
The Role of Chaperone-Mediated Autophagy in Tissue Homeostasis and Disease Pathogenesis



{kind=link}
{kind=link}
Abstract
:1. Introduction
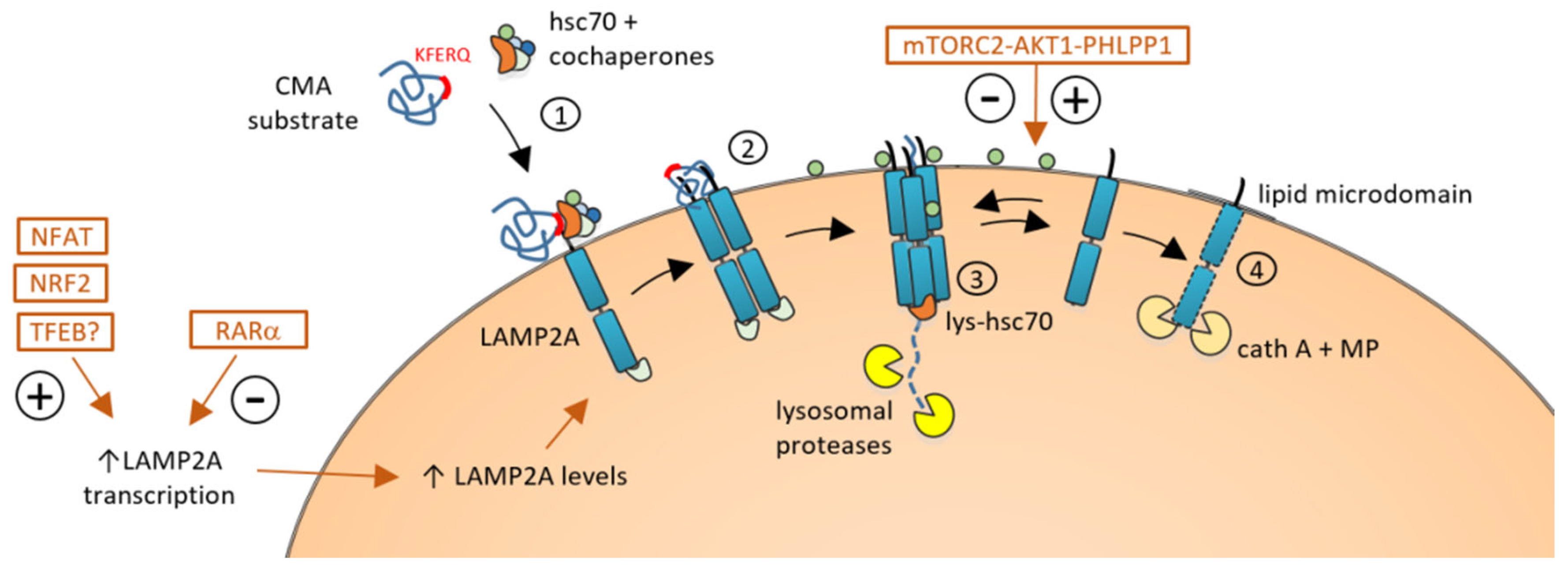
2. Chaperone-Mediated Autophagy
2.1. Mechanisms of CMA
2.2. Selective Regulation in Response to Stress and Cellular Conditions
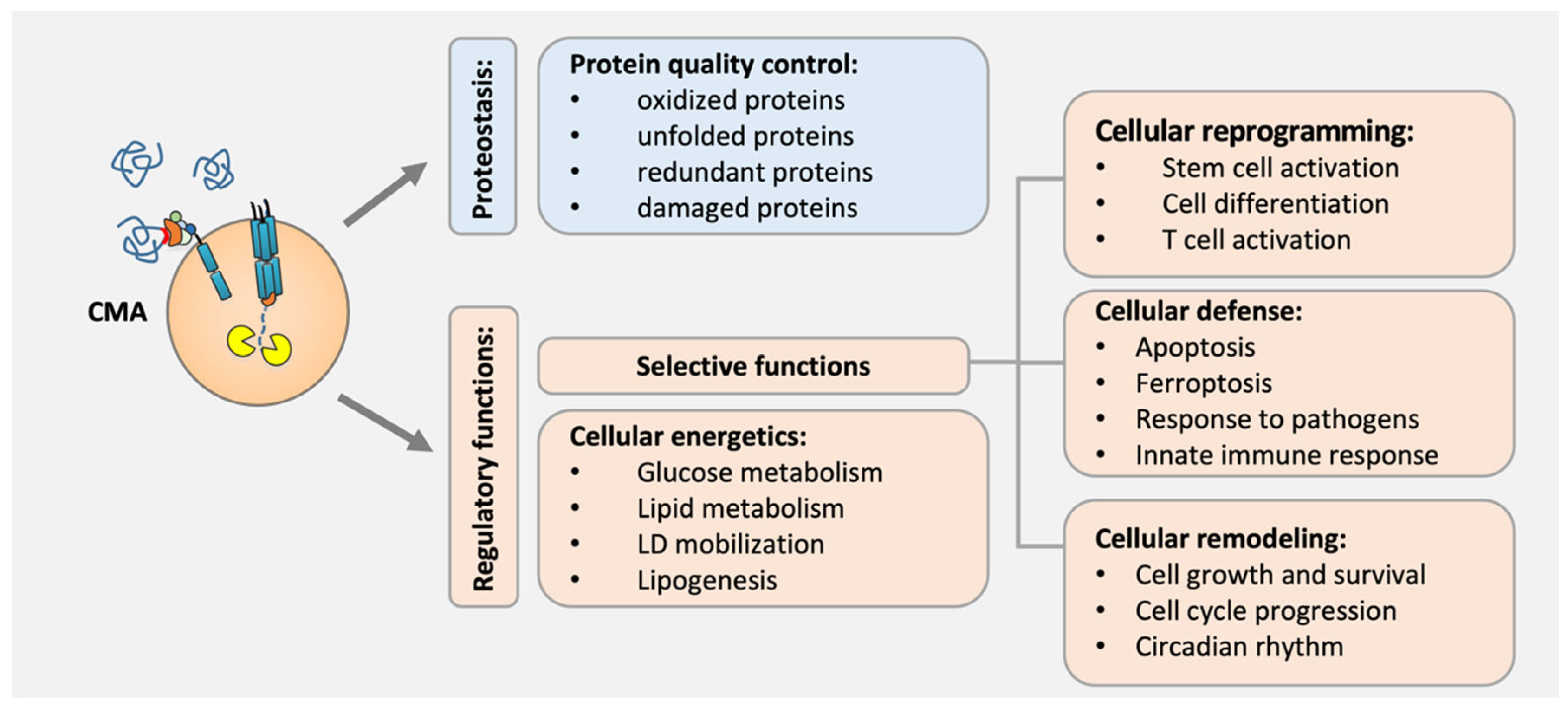
3. Biological Functions of CMA
3.1. Protein Quality Control
3.2. Cellular Energetics
3.3. Cellular Reprogramming
3.4. Other Cellular Processes
3.5. Cellular Defense
4. CMA and Human Diseases: Role in Pathogenesis
4.1. Neurodegenerative Diseases
4.1.1. Parkinson’s Disease (PD)
4.1.2. Alzheimer’s Disease (AD)
4.1.3. Other Neurodegenerative Diseases
4.2. Cancer
4.2.1. CMA Regulation: Pro-Tumor versus Anti-Tumor Roles
4.2.2. Identified Substrates in Different Types of Cancers
4.2.3. Targeting CMA in the Tumor Microenvironment
4.3. Aging-Associated Diseases and Other Pathologies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy Fights Disease through Cellular Self-Digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Altered Degradation of Proteins Microinjected into Senescent Human Fibroblasts. J. Biol. Chem. 1982, 257, 14624–14627. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Peptide Sequences That Target Cytosolic Proteins for Lysosomal Proteolysis. Trends Biochem. Sci. 1990, 15, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. AMPK-Dependent Phosphorylation of Lipid Droplet Protein PLIN2 Triggers Its Degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Quintavaller, C.; Costanzo, S.; Zanca, C.; Tasset, I.; Fraldi, A.; Incoronato, M.; Mirabelli, P.; Monti, M.; Ballabio, A.; Pucci, P.; et al. Phosphorylation-Regulated Degradation of the Tumor-Suppressor Form of PED by Chaperone-Mediated Autophagy in Lung Cancer Cells. J. Cell Physiol. 2014, 229, 1359–1368. [Google Scholar] [CrossRef]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated Degradation of Chk1 by Chaperone-Mediated Autophagy in Response to DNA Damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, J.; Fan, X.; Hu, S.; Zhou, F.; Dong, J.; Zhang, S.; Shang, Y.; Jiang, X.; Guo, H.; et al. Chaperone-Mediated Autophagy Regulates Proliferation by Targeting RND3 in Gastric Cancer. Autophagy 2016, 12, 515–528. [Google Scholar] [CrossRef]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation Targets the M2 Isoform of Pyruvate Kinase for Degradation through Chaperone-Mediated Autophagy and Promotes Tumor Growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef]
- Chiang, H.; Dice, J.F. A Role for a 70 KDa Heat Shock Protein in Lysosomal Degradation of Intracellular Protein. Science 1989, 246, 382–385. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A Receptor for the Selective Uptake and Degradation of Proteins by Lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef]
