

Treatment with Cobra Venom Factor Decreases Ischemic Tissue Damage in Mice

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
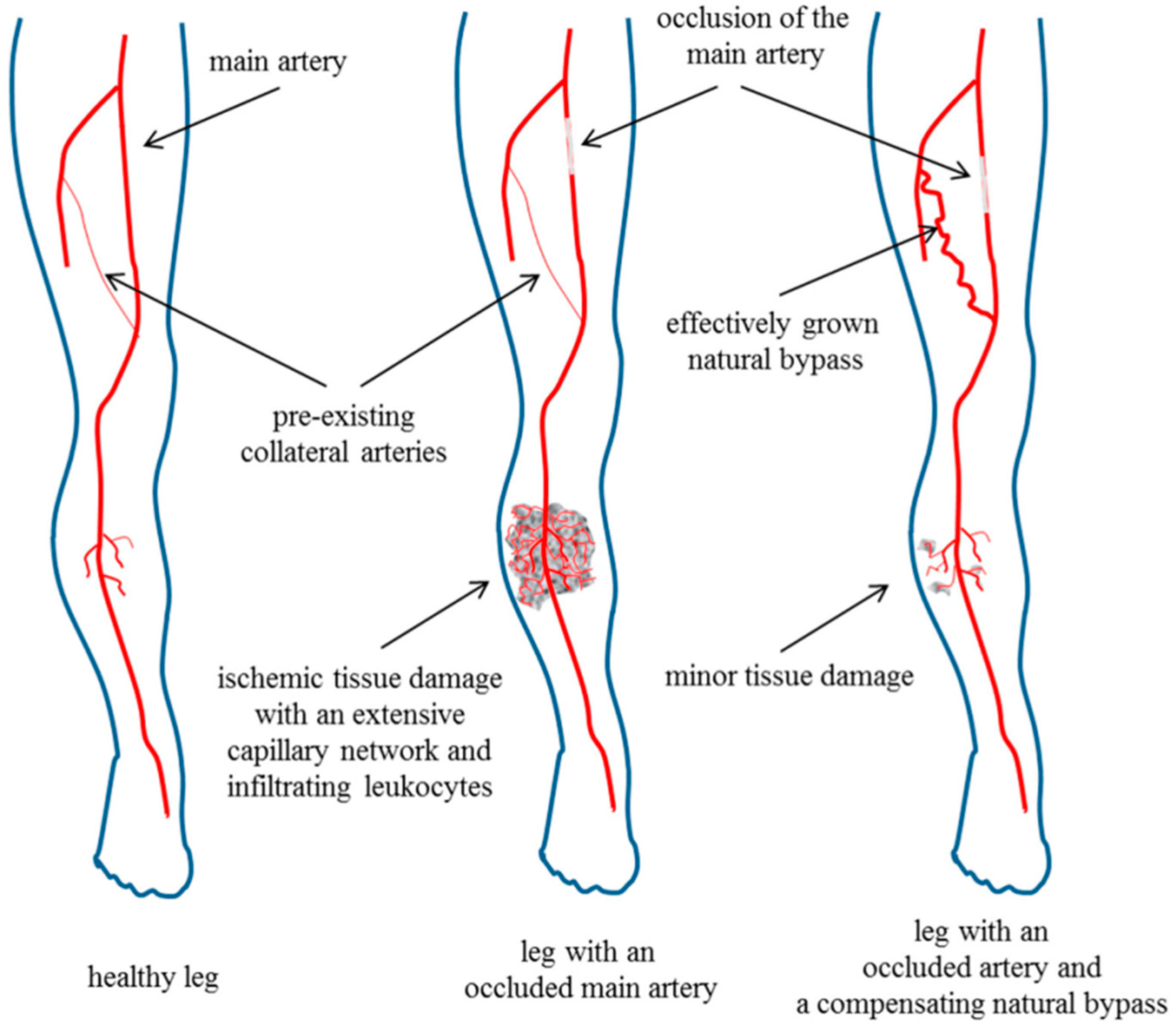
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Procedures
2.2. Histological and Immunofluorescence Analysis
3. Results
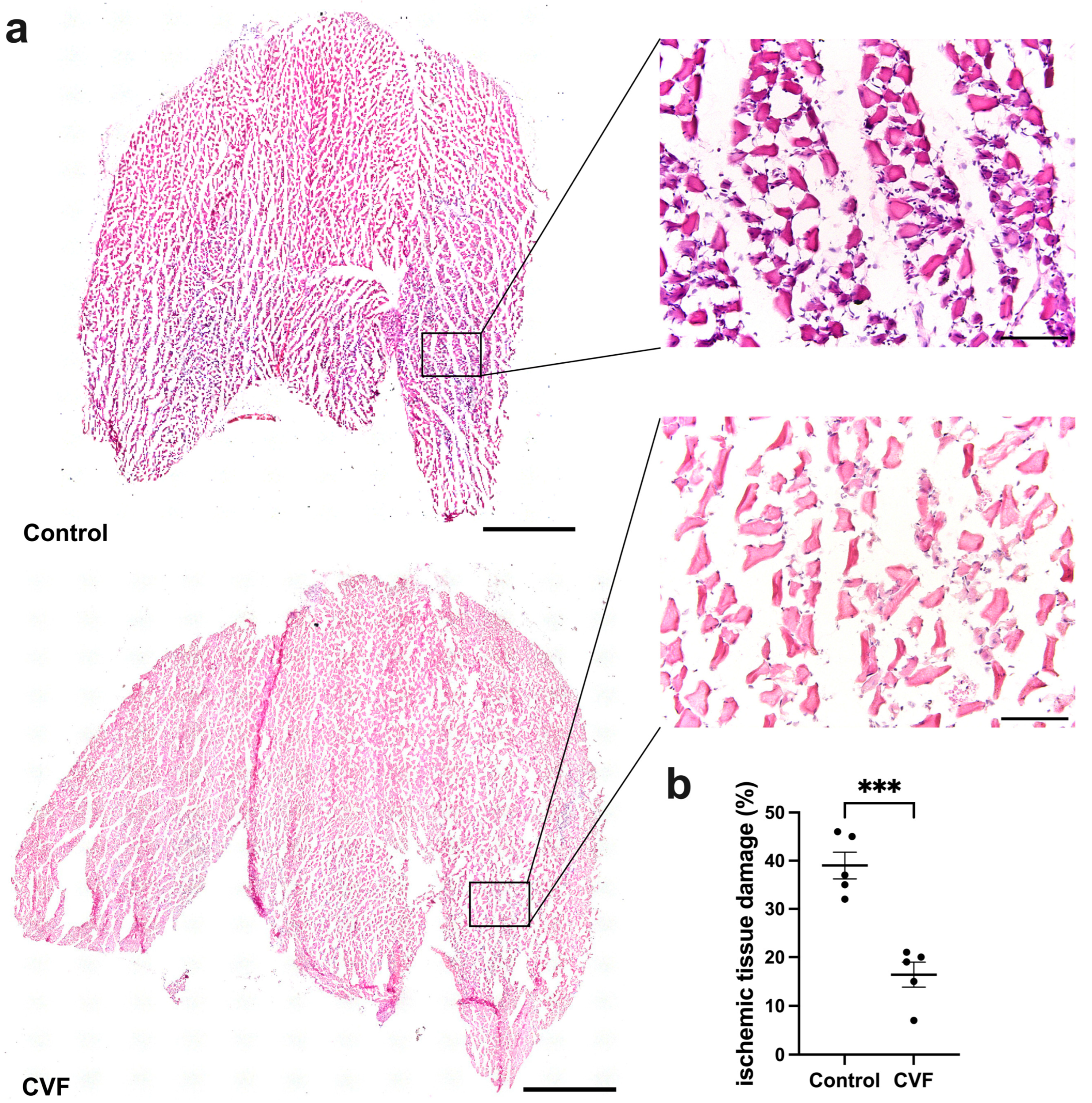
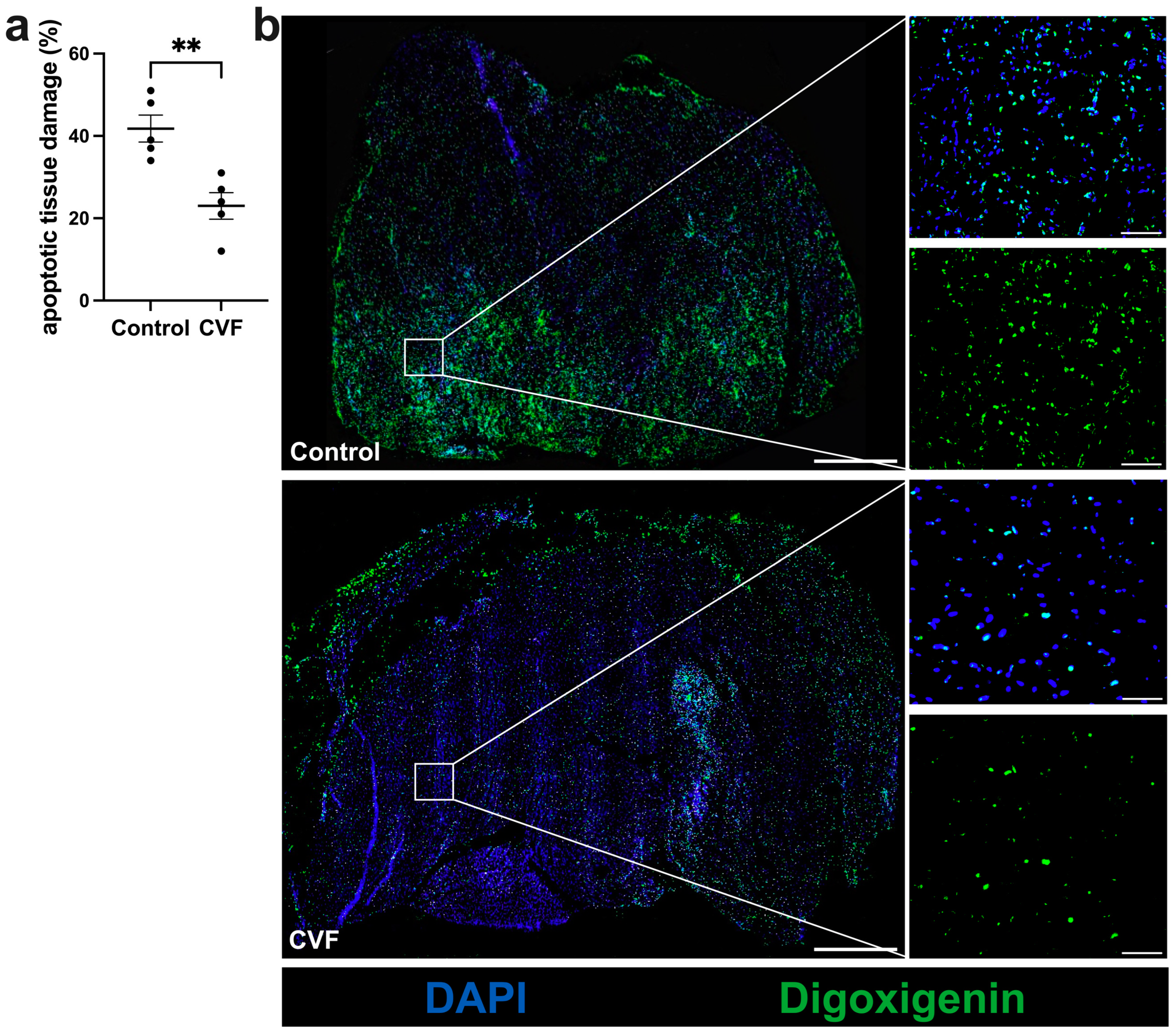
3.1. CVF Treatment Leads to Reduced Ischemic Tissue Damage
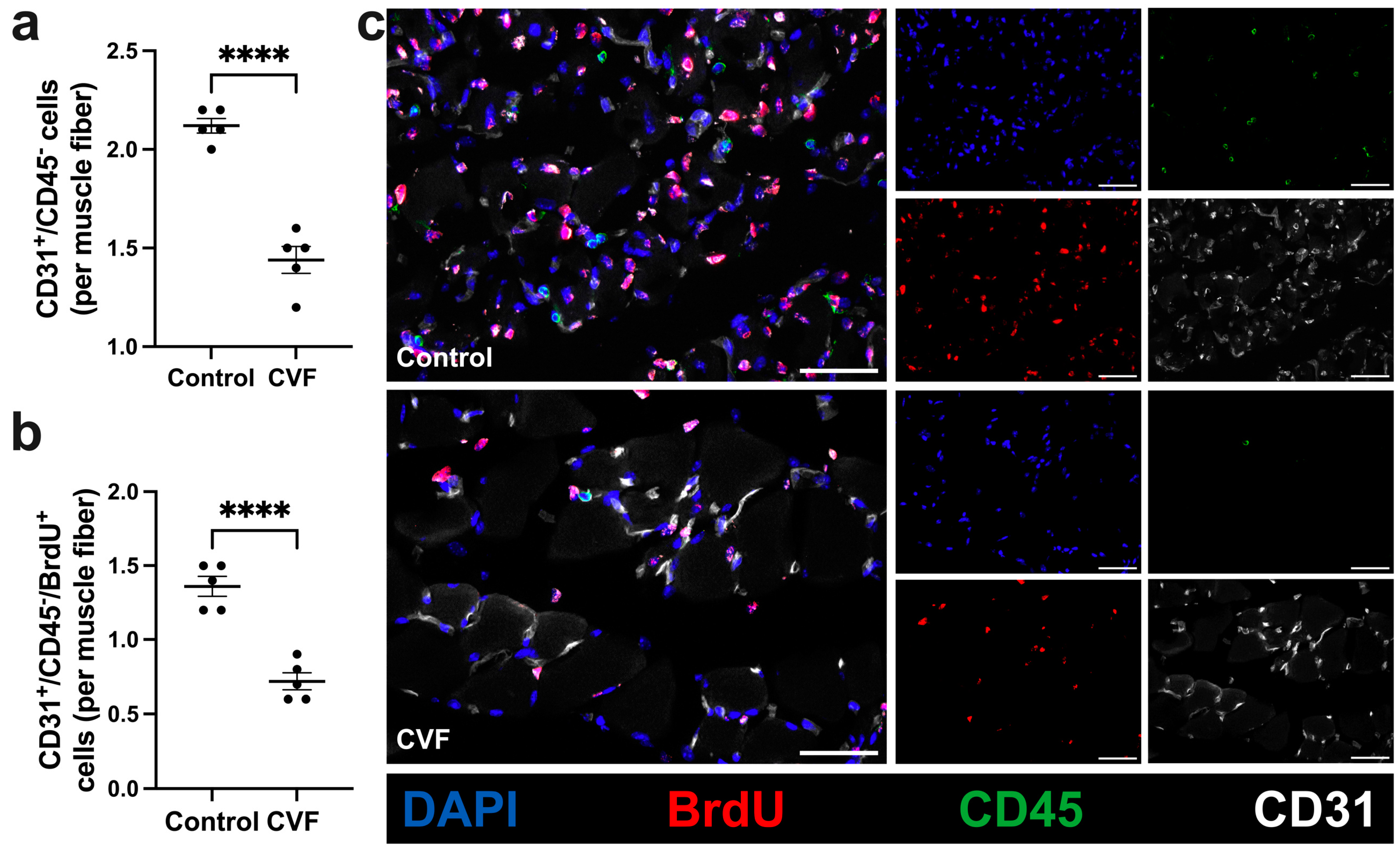
3.2. CVF-Treated Mice Show a Reduced Capillarity
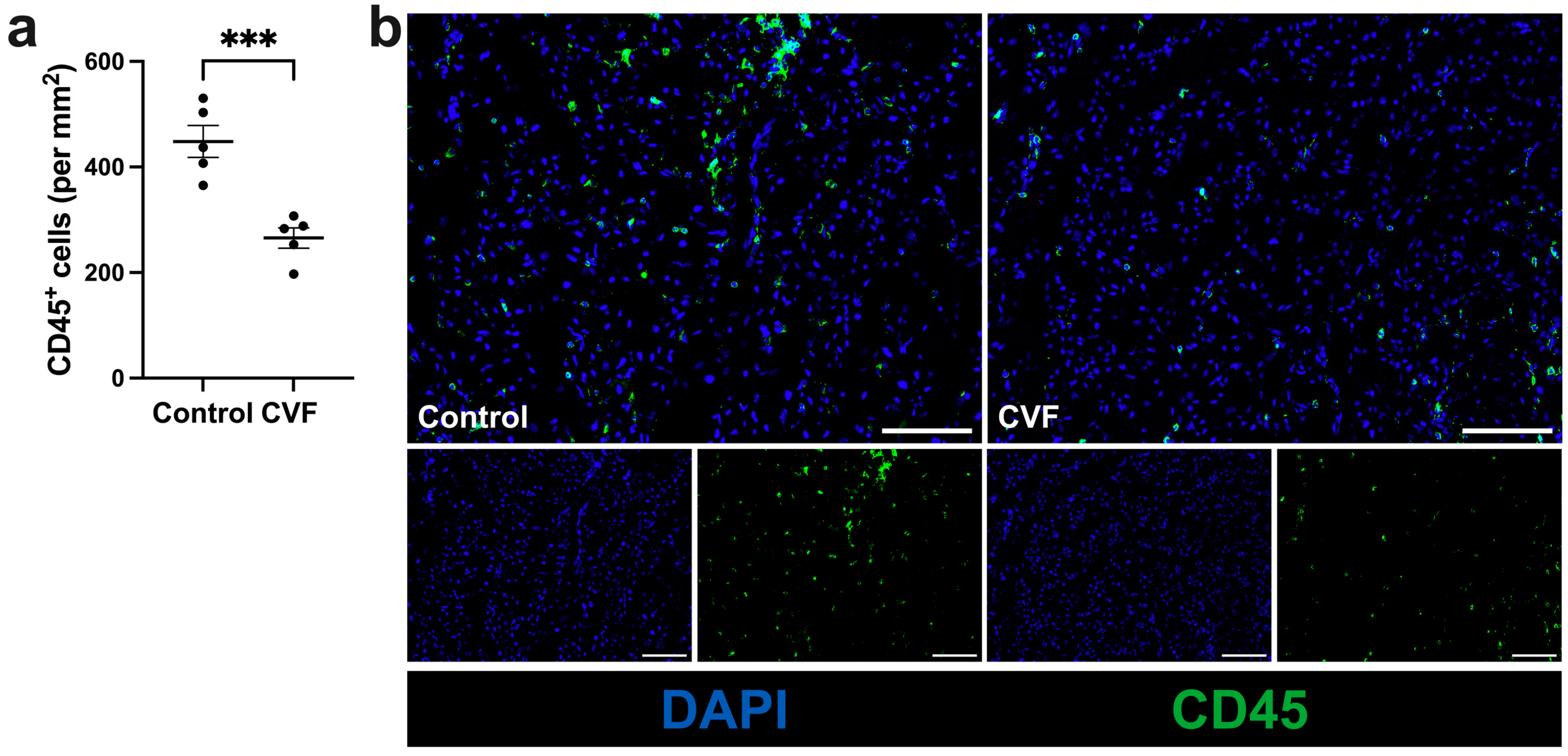
3.3. Treatment with CVF Leads to Reduced Leukocyte Infiltration
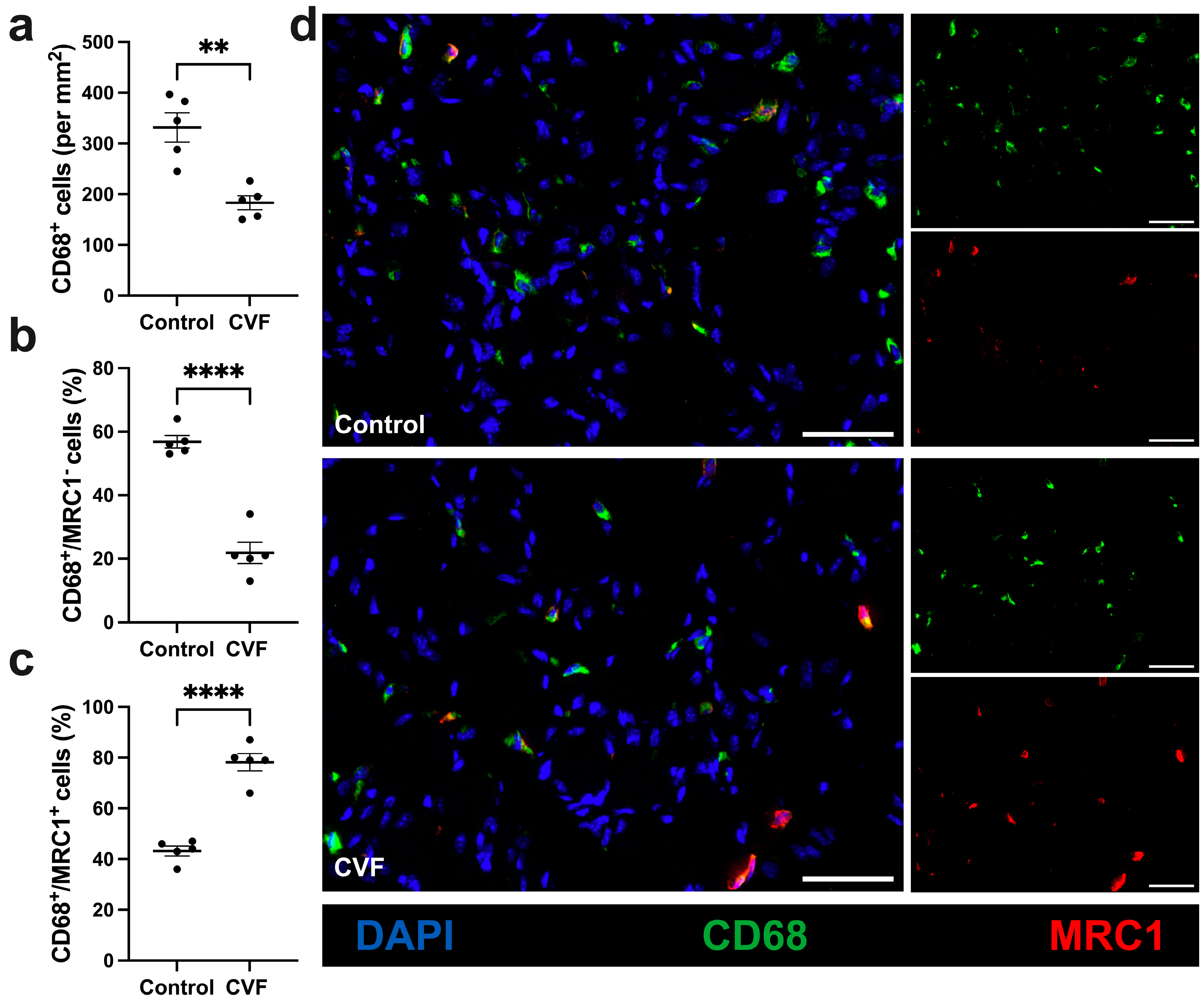
3.4. CVF-Treated Mice Show a Lower Number of Macrophages with a Higher Percentage of M2-like Polarization
4. Discussion
5. Conclusions
6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics—2023 Update: A Report From the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Novel mechanisms and functions of complement. Nat. Immunol. 2017, 18, 1288–1298. [Google Scholar] [CrossRef]
- Nesargikar, P.; Spiller, B.; Chavez, R. The complement system: History, pathways, cascade and inhibitors. Eur. J. Microbiol. Immunol. (Bp) 2012, 2, 103–111. [Google Scholar] [CrossRef]
- Ricklin, D.; Lambris, J.D. Complement in immune and inflammatory disorders: Pathophysiological mechanisms. J. Immunol. 2013, 190, 3831–3838. [Google Scholar] [CrossRef]
- Kolev, M.; Le Friec, G.; Kemper, C. Complement--tapping into new sites and effector systems. Nat. Rev. Immunol. 2014, 14, 811–820. [Google Scholar] [CrossRef]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef]
