


Cutaneous Redox Senescence




Abstract
:1. Introduction
2. Aging and Senescence
3. Senescence Mechanisms
4. Skin Senescence
5. Contributors (Extrinsic and Intrinsic Factors)
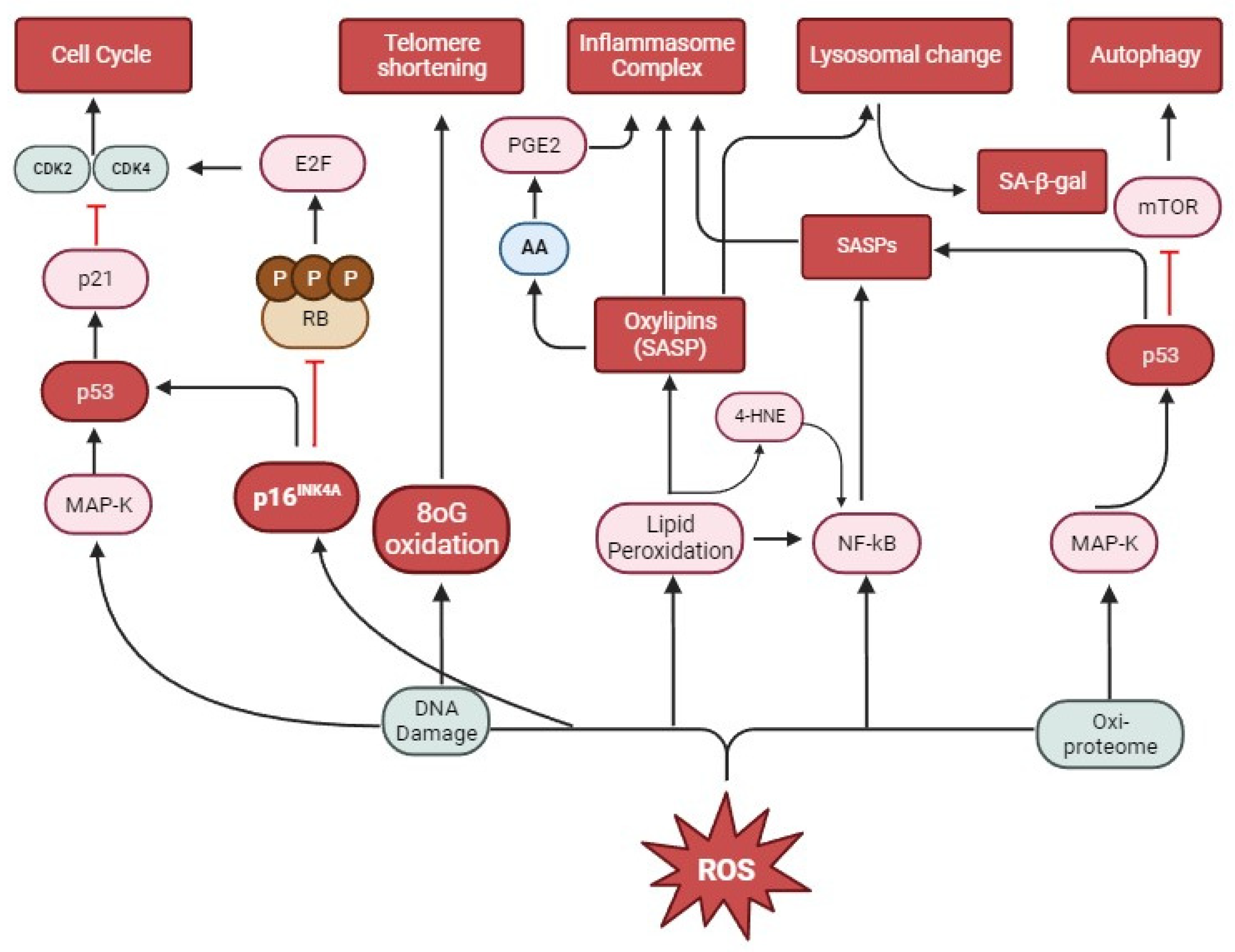
6. Oxidative Stress and Senescence
7. Oxylipins and Senescence
8. Strategies to Prevent Skin Senescence
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- de Bengy, A.-F.; Decorps, J.; Martin, L.S.; Pagnon, A.; Chevalier, F.P.; Sigaudo-Roussel, D.; Fromy, B. Alpha-Lipoic Acid Supplementation Restores Early Age-Related Sensory and Endothelial Dysfunction in the Skin. Biomedicines 2022, 10, 2887. [Google Scholar] [CrossRef]
- Csekes, E.; Račková, L. Skin Aging, Cellular Senescence and Natural Polyphenols. Int. J. Mol. Sci. 2021, 22, 12641. [Google Scholar] [CrossRef] [PubMed]
- Papaconstantinou, J. The Role of Signaling Pathways of Inflammation and Oxidative Stress in Development of Senescence and Aging Phenotypes in Cardiovascular Disease. Cells 2019, 8, 1383. [Google Scholar] [CrossRef]
- North, B.J.; Sinclair, D.A. The Intersection Between Aging and Cardiovascular Disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef]
- Merkt, W.; Bueno, M.; Mora, A.L.; Lagares, D. Senotherapeutics: Targeting Senescence in Idiopathic Pulmonary Fibrosis. Semin. Cell Dev. Biol. 2020, 101, 104–110. [Google Scholar] [CrossRef]
- Tucker-Drob, E.M. Cognitive Aging and Dementia: A Life-Span Perspective. Annu. Rev. Dev. Psychol. 2019, 1, 177–196. [Google Scholar] [CrossRef]
- Lutshumba, J.; Nikolajczyk, B.S.; Bachstetter, A.D. Dysregulation of Systemic Immunity in Aging and Dementia. Front. Cell. Neurosci. 2021, 15, 652111. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, N.; Curtis, A.J.; Heritier, S.; Gadowski, A.M.; Pavkov, M.E.; Kenealy, T.; Owens, D.R.; Thomas, R.L.; Song, S.; Wong, J.; et al. Impact of Age at Type 2 Diabetes Mellitus Diagnosis on Mortality and Vascular Complications: Systematic Review and Meta-Analyses. Diabetologia 2021, 64, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Bellary, S.; Kyrou, I.; Brown, J.E.; Bailey, C.J. Type 2 Diabetes Mellitus in Older Adults: Clinical Considerations and Management. Nat. Rev. Endocrinol. 2021, 17, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; Law, S.F.; Chandra, A. Bone Aging, Cellular Senescence, and Osteoporosis. JBMR Plus 2021, 5, e10488. [Google Scholar] [CrossRef] [PubMed]