- Eskelinen, E.-L.L.; Cuervo, A.M.; Taylor, M.R.G.; Nishino, I.; Blum, J.S.; Dice, J.F.; Sandoval, I.V.; Lippincott-Schwartz, J.; August, J.T.; Saftig, P. Unifying Nomenclature for the Isoforms of the Lysosomal Membrane Protein LAMP-2. Traffic 2005, 6, 1058–1061. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The Chaperone-Mediated Autophagy Receptor Organizes in Dynamic Protein Complexes at the Lysosomal Membrane. Mol. Cell Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Navarro, J.A.; Kaushik, S.; Koga, H.; Dall’Armi, C.; Shui, G.; Wenk, M.R.; Di Paolo, G.; Cuervo, A.M. Inhibitory Effect of Dietary Lipids on Chaperone-Mediated Autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, E705–E714. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Massey, A.C.; Cuervo, A.M. Lysosome Membrane Lipid Microdomains: Novel Regulators of Chaperone-Mediated Autophagy. EMBO J. 2006, 25, 3921–3933. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Romero, A.; Fernandez-Gonzalez, I.; Riera, J.; Montpeyo, M.; Albert-Bayo, M.; Lopez-Royo, T.; Castillo-Sanchez, P.; Carnicer-Caceres, C.; Arranz-Amo, J.A.; Castillo-Ribelles, L.; et al. Lysosomal Lipid Alterations Caused by Glucocerebrosidase Deficiency Promote Lysosomal Dysfunction, Chaperone-Mediated-Autophagy Deficiency, and Alpha-Synuclein Pathology. NPJ Park. Dis. 2022, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Johnson, J.L.; He, J.; Napolitano, G.; Ramadass, M.; Rocca, C.; Kiosses, W.B.; Bucci, C.; Xin, Q.; Gavathiotis, E.; et al. Cystinosin, the Small GTPase Rab11, and the Rab7 Effector RILP Regulate Intracellular Trafficking of the Chaperone-Mediated Autophagy Receptor LAMP2A. J. Biol. Chem. 2017, 292, 10328–10346. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sintes, R.; Xin, Q.; Jimenez-Loygorri, J.I.; McCabe, M.; Diaz, A.; Garner, T.P.; Cotto-Rios, X.M.; Wu, Y.; Dong, S.; Reynolds, C.A.; et al. Targeting Retinoic Acid Receptor Alpha-Corepressor Interaction Activates Chaperone-Mediated Autophagy and Protects against Retinal Degeneration. Nat. Commun. 2022, 13, 4220. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Tasset, I.; Diaz, A.; Anguiano, J.; Tas, E.; Cui, L.; Kuliawat, R.; Liu, H.; Kühn, B.; Cuervo, A.M.; et al. Humanin Is an Endogenous Activator of Chaperonemediated Autophagy. J. Cell Biol. 2018, 217, 635–647. [Google Scholar] [CrossRef]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical Modulation of Chaperone-Mediated Autophagy by Retinoic Acid Derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef]
- Bourdenx, M.; Martín-Segura, A.; Scrivo, A.; Rodriguez-Navarro, J.A.; Kaushik, S.; Tasset, I.; Diaz, A.; Storm, N.J.; Xin, Q.; Juste, Y.R.; et al. Chaperone-Mediated Autophagy Prevents Collapse of the Neuronal Metastable Proteome. Cell 2021, 184, 2696–2714.e25. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Mao, K.; Yu, H.; Wen, Y.; She, H.; Zhang, H.; Liu, L.; Li, M.; Li, W.; Zou, F. P38-TFEB Pathways Promote Microglia Activation through Inhibiting CMA-Mediated NLRP3 Degradation in Parkinson’s Disease. J. Neuroinflamm. 2021, 18, 295. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.J.; Kim, J.H.; Oh, S.-O.; Kim, J.-Y. Regulation of Self-Renewal in Ovarian Cancer Stem Cells by Fructose via Chaperone-Mediated Autophagy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2023, 1869, 166723. [Google Scholar] [CrossRef]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-Mediated Autophagy Regulates T Cell Responses through Targeted Degradation of Negative Regulators of T Cell Activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of Chaperone-Mediated Autophagy during Oxidative Stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Kshitiz; Ahmed, I.; Semenza, G.L. Cyclin-Dependent Kinases Regulate Lysosomal Degradation of Hypoxia-Inducible Factor 1α to Promote Cell-Cycle Progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3325–E3334. [Google Scholar] [CrossRef]