- Bora, P.S.; Sohn, J.-H.; Cruz, J.M.C.; Jha, P.; Nishihori, H.; Wang, Y.; Kaliappan, S.; Kaplan, H.J.; Bora, N.S. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J. Immunol. 2005, 174, 491–497. [Google Scholar] [CrossRef]
- Ambati, J.; Anand, A.; Fernandez, S.; Sakurai, E.; Lynn, B.C.; A Kuziel, W.; Rollins, B.J.; Ambati, B.K. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat. Med. 2003, 9, 1390–1397. [Google Scholar] [CrossRef]
- Capitão, M.; Soares, R. Angiogenesis and Inflammation Crosstalk in Diabetic Retinopathy. J. Cell. Biochem. 2016, 117, 2443–2453. [Google Scholar] [CrossRef]
- Langer, H.F.; Chung, K.-J.; Orlova, V.V.; Choi, E.Y.; Kaul, S.; Kruhlak, M.J.; Alatsatianos, M.; DeAngelis, R.A.; Roche, P.A.; Magotti, P.; et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood 2010, 116, 4395–4403. [Google Scholar] [CrossRef]
- Nording, H.; Baron, L.; Haberthür, D.; Emschermann, F.; Mezger, M.; Sauter, M.; Sauter, R.; Patzelt, J.; Knoepp, K.; Nording, A.; et al. The C5a/C5a receptor 1 axis controls tissue neovascularization through CXCL4 release from platelets. Nat. Commun. 2021, 12, 3352. [Google Scholar] [CrossRef]
- Götz, P.; Braumandl, A.; Kübler, M.; Kumaraswami, K.; Ishikawa-Ankerhold, H.; Lasch, M.; Deindl, E. C3 Deficiency Leads to Increased Angiogenesis and Elevated Pro-Angiogenic Leukocyte Recruitment in Ischemic Muscle Tissue. Int. J. Mol. Sci. 2021, 22, 5800. [Google Scholar] [CrossRef]
- Vogel, C.-W.; Bredehorst, R.; Fritzinger, D.C.; Grunwald, T.; Ziegelmüller, P.; Kock, M.A. Structure and function of cobra venom factor, the complement-activating protein in cobra venom. Adv. Exp. Med. Biol. 1996, 391, 97–114. [Google Scholar] [PubMed]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Deindl, E.; Schaper, W. The art of arteriogenesis. Cell Biochem. Biophys. 