrossRef]
- Hurst, D.R. Metastasis suppression by BRMS1 associated with SIN3 chromatin remodeling complexes. Cancer Metastasis Rev. 2012, 31, 641–651. [Google Scholar] [CrossRef]
- Meehan, W.J.; Samant, R.S.; Hopper, J.E.; Carrozza, M.J.; Shevde, L.A.; Workman, J.L.; Eckert, K.A.; Verderame, M.F.; Welch, D.R. Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J. Biol. Chem. 2004, 279, 1562–1569. [Google Scholar] [CrossRef]
- Meehan, W.J.; Welch, D.R. Breast cancer metastasis suppressor 1: Update. Clin. Exp. Metastasis 2003, 20, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Petrie, K.; Christova, R.; Chung, C.Y.; Leibovitch, B.A.; Howell, L.; Gil, V.; Sbirkov, Y.; Lee, E.; Wexler, J.; et al. Targeting the SIN3A-PF1 interaction inhibits epithelial to mesenchymal transition and maintenance of a stem cell phenotype in triple negative breast cancer. Oncotarget 2015, 6, 34087–34105. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, Z.; Liu, X.; Cheng, X.; Zhang, Y.; Han, X.; Zhang, Y.; Liu, S.; Yang, J.; Xu, B.; et al. The FOXN3-NEAT1-SIN3A repressor complex promotes progression of hormonally responsive breast cancer. J. Clin. Investig. 2017, 127, 3421–3440. [Google Scholar] [CrossRef] [PubMed]
- Gambi, G.; Di Simone, E.; Basso, V.; Ricci, L.; Wang, R.; Verma, A.; Elemento, O.; Ponzoni, M.; Inghirami, G.; Icardi, L.; et al. The Transcriptional Regulator Sin3A Contributes to the Oncogenic Potential of STAT3. Cancer Res. 2019, 79, 3076–3087. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.; Li, W.; Zhao, W.; Wang, R. Targeting epigenetic regulations in cancer. Acta Biochim. Biophys. Sin. 2016, 48, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Carraro, G.; Konda, B.; Guan, X.; Mizuno, T.; Chiba, N.; Kostelny, M.; Kurkciyan, A.; David, G.; McQualter, J.L.; et al. Sin3a regulates epithelial progenitor cell fate during lung development. Development 2017, 144, 2618–2628. [Google Scholar] [CrossRef]
- Suzuki, H.; Ouchida, M.; Yamamoto, H.; Yano, M.; Toyooka, S.; Aoe, M.; Shimizu, N.; Date, H.; Shimizu, K. Decreased expression of the SIN3A gene, a candidate tumor suppressor located at the prevalent allelic loss region 15q23 in non-small cell lung cancer. Lung Cancer 2008, 59, 24–31. [Google Scholar] [CrossRef]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Nayakanti, S.; Chelladurai, P.; Mamazhakypov, A.; Mansouri, S.; Savai, R.; Seeger, W. Cancer and pulmonary hypertension: Learning lessons and real-life interplay. Glob. Cardiol. Sci. Pract. 2020, 2020, e202010. [Google Scholar] [CrossRef]
- El Chami, H.; Hassoun, P.M. Immune and inflammatory mechanisms in pulmonary arterial hypertension. Prog. Cardiovasc. Dis. 2012, 55, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Maston, L.D.; Jones, D.T.; Giermakowska, W.; Resta, T.C.; Ramiro-Diaz, J.; Howard, T.A.; Jernigan, N.L.; Herbert, L.; Maurice, A.A.; Gonzalez Bosc, L.V. Interleukin-6 trans-signaling contributes to chronic hypoxia-induced pulmonary hypertension. Pulm. Circ. 2018, 8, 2045894018780734. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zhu, T.; Fang, Z. The Role and Regulation of Pulmonary Artery Smooth Muscle Cells in Pulmonary Hypertension. Int. J. Hypertens. 2020, 2020, 1478291. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Meloche, J.; Bonnet, S. STAT3 signaling in pulmonary arterial hypertension. JAK-STAT 2012, 1, 223–233. [Google Scholar] [CrossRef]
- Bisserier, M.; Katz, M.G.; Bueno-Beti, C.; Brojakowska, A.; Zhang, S.; Gubara, S.; Kohlbrenner, E.; Fazal, S.; Fargnoli, A.; Dorfmuller, P.; et al. Combination Therapy with STAT3 Inhibitor Enhances SERCA2a-Induced BMPR2 Expression and Inhibits Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2021, 22, 9105. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Meloche, J.; Jacob, M.H.; Bisserier, M.; Courboulin, A.; Bonnet, S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1798–H1809. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Courboulin, A.; Meloche, J.; Mainguy, V.; Dumas de la Roque, E.; Saksouk, N.; Cote, J.; Provencher, S.; Sussman, M.A.; Bonnet, S. Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation 2011, 123, 1205–1215. [Google Scholar] [CrossRef]
- Meloche, J.; Courchesne, A.; Barrier, M.; Carter, S.; Bisserier, M.; Paulin, R.; Lauzon-Joset, J.F.; Breuils-Bonnet, S.; Tremblay, E.; Biardel, S.; et al. Critical role for the advanced glycation end-products receptor in pulmonary arterial hypertension etiology. J. Am. Heart Assoc. 2013, 2, e005157. [Google Scholar] [CrossRef]