- Ferreira, J.V.; Fôfo, H.; Bejarano, E.; Bento, C.F.; Ramalho, J.S.; Girão, H.; Pereira, P. STUB1/CHIP Is Required for HIF1A Degradation by Chaperone-Mediated Autophagy. Autophagy 2013, 9, 1349–1366. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant Response Elements: Discovery, Classes, Regulation and Potential Applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Arias, E.; Díaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription Factor NFE2L2/NRF2 Modulates Chaperone-Mediated Autophagy through the Regulation of LAMP2A. Autophagy 2018, 8627, 1310–1322. [Google Scholar] [CrossRef]
- Arias, E.; Koga, H.; Diaz, A.; Mocholi, E.; Patel, B.; Cuervo, A.M.M. Lysosomal MTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef]
- Ebner, M.; Sinkovics, B.; Szczygieł, M.; Ribeiro, D.W.; Yudushkin, I. Localization of MTORC2 Activity inside Cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Juste, Y.R.; Kaushik, S.; Bourdenx, M.; Aflakpui, R.; Bandyopadhyay, S.; Garcia, F.; Diaz, A.; Lindenau, K.; Tu, V.; Krause, G.J.; et al. Reciprocal Regulation of Chaperone-Mediated Autophagy and the Circadian Clock. Nat. Cell Biol. 2021, 23, 1255–1270. [Google Scholar] [CrossRef]
- Hormazabal, J.; Saavedra, F.; Espinoza-Arratia, C.; Martinez, N.W.; Cruces, T.; Alfaro, I.E.; Loyola, A. Chaperone mediated autophagy contributes to the newly synthesized histones H3 and H4 quality control. Nucleic Acids Res. 2022, 50, 1875–1887. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.-H.; Tasset, I.; Cheng, M.M.; Diaz, A.; Pan, M.-K.; Lieberman, O.J.; Hutten, S.J.; Alcalay, R.N.; Kim, S.; Ximénez-Embún, P.; et al. Mutant Glucocerebrosidase Impairs α-Synuclein Degradation by Blockade of Chaperone-Mediated Autophagy. Sci. Adv. 2022, 8, eabm6393. [Google Scholar] [CrossRef] [PubMed]
- Finn, P.F.; Dice, J.F. Ketone Bodies Stimulate Chaperone-MediatedAutophagy. J. Biol. Chem. 2005, 280, 25864–25870. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Villarroya, J.; Diaz-Carretero, A.; Patel, B.; Urbanska, A.M.; Thi, M.M.; Villarroya, F.; Santambrogio, L.; Cuervo, A.M. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 2015, 14, 249–264. [Google Scholar] [CrossRef]
- Kaushik, S.; Massey, A.C.; Mizushima, N.; Cuervo, A.M. Constitutive Activation of Chaperone-Mediated Autophagy in Cells with Impaired Macroautophagy. Mol. Biol. Cell 2008, 19, 2179–2192. [Google Scholar] [CrossRef]
- Cui, H.; Norrbacka, S.; Myöhänen, T.T. Prolyl Oligopeptidase Acts as a Link between Chaperone-Mediated Autophagy and Macroautophagy. Biochem. Pharmacol. 2022, 197, 114899. [Google Scholar] [CrossRef]
- Li, W.; Zhu, J.; Dou, J.; She, H.; Tao, K.; Xu, H.; Yang, Q.; Mao, Z. Phosphorylation of LAMP2A by P38 MAPK Couples ER Stress to Chaperone-Mediated Autophagy. Nat. Commun. 2017, 8, 1763. [Google Scholar] [CrossRef]