2005, 43, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Deindl, E.; Zaruba, M.-M.; Brunner, S.; Huber, B.; Mehl, U.; Assmann, G.; Hoefer, I.E.; Mueller-Hoecker, J.; Franz, W.-M. G-CSF administration after myocardial infarction in mice attenuates late ischemic cardiomyopathy by enhanced arteriogenesis. FASEB J. 2006, 20, 956–958. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar]
- Shima, D.T.; Adamis, A.P.; Ferrara, N.; Yeo, K.-T.; Yeo, T.-K.; Allende, R.; Folkman, J.; D’amore, P.A. Hypoxic induction of endothelial cell growth factors in retinal cells: Identification and characterization of vascular endothelial growth factor (VEGF) as the mitogen. Mol. Med. 1995, 1, 182–193. [Google Scholar] [CrossRef]
- Du Cheyne, C.; Tay, H.; De Spiegelaere, W. The complex TIE between macrophages and angiogenesis. Anat. Histol. Embryol. 2020, 49, 585–596. [Google Scholar] [CrossRef]
- Scapini, P.; Morini, M.; Tecchio, C.; Minghelli, S.; Di Carlo, E.; Tanghetti, E.; Albini, A.; Lowell, C.; Berton, G.; Noonan, D.M.; et al. CXCL1/macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J. Immunol. 2004, 172, 5034–5040. [Google Scholar] [CrossRef]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Hoefer, I.E.; van Royen, N.; Rectenwald, J.E.; Deindl, E.; Hua, J.; Jost, M.; Grundmann, S.; Voskuil, M.; Ozaki, C.K.; Piek, J.J.; et al. Arteriogenesis proceeds via ICAM-1/Mac-1- mediated mechanisms. Circ. Res. 2004, 94, 1179–1185. [Google Scholar] [CrossRef]
- Arnholdt, C.; Kumaraswami, K.; Götz, P.; Kübler, M.; Lasch, M.; Deindl, E. Depletion of γδ T Cells Leads to Reduced Angiogenesis and Increased Infiltration of Inflammatory M1-like Macrophages in Ischemic Muscle Tissue. Cells 2022, 11, 1490. [Google Scholar] [CrossRef]