- Fessel, J.P.; Loyd, J.E.; Austin, E.D. The genetics of pulmonary arterial hypertension in the post-BMPR2 era. Pulm. Circ. 2011, 1, 305–319. [Google Scholar] [CrossRef]
- Evans, J.D.; Girerd, B.; Montani, D.; Wang, X.J.; Galie, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grunig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Kim, M.J.; Park, S.Y.; Chang, H.R.; Jung, E.Y.; Munkhjargal, A.; Lim, J.S.; Lee, M.S.; Kim, Y. Clinical significance linked to functional defects in bone morphogenetic protein type 2 receptor, BMPR2. BMB Rep. 2017, 50, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Loyd, J.E. The genetics of pulmonary arterial hypertension. Circ. Res. 2014, 115, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.H.; Lee, Y.J.; Lee, E.; Park, S.O.; Han, C.; Beppu, H.; Li, E.; Raizada, M.K.; Bloch, K.D.; Oh, S.P. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 2008, 118, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Andruska, A.; Spiekerkoetter, E. Consequences of BMPR2 Deficiency in the Pulmonary Vasculature and Beyond: Contributions to Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2499. [Google Scholar] [CrossRef] [PubMed]
- Fazal, S.; Bisserier, M.; Hadri, L. Molecular and Genetic Profiling for Precision Medicines in Pulmonary Arterial Hypertension. Cells 2021, 10, 638. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, P.M. Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 385, 2361–2376. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Chandel, N.S. Lessons from Cancer Metabolism for Pulmonary Arterial Hypertension and Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 65, 134–145. [Google Scholar] [CrossRef]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Bisserier, M.; Wajapeyee, N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood 2018, 131, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Wang, Y.; Zhou, F.; Gao, G.; Ren, S.; Zhou, C. Prognostic value of high EZH2 expression in patients with different types of cancer: A systematic review with meta-analysis. Oncotarget 2016, 7, 4584–4597. [Google Scholar] [CrossRef] [PubMed]
- Pietersen, A.M.; Horlings, H.M.; Hauptmann, M.; Langerod, A.; Ajouaou, A.; Cornelissen-Steijger, P.; Wessels, L.F.; Jonkers, J.; van de Vijver, M.J.; van Lohuizen, M. EZH2 and BMI1 inversely correlate with prognosis and TP53 mutation in breast cancer. Breast Cancer Res. 2008, 10, R109. [Google Scholar] [CrossRef] [PubMed]
- Sudo, T.; Utsunomiya, T.; Mimori, K.; Nagahara, H.; Ogawa, K.; Inoue, H.; Wakiyama, S.; Fujita, H.; Shirouzu, K.; Mori, M. Clinicopathological significance of EZH2 mRNA expression in patients with hepatocellular carcinoma. Br. J. Cancer 2005, 92, 1754–1758. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dai, H.; Wang, Q.; Wang, Q.; Xu, Y.; Wang, Y.; Sun, A.; Ruan, J.; Chen, S.; Wu, D. EZH2 mutations are related to low blast percentage in bone marrow and -7/del(7q) in de novo acute myeloid leukemia. PLoS ONE 2013, 8, e61341. [Google Scholar] [CrossRef]
- Aljubran, S.A.; Cox, R., Jr.; Tamarapu Parthasarathy, P.; Kollongod Ramanathan, G.; Rajanbabu, V.; Bao, H.; Mohapatra, S.S.; Lockey, R.; Kolliputi, N. Enhancer of zeste homolog 2 induces pulmonary artery smooth muscle cell proliferation. PLoS ONE 2012, 7, e37712. [Google Scholar] [CrossRef]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jankowski, K.; Jagana, V.; Bisserier, M.; Hadri, L. Switch-Independent 3A: An Epigenetic Regulator in Cancer with New Implications for Pulmonary Arterial Hypertension. Biomedicines 2024, 12, 10. https://doi.org/10.3390/biomedicines12010010
Jankowski K, Jagana V, Bisserier M, Hadri L. Switch-Independent 3A: An Epigenetic Regulator in Cancer with New Implications for Pulmonary Arterial Hypertension. Biomedicines. 2024; 12(1):10. https://doi.org/10.3390/biomedicines12010010
Chicago/Turabian StyleJankowski, Katherine, Vineeta Jagana, Malik Bisserier, and Lahouaria Hadri. 2024. "Switch-Independent 3A: An Epigenetic Regulator in Cancer with New Implications for Pulmonary Arterial Hypertension" Biomedicines 12, no. 1: 10. https://doi.org/10.3390/biomedicines12010010
APA StyleJankowski, K., Jagana, V., Bisserier, M., & Hadri, L. (2024). Switch-Independent 3A: An Epigenetic Regulator in Cancer with New Implications for Pulmonary Arterial Hypertension. Biomedicines, 12(1), 10. https://doi.org/10.3390/biomedicines12010010