- Hosaka, Y.; Araya, J.; Fujita, Y.; Kadota, T.; Tsubouchi, K.; Yoshida, M.; Minagawa, S.; Hara, H.; Kawamoto, H.; Watanabe, N.; et al. Chaperone-Mediated Autophagy Suppresses Apoptosis via Regulation of the Unfolded Protein Response during Chronic Obstructive Pulmonary Disease Pathogenesis. J. Immunol. 2020, 205, 1256–1267. [Google Scholar] [CrossRef]
- Dong, S.; Wang, Q.; Kao, Y.R.; Diaz, A.; Tasset, I.; Kaushik, S.; Thiruthuvanathan, V.; Zintiridou, A.; Nieves, E.; Dzieciatkowska, M.; et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 2021, 591, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Martín-Segura, A.; Puri, C.; Scrivo, A.; Skidmore, J.; et al. The different autophagy degradation pathways and neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef]
- Endicott, S.J.; Monovich, A.C.; Huang, E.L.; Henry, E.I.; Boynton, D.N.; Beckmann, L.J.; MacCoss, M.J.; Miller, R.A. Lysosomal Targetomics of Ghr KO Mice Shows Chaperone-Mediated Autophagy Degrades Nucleocytosolic Acetyl-CoA Enzymes. Autophagy 2022, 18, 1551–1571. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Ma, J.; Zhang, Z.; Sui, W.; Zhai, C.; Xu, D.; Wang, Z.; Lu, H.; Zhang, M.; Zhang, C.; et al. Deficient Chaperone-Mediated Autophagy Promotes Inflammation and Atherosclerosis. Circ. Res. 2021, 129, 1141–1157. [Google Scholar] [CrossRef] [PubMed]
- Madrigal-Matute, J.; de Bruijn, J.; van Kuijk, K.; Riascos-Bernal, D.F.; Diaz, A.; Tasset, I.; Martín-Segura, A.; Gijbels, M.J.J.; Sander, B.; Kaushik, S.; et al. Protective Role of Chaperone-Mediated Autophagy against Atherosclerosis. Proc. Natl. Acad. Sci. USA 2022, 119, 119. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Degradation of Lipid Droplet-Associated Proteins by Chaperone-Mediated Autophagy Facilitates Lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef]
- Nie, T.; Tao, K.; Zhu, L.; Huang, L.; Hu, S.; Yang, R.; Xu, P.; Mao, Z.; Yang, Q. Chaperone-Mediated Autophagy Controls the Turnover of E3 Ubiquitin Ligase MARCHF5 and Regulates Mitochondrial Dynamics. Autophagy 2021, 17, 2923–2938. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.; García-Cañaveras, J.C.; Guo, L.; Kan, M.; Yu, S.; Blair, I.A.; Rabinowitz, J.D.; Yang, X. Chaperone-Mediated Autophagy Regulates the Pluripotency of Embryonic Stem Cells. Science 2020, 369, 397–403. [Google Scholar] [CrossRef]
- He, Q.; Qin, R.; Glowacki, J.; Zhou, S.; Shi, J.; Wang, S.; Gao, Y.; Cheng, L. Synergistic Stimulation of Osteoblast Differentiation of Rat Mesenchymal Stem Cells by Leptin and 25(OH)D3 Is Mediated by Inhibition of Chaperone-Mediated Autophagy. Stem Cell Res. Ther. 2021, 12, 557. [Google Scholar] [CrossRef]
- Wang, F.; Tasset, I.; Cuervo, A.M.; Muller, S. In Vivo Remodeling of Altered Autophagy-Lysosomal Pathway by a Phosphopeptide in Lupus. Cells 2020, 9, 2328. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; McCabe, M.; Cuervo, A.M. Chaperone-mediated autophagy: Mechanisms and physiological relevance. Curr. Opin. Physiol. 2022, 30, 100597. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Nguyen, D.; Yang, K.; Chiao, L.; Deng, Y.; Zhou, X.; Zhang, Z.; Zeng, S.X.; Lu, H. Inhibition of Tumor Suppressor P73 by Nerve Growth Factor Receptor via Chaperone-Mediated Autophagy. J. Mol. Cell Biol. 2020, 12, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.; Johnson, J.L.; Zhang, J.; He, J.; Pestonjamasp, K.; Cherqui, S.; Catz, S.D. DYNC1LI2 Regulates Localization of the Chaperone-Mediated Autophagy Receptor LAMP2A and Improves Cellular Homeostasis in Cystinosis. Autophagy 2022, 18, 1108–1126. [Google Scholar] [CrossRef]