- Kübler, M.; Beck, S.; Peffenköver, L.L.; Götz, P.; Ishikawa-Ankerhold, H.; Preissner, K.T.; Fischer, S.; Lasch, M.; Deindl, E. The Absence of Extracellular Cold-Inducible RNA-Binding Protein (eCIRP) Promotes Pro-Angiogenic Microenvironmental Conditions and Angiogenesis in Muscle Tissue Ischemia. Int. J. Mol. Sci. 2021, 22, 9484. [Google Scholar] [CrossRef]
- Götz, P.; Azubuike-Osu, S.O.; Braumandl, A.; Arnholdt, C.; Kübler, M.; Richter, L.; Lasch, M.; Bobrowski, L.; Preissner, K.T.; Deindl, E. Cobra Venom Factor Boosts Arteriogenesis in Mice. Int. J. Mol. Sci. 2022, 23, 8454. [Google Scholar] [CrossRef] [PubMed]
- Olfert, I.M.; Baum, O.; Hellsten, Y.; Egginton, S. Advances and challenges in skeletal muscle angiogenesis. Am. J. Physiol.-Heart Circ. Physiol. 2016, 310, H326–H336. [Google Scholar] [CrossRef] [PubMed]
- Deindl, E.; Ziegelhöffer, T.; Kanse, S.M.; Fernandez, B.; Neubauer, E.; Carmeliet, P.; Preissner, K.T.; Schaper, W. Receptor-independent role of the urokinase-type plasminogen activator during arteriogenesis. FASEB J. 2003, 17, 1174–1176. [Google Scholar] [CrossRef] [PubMed]
- Kübler, M.; Beck, S.; Fischer, S.; Götz, P.; Kumaraswami, K.; Ishikawa-Ankerhold, H.; Lasch, M.; Deindl, E. Absence of Cold-Inducible RNA-Binding Protein (CIRP) Promotes Angiogenesis and Regeneration of Ischemic Tissue by Inducing M2-Like Macrophage Polarization. Biomedicines 2021, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.-I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A.; et al. Perivascular Mast Cells Govern Shear Stress-Induced Arteriogenesis by Orchestrating Leukocyte Function. Cell Rep. 2016, 16, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; Daugherity, E.; Reese, B.; Karbowniczek, M. The Role