- Nagira, K.; Ikuta, Y.; Shinohara, M.; Sanada, Y.; Omoto, T.; Kanaya, H.; Nakasa, T.; Ishikawa, M.; Adachi, N.; Miyaki, S.; et al. Histological Scoring System for Subchondral Bone Changes in Murine Models of Joint Aging and Osteoarthritis. Sci. Rep. 2020, 10, 10077. [Google Scholar] [CrossRef]
- Xia, C.; Dong, X.; Li, H.; Cao, M.; Sun, D.; He, S.; Yang, F.; Yan, X.; Zhang, S.; Li, N.; et al. Cancer Statistics in China and United States, 2022: Profiles, Trends, and Determinants. Chin. Med. J. 2022, 135, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Jammal, A.A.; Berchuck, S.I.; Thompson, A.C.; Costa, V.P.; Medeiros, F.A. The Effect of Age on Increasing Susceptibility to Retinal Nerve Fiber Layer Loss in Glaucoma. Investig. Opthalmol. Vis. Sci. 2020, 61, 8. [Google Scholar] [CrossRef] [PubMed]
- Parrado, C.; Mercado-Saenz, S.; Perez-Davo, A.; Gilaberte, Y.; Gonzalez, S.; Juarranz, A. Environmental Stressors on Skin Aging. Mechanistic Insights. Front. Pharmacol. 2019, 10, 759. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16INK4A -Positive Senescent Cells Delays Ageing-Associated Disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Sheaff, M. The Pathology of Ageing: Concepts and Mechanisms. J. Pathol. 2007, 211, 111–113. [Google Scholar] [CrossRef]
- Duan, R.; Fu, Q.; Sun, Y.; Li, Q. Epigenetic Clock: A Promising Biomarker and Practical Tool in Aging. Ageing Res. Rev. 2022, 81, 101743. [Google Scholar] [CrossRef]
- Wagner, W. The Link Between Epigenetic Clocks for Aging and Senescence. Front. Genet. 2019, 10, 303. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and Aging: Causes, Consequences, and Therapeutic Avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Valacchi, G.; Virgili, F.; Cervellati, C.; Pecorelli, A. OxInflammation: From Subclinical Condition to Pathological Biomarker. Front. Physiol. 2018, 9, 858. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Gladyshev, V.N. The Meaning of Adaptation in Aging: Insights from Cellular Senescence, Epigenetic Clocks and Stem Cell Alterations. Nat. Aging 2023, 3, 766–775. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular Senescence in Ageing: From Mechanisms to Therapeutic Opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Schmeer, C.; Kretz, A.; Wengerodt, D.; Stojiljkovic, M.; Witte, O.W. Dissecting Aging and Senescence—Current Concepts and Open Lessons. Cells 2019, 8, 1446. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [PubMed]
- UN World Population Prospects 2022: Summary of Results Ten Key Messages; United Nations, Department of Economic and Social Affairs, Population Division: New York, NY, USA, 2022; pp. 2–3.