- Wu, Z.; Geng, Y.; Lu, X.; Shi, Y.; Wu, G.; Zhang, M.; Shan, B.; Pan, H.; Yuan, J. Chaperone-Mediated Autophagy Is Involved in the Execution of Ferroptosis. Proc. Natl. Acad. Sci. USA 2019, 116, 2996–3005. [Google Scholar] [CrossRef]
- Hos, N.J.; Fischer, J.; Hos, D.; Hejazi, Z.; Calabrese, C.; Ganesan, R.; Murthy, A.M.V.; Rybniker, J.; Kumar, S.; Krönke, M.; et al. TRIM21 Is Targeted for Chaperone-Mediated Autophagy during Salmonella typhimurium Infection. J. Immunol. 2020, 205, 2456–2467. [Google Scholar] [CrossRef]
- Zhao, X.; Di, Q.; Yu, J.; Quan, J.; Xiao, Y.; Zhu, H.; Li, H.; Ling, J.; Chen, W. USP19 (Ubiquitin Specific Peptidase 19) Promotes TBK1 (TANK-Binding Kinase 1) Degradation via Chaperone-Mediated Autophagy. Autophagy 2022, 18, 891–908. [Google Scholar] [CrossRef]
- Hu, M.-M.; Yang, Q.; Xie, X.-Q.; Liao, C.-Y.; Lin, H.; Liu, T.-T.; Yin, L.; Shu, H.-B. Sumoylation Promotes the Stability of the DNA Sensor CGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef]
- Matsui, C.; Yuliandari, P.; Deng, L.; Abe, T.; Shoji, I. The Role of Chaperone-Mediated Autophagy in Hepatitis C Virus-Induced Pathogenesis. Front. Cell Infect. Microbiol. 2021, 11, 1222. [Google Scholar] [CrossRef]
- Rodríguez-Muela, N.; Koga, H.; García-Ledo, L.; de la Villa, P.; de la Rosa, E.J.; Cuervo, A.M.; Boya, P. Balance between Autophagic Pathways Preserves Retinal Homeostasis. Aging Cell 2013, 12, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cuervo, A.M. Restoration of Chaperone-Mediated Autophagy in Aging Liver Improves Cellular Maintenance and Hepatic Function. Nat. Med. 2008, 14, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting α-Synuclein for Treatment of Parkinson’s Disease: Mechanistic and Therapeutic Considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-Mediated Autophagy Markers in Parkinson Disease Brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in Lysosomal and Proteasomal Markers in Parkinson’s Disease: Relationship to Alpha-Synuclein Inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic Lysosomal Depletion in Parkinson’s Disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stafanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-Modified α-Synuclein Blocks Chaperone-Mediated Autophagy. J. Clin. Investig. 2008, 118, 777–778. [Google Scholar] [CrossRef]
- Tang, F.-L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.-M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.-C. VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for α-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628. [Google Scholar] [CrossRef]
- Neumann, J.; Bras, J.; Deas, E.; O’sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase Mutations in Clinical and Pathologically Proven Parkinson’s Disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.A.; Aasly, J.O.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting Chaperone-Mediated Autophagy in Vivo Mitigates α-Synuclein-Induced Neurodegeneration. Brain 2013, 136, 2130–2146. [Google Scholar] [CrossRef]