of Complement in Angiogenesis. Antibodies 2020, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Kotimaa, J.P.; van Werkhoven, M.B.; O’Flynn, J.; Klar-Mohamad, N.; van Groningen, J.; Schilders, G.; Rutjes, H.; Daha, M.R.; Seelen, M.A.; van Kooten, C. Functional assessment of mouse complement pathway activities and quantification of C3b/C3c/iC3b in an experimental model of mouse renal ischaemia/reperfusion injury. J. Immunol. Methods 2015, 419, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.N.; Pavlov, V.; Takahashi, K.; Stahl, G.L.; Keppner, L.; Heinrichs, M.; Rieckmann, M.; Demengeot, J.; Frantz, S.; Hofmann, U.; et al. Myocardial ischemia and reperfusion injury is dependent on both IgM and mannose-binding lectin. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1853–H1859. [Google Scholar] [CrossRef]
- Gorsuch, W.B.; Guikema, B.J.; Fritzinger, D.C.; Vogel, C.-W.; Stahl, G.L. Humanized cobra venom factor decreases myocardial ischemia-reperfusion injury. Mol. Immunol. 2009, 47, 506–510. [Google Scholar] [CrossRef]
- Lin, Z.; Lin, H.; Li, W.; Huang, Y.; Dai, H. Complement Component C3 Promotes Cerebral Ischemia/Reperfusion Injury Mediated by TLR2/NFκB Activation in Diabetic Mice. Neurochem. Res. 2018, 43, 1599–1607. [Google Scholar] [CrossRef]
- Zhou, W.; Farrar, C.A.; Abe, K.; Pratt, J.R.; Marsh, J.E.; Wang, Y.; Stahl, G.L.; Sacks, S.H. Predominant role for C5b-9 in renal ischemia/reperfusion injury. J. Clin. Investig. 2000, 105, 1363–1371. [Google Scholar] [CrossRef]
- Moore, E.M.; West, J.L. Harnessing Macrophages for Vascularization in Tissue Engineering. Ann. Biomed. Eng. 2019, 47, 354–365. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Klos, A.; Tenner, A.J.; Johswich, K.-O.; Ager, R.R.; Reis, E.S.; Köhl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef]
- Vestweber, D. Adhesion and signaling molecules controlling the transmigration of leukocytes through endothelium. Immunol. Rev. 2007, 218, 178–196. [Google Scholar] [CrossRef]
- Homeister, J.W.; Lucchesi, B.R. Complement activation and inhibition in myocardial ischemia and reperfusion injury. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 17–40. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Dort, J.; Fabre, P.; Molina, T.; Dumont, N.A. Macrophages Are Key Regulators of Stem Cells during Skeletal Muscle Regeneration and Diseases. Stem Cells Int. 2019, 2019, 4761427. [Google Scholar] [CrossRef]
- Gurevich, D.B.; Severn, C.E.; Twomey, C.; Greenhough, A.; Cash, J.; Toye, A.M.; Mellor, H.; Martin, P. Live imaging of wound angiogenesis reveals macrophage orchestrated vessel sprouting and regression. Embo J. 2018, 37, e97786. [Google Scholar] [CrossRef] [PubMed]
- Zajac, E.; Schweighofer, B.; Kupriyanova, T.A.; Juncker-Jensen, A.; Minder, P.; Quigley, J.P.; Deryugina, E.I. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood 2013, 122, 4054–4067. [Google Scholar] [CrossRef] [PubMed]
- Asgari, E.; Le Friec, G.; Yamamoto, H.; Perucha, E.; Sacks, S.S.; Köhl, J.; Cook, H.T.; Kemper, C. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 2013, 122, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Takabayashi, T.; Vannier, E.; Clark, B.D.; Margolis, N.H.; A Dinarello, C.; Burke, J.F.; A Gelfand, J. A new biologic role for C3a and C3a desArg: Regulation of TNF-alpha and IL-1 beta synthesis. J. Immunol. 1996, 156, 3455–3460. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.W.; Fritzinger, D.C.; Hew, B.E.; Thorne, M.; Bammert, H. Recombinant cobra venom factor. Mol. Immunol. 