- Liu, H.; Wang, P.; Song, W.; Sun, X. Degradation of Regulator of Calcineurin 1 (RCAN1) Is Mediated by Both Chaperone-mediated Autophagy and Ubiquitin Proteasome Pathways. FASEB J. 2009, 23, 3383–3392. [Google Scholar] [CrossRef] [PubMed]
- Ermak, G.; Sojitra, S.; Yin, F.; Cadenas, E.; Cuervo, A.M.; Davies, K.J. Chronic Expression of RCAN1-1L Protein Induces Mitochondrial Autophagy and Metabolic Shift from Oxidative Phosphorylation to Glycolysis in Neuronal Cells. J. Biol. Chem. 2012, 287, 14088–14098. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, Y.; Cen, X.; Shan, B.; Zhao, Q.; Xie, T.; Wang, Z.; Hou, T.; Xue, Y.; Zhang, M.; et al. Metformin Activates Chaperone-Mediated Autophagy and Improves Disease Pathologies in an Alzheimer Disease Mouse Model. Protein Cell 2021, 12, 769–787. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Martinez-Vicente, M.; Arias, E.; Kaushik, S.; Sulzer, D.; Cuervo, A.M.A.M. Constitutive Upregulation of Chaperone-Mediated Autophagy in Huntington’s Disease. J. Neurosci. 2011, 31, 18492–18505. [Google Scholar] [CrossRef]
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y.; et al. Harnessing Chaperone-Mediated Autophagy for the Selective Degradation of Mutant Huntingtin Protein. Nat. Biotechnol. 2010, 28, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Zhang, X.-D.; Wu, J.-C.; Lin, F.; Wang, J.; DiFiglia, M.; Qin, Z.-H. The Role of Chaperone-Mediated Autophagy in Huntingtin Degradation. PLoS ONE 2012, 7, e46834. [Google Scholar] [CrossRef]
- Huang, C.-C.; Bose, J.K.; Majumder, P.; Lee, K.-H.; Huang, J.-T.J.; Huang, J.K.; Shen, C.-K.J. Metabolism and Mis-Metabolism of the Neuropathological Signature Protein TDP-43. J. Cell Sci. 2014, 127, 3024–3038. [Google Scholar] [CrossRef]
- Wang, H.; Tian, C.; Sun, J.; Chen, L.-N.; Lv, Y.; Yang, X.-D.; Xiao, K.; Wang, J.; Chen, C.; Shi, Q.; et al. Overexpression of PLK3 Mediates the Degradation of Abnormal Prion Proteins Dependent on Chaperone-Mediated Autophagy. Mol. Neurobiol. 2017, 54, 4401–4413. [Google Scholar] [CrossRef]
- Kon, M.; Cuervo, A.M. Chaperone-mediated Autophagy in Health and Disease. FEBS Lett. 2010, 584, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Cuervo, A.M. Pros and Cons of Chaperone-Mediated Autophagy in Cancer Biology. Trends Endocrinol. Metab. 2020, 31, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Sintes, R.; Arias, E. Chaperone-Mediated Autophagy and Disease: Implications for Cancer and Neurodegeneration. Mol. Asp. Med. 2021, 82, 101025. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Menck, C.F.M.; Cuervo, A.M. Chaperone-Mediated Autophagy Prevents Cellular Transformation by Regulating MYC Proteasomal Degradation. Autophagy 2017, 13, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-Mediated Autophagy Is Required for Tumor Growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef] [PubMed]
- Molina, M.L.; García-Bernal, D.; Martinez, S.; Valdor, R. Autophagy in the Immunosuppressive Perivascular Microenvironment of Glioblastoma. Cancers 2019, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Kacal, M.; Ouchida, A.T.; Zhang, B.; Norberg, E.; Vakifahmetoglu-Norberg, H. Targetome Analysis of Chaperone-Mediated Autophagy in Cancer Cells. Autophagy 2019, 15, 1558–1571. [Google Scholar] [CrossRef]