2004, 41, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Alper, C.A.; Balavitch, D. Cobra Venom Factor: Evidence for Its Being Altered Cobra C3 (the Third Component of Complement). Science 1976, 191, 1275–1276. [Google Scholar] [CrossRef] [PubMed]
- Eggertsen, G.; Lind, P.; Sjöquist, J. Molecular characterization of the complement activating protein in the venom of the Indian cobra (Naja N. siamensis). Mol. Immunol. 1981, 18, 125–133. [Google Scholar] [CrossRef]
- Vogel, C.W.; Müller-Eberhard, H.J. The cobra complement system: I. The alternative pathway of activation. Dev. Comp. Immunol. 1985, 9, 311–325. [Google Scholar] [CrossRef]
- Cochrane, C.G.; Müller-Eberhard, H.J.; Aikin, B.S. Depletion of Plasma Complement in Vivo by a Protein of Cobra Venom: Its Effect on Various Immunologic Reactions1. J. Immunol. 1970, 105, 55–69. [Google Scholar] [CrossRef]
- Mulligan, M.S.; Schmid, E.; Beck-Schimmer, B.; O Till, G.; Friedl, H.P.; Brauer, R.B.; E Hugli, T.; Miyasaka, M.; Warner, R.L.; Johnson, K.J.; et al. Requirement and role of C5a in acute lung inflammatory injury in rats. J. Clin. Investig. 1996, 98, 503–512. [Google Scholar] [CrossRef] [PubMed]
- O Till, G.; Johnson, K.J.; Kunkel, R.; A Ward, P. Intravascular activation of complement and acute lung injury. Dependency on neutrophils and toxic oxygen metabolites. J. Clin. Investig. 1982, 69, 1126–1135. [Google Scholar]
- Fritzinger, D.C.; Dean, R.; Meschter, C.; Wong, K.; Halter, R.; Borlak, J.; John, W.D.S.; Vogel, C.-W. Complement depletion with humanized cobra venom factor in a mouse model of age-related macular degeneration. Adv. Exp. Med. Biol. 2010, 703, 151–162. [Google Scholar]