- Lu, T.-L.; Huang, G.-J.; Wang, H.-J.; Chen, J.-L.; Hsu, H.-P.; Lu, T.-J. Hispolon Promotes MDM2 Downregulation through Chaperone-Mediated Autophagy. Biochem. Biophys. Res. Commun. 2010, 398, 26–31. [Google Scholar] [CrossRef]
- Bonhoure, A.; Vallentin, A.; Martin, M.; Senff-Ribeiro, A.; Amson, R.; Telerman, A.; Vidal, M. Acetylation of Translationally Controlled Tumor Protein Promotes Its Degradation through Chaperone-Mediated Autophagy. Eur. J. Cell Biol. 2017, 96, 83–98. [Google Scholar] [CrossRef]
- Franch, H.A.; Sooparb, S.; Du, J.; Brown, N.S. A Mechanism Regulating Proteolysis of Specific Proteins during Renal Tubular Cell Growth. J. Biol. Chem. 2001, 276, 19126–19131. [Google Scholar] [CrossRef]
- Tang, J.; Zhan, M.-N.; Yin, Q.-Q.; Zhou, C.-X.; Wang, C.-L.; Wo, L.-L.; He, M.; Chen, G.-Q.; Zhao, Q. Impaired P65 Degradation by Decreased Chaperone-Mediated Autophagy Activity Facilitates Epithelial-to-Mesenchymal Transition. Oncogenesis 2017, 6, e387. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Ren, C.; Qiao, P.; Han, X.; Wang, L.; Lv, S.; Sun, Y.; Liu, Z.; Du, Y.; Yu, Z. PIM2-Mediated Phosphorylation of Hexokinase 2 Is Critical for Tumor Growth and Paclitaxel Resistance in Breast Cancer. Oncogene 2018, 37, 5997–6009. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Huang, S.; Wang, J.; Zhang, Y.; Xiong, Y.; Zeng, S.X.; Lu, H. Inactivating P53 Is Essential for Nerve Growth Factor Receptor to Promote Melanoma-Initiating Cell-Stemmed Tumorigenesis. Cell Death Dis. 2020, 11, 550. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; He, H.; Liu, Y.; Jiang, Y.; Yang, J. The Complex Role of Chaperone-Mediated Autophagy in Cancer Diseases. Biomedicines 2023, 11, 2050. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Xu, G.; Wang, Z.; Chong, T. Chaperon-mediated Autophagy Can Promote Proliferation and Invasion of Renal Carcinoma Cells and Inhibit Apoptosis through PKM2. Oncol. Rep. 2021, 46, 214. [Google Scholar] [CrossRef]
- Auzmendi-Iriarte, J.; Otaegi-Ugartemendia, M.; Carrasco-Garcia, E.; Azkargorta, M.; Diaz, A.; Saenz-Antoñanzas, A.; Andermatten, J.A.; Garcia-Puga, M.; Garcia, I.; Elua-Pinin, A.; et al. Chaperone-Mediated Autophagy Controls Proteomic and Transcriptomic Pathways to Maintain Glioma Stem Cell Activity. Cancer Res. 2022, 82, 1283–1297. [Google Scholar] [CrossRef]
- Valdor, R.; García-Bernal, D.; Riquelme, D.; Martinez, C.M.; Moraleda, J.M.; Cuervo, A.M.; Macian, F.; Martinez, S. Glioblastoma Ablates Pericytes Antitumor Immune Function through Aberrant Up-Regulation of Chaperone-Mediated Autophagy. Proc. Natl. Acad. Sci. USA 2019, 116, 20655–20665. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhou, J.; Li, L.; Liao, S.; He, J.; Zhou, S.; Zhou, Y. Pericytes in the Tumor Microenvironment. Cancer Lett. 2023, 556, 216074. [Google Scholar] [CrossRef]
- Salinas, M.D.; Valdor, R. Chaperone-Mediated Autophagy in Pericytes: A Key Target for the Development of New Treatments against Glioblastoma Progression. Int. J. Mol. Sci. 2022, 23, 8886. [Google Scholar] [CrossRef]