- Kerwar, S.S.; Bauman, N.; Oronsky, A.L.; Sloboda, A.E. Studies on type II collagen induced polyarthritis in rats. Effect of complement depletion. J. Immunopharmacol. 1981, 3, 323–337. [Google Scholar]
- Vogel, C.-W.; Finnegan, P.W.; Fritzinger, D.C. Humanized cobra venom factor: Structure, activity, and therapeutic efficacy in preclinical disease models. Mol. Immunol. 2014, 61, 191–203. [Google Scholar]
- Fritzinger, D.C.; Hew, B.E.; Thorne, M.; Pangburn, M.K.; Janssen, B.J.C.; Gros, P.; Vogel, C.-W. Functional characterization of human C3/cobra venom factor hybrid proteins for therapeutic complement depletion. Dev. Comp. Immunol. 2009, 33, 105–116. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azubuike-Osu, S.O.; Kuhs, A.; Götz, P.; Faro, A.; Preissner, K.T.; Arnholdt, C.; Deindl, E. Treatment with Cobra Venom Factor Decreases Ischemic Tissue Damage in Mice. Biomedicines 2024, 12, 309. https://doi.org/10.3390/biomedicines12020309
Azubuike-Osu SO, Kuhs A, Götz P, Faro A, Preissner KT, Arnholdt C, Deindl E. Treatment with Cobra Venom Factor Decreases Ischemic Tissue Damage in Mice. Biomedicines. 2024; 12(2):309. https://doi.org/10.3390/biomedicines12020309
Chicago/Turabian StyleAzubuike-Osu, Sharon O., Amelie Kuhs, Philipp Götz, Anna Faro, Klaus T. Preissner, Christoph Arnholdt, and Elisabeth Deindl. 2024. "Treatment with Cobra Venom Factor Decreases Ischemic Tissue Damage in Mice" Biomedicines 12, no. 2: 309. https://doi.org/10.3390/biomedicines12020309
APA StyleAzubuike-Osu, S. O., Kuhs, A., Götz, P., Faro, A., Preissner, K. T., Arnholdt, C., & Deindl, E. (2024). Treatment with Cobra Venom Factor Decreases Ischemic Tissue Damage in Mice. Biomedicines, 12(2), 309. https://doi.org/10.3390/biomedicines12020309