- Molina, M.L.; García-Bernal, D.; Salinas, M.D.; Rubio, G.; Aparicio, P.; Moraleda, J.M.; Martínez, S.; Valdor, R. Chaperone-Mediated Autophagy Ablation in Pericytes Reveals New Glioblastoma Prognostic Markers and Efficient Treatment Against Tumor Progression. Front. Cell Dev. Biol. 2022, 10, 797945. [Google Scholar] [CrossRef]
- Hubert, V.; Weiss, S.; Rees, A.J.; Kain, R. Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells 2022, 11, 2562. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.J.; Lee, K.H.; Woo, J.; Kim, J.Y.; Lee, C.H.; Yoo, C.G. Downregulation of Lysosome-Associated Membrane Protein-2A Contributes to the Pathogenesis of COPD. Int. J. COPD 2023, 18, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.Y.; Sun, K.S.; Zhang, M.; Zhou, X.; Zheng, X.H.; Tian, S.Y.; Liu, Y.S.; Chen, L.; Gao, X.; Ye, J.; et al. Disruption of Plin5 Degradation by CMA Causes Lipid Homeostasis Imbalance in NAFLD. Liver Int. 2020, 40, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Li, W.Z.; Zhang, S.; Hu, B.; Li, Y.X.; Li, H.D.; Tang, H.H.; Li, Q.W.; Guan, Y.Y.; Liu, L.X.; et al. SNX10 Mediates Alcohol-Induced Liver Injury and Steatosis by Regulating the Activation of Chaperone-Mediated Autophagy. J. Hepatol. 2018, 69, 129–141. [Google Scholar] [CrossRef]
- Lee, W.; Kim, H.Y.; Choi, Y.J.; Jung, S.H.; Nam, Y.A.; Zhang, Y.; Yun, S.H.; Chang, T.S.; Lee, B.H. SNX10-Mediated Degradation of LAMP2A by NSAIDs Inhibits Chaperone-Mediated Autophagy and Induces Hepatic Lipid Accumulation. Theranostics 2022, 12, 2351. [Google Scholar] [CrossRef]
- Yuan, Z.; Wang, S.; Tan, X.; Wang, D. New Insights into the Mechanisms of Chaperon-Mediated Autophagy and Implications for Kidney Diseases. Cells 2022, 11, 406. [Google Scholar] [CrossRef]
- Sooparb, S.; Price, S.R.; Shaoguang, J.; Franch, H.A. Suppression of Chaperone-Mediated Autophagy in the Renal Cortex during Acute Diabetes Mellitus. Kidney Int. 2004, 65, 2135–2144. [Google Scholar] [CrossRef]
- Venugopal, B.; Mesires, N.T.; Kennedy, J.C.; Curcio-Morelli, C.; Laplante, J.M.; Dice, J.F.; Slaugenhaupt, S.A. Chaperone-Mediated Autophagy Is Defective in Mucolipidosis Type IV. J. Cell Physiol. 2009, 219, 344–353. [Google Scholar] [CrossRef]
- Retnakumar, S.V.; Geesala, R.; Bretin, A.; Tourneur-Marsille, J.; Ogier-Denis, E.; Maretzky, T.; Thu Nguyen, H.T.; Muller, S. Targeting the Endo-Lysosomal Autophagy Pathway to Treat Inflammatory Bowel Diseases. J. Autoimmun. 2022, 128, 102814. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdor, R.; Martinez-Vicente, M. The Role of Chaperone-Mediated Autophagy in Tissue Homeostasis and Disease Pathogenesis. Biomedicines 2024, 12, 257. https://doi.org/10.3390/biomedicines12020257
Valdor R, Martinez-Vicente M. The Role of Chaperone-Mediated Autophagy in Tissue Homeostasis and Disease Pathogenesis. Biomedicines. 2024; 12(2):257. https://doi.org/10.3390/biomedicines12020257
Chicago/Turabian StyleValdor, Rut, and Marta Martinez-Vicente. 2024. "The Role of Chaperone-Mediated Autophagy in Tissue Homeostasis and Disease Pathogenesis" Biomedicines 12, no. 2: 257. https://doi.org/10.3390/biomedicines12020257
APA StyleValdor, R., & Martinez-Vicente, M. (2024). The Role of Chaperone-Mediated Autophagy in Tissue Homeostasis and Disease Pathogenesis. Biomedicines, 12(2), 257. https://doi.org/10.3390/biomedicines12020257