- Krutmann, J.; Schikowski, T.; Morita, A.; Berneburg, M. Environmentally-Induced (Extrinsic) Skin Aging: Exposomal Factors and Underlying Mechanisms. J. Investig. Dermatol. 2021, 141, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Augello, F.R.; Lombardi, F.; Artone, S.; Ciafarone, A.; Altamura, S.; Di Marzio, L.; Cifone, M.G.; Palumbo, P.; Giuliani, M.; Cinque, B. Evaluation of the Effectiveness of an Innovative Polycomponent Formulation on Adult and Aged Human Dermal Fibroblasts. Biomedicines 2023, 11, 2410. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B.K.; Bishop, C.L. Current Understanding of the Role of Senescent Melanocytes in Skin Ageing. Biomedicines 2022, 10, 3111. [Google Scholar] [CrossRef]
- Bulbiankova, D.; Díaz-Puertas, R.; Álvarez-Martínez, F.J.; Herranz-López, M.; Barrajón-Catalán, E.; Micol, V. Hallmarks and Biomarkers of Skin Senescence: An Updated Review of Skin Senotherapeutics. Antioxidants 2023, 12, 444. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Wlaschek, M.; Maity, P.; Makrantonaki, E.; Scharffetter-Kochanek, K. Connective Tissue and Fibroblast Senescence in Skin Aging. J. Investig. Dermatol. 2021, 141, 985–992. [Google Scholar] [CrossRef]
- Epel, E.S.; Merkin, S.S.; Cawthon, R.; Blackburn, E.H.; Adler, N.E.; Pletcher, M.J.; Seeman, T.E. The Rate of Leukocyte Telomere Shortening Predicts Mortality from Cardiovascular Disease in Elderly Men: A Novel Demonstration. Aging 2008, 1, 81–88. [Google Scholar] [CrossRef]
- Aubert, G.; Lansdorp, P.M. Telomeres and Aging. Physiol. Rev. 2008, 88, 557–579. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.T.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small Molecule Compounds That Induce Cellular Senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A Guide to Assessing Cellular Senescence In Vitro and In Vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef]
- Itahana, K.; Campisi, J.; Dimri, G.P. Methods to Detect Biomarkers of Cellular Senescence. Methods Mol. Biol. 2007, 371, 21–31. [Google Scholar]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated Β-galactosidase Is Lysosomal Β-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Meljem, J.M.; Apps, J.R.; Fraser, H.C.; Martinez-Barbera, J.P. Paracrine Roles of Cellular Senescence in Promoting Tumourigenesis. Br. J. Cancer 2018, 118, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.I.; Choi, S.; Roh, W.S.; Lee, J.H.; Kim, T.-G. Cellular Senescence and Inflammaging in the Skin Microenvironment. Int. J. Mol. Sci. 2021, 22, 3849. [Google Scholar] [CrossRef]
- Aioi, A. Inflammaging in Skin and Intrinsic Underlying Factors. Trends Immunother. 2021, 5, 44–53. [Google Scholar] [CrossRef]
- D’Arcangelo, D.; Tinaburri, L.; Dellambra, E. The Role of p16INK4A Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer. Int. J. Mol. Sci. 2017, 18, 1591. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.C.d.O.; Costa, T.F.; Andrade, Z.d.A.; Medrado, A.R.A.P. Wound Healing—A Literature Review. An. Bras. Dermatol. 2016, 91, 614–620. [Google Scholar] [CrossRef]
- Pullar, J.; Carr, A.; Vissers, M. The Roles of Vitamin C in Skin Health. Nutrients 2017, 9, 866. [Google Scholar] [CrossRef]
- Ho, C.Y.; Dreesen, O. Faces of Cellular Senescence in Skin Aging. Mech. Ageing Dev. 2021, 198, 111525. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Y.; Zhao, Z.; Qiu, J. Oxidative Stress in the Skin: Impact and Related Protection. Int. J. Cosmet. Sci. 2021, 43, 495–509. [Google Scholar] [CrossRef]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent Human Melanocytes Drive Skin Ageing via Paracrine Telomere Dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef]
- Krunic, D.; Moshir, S.; Greulich-Bode, K.M.; Figueroa, R.; Cerezo, A.; Stammer, H.; Stark, H.-J.; Gray, S.G.; Nielsen, K.V.; Hartschuh, W.; et al. Tissue Context-Activated Telomerase in Human Epidermis Correlates with Little Age-Dependent Telomere Loss. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2009, 1792, 297–308. [Google Scholar] [CrossRef]
- Low, E.; Alimohammadiha, G.; Smith, L.A.; Costello, L.F.; Przyborski, S.A.; von Zglinicki, T.; Miwa, S. How Good Is the Evidence That Cellular Senescence Causes Skin Ageing? Ageing Res. Rev. 2021, 71, 101456. [Google Scholar] [CrossRef]
- Laethem, A.V.; Claerhout, S.; Garmyn, M.; Agostinis, P. The Sunburn Cell: Regulation of Death and Survival of the Keratinocyte. Int. J. Biochem. Cell Biol. 2005, 37, 1547–1553. [Google Scholar] [CrossRef]
- Velarde, M.C.; Flynn, J.M.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial Oxidative Stress Caused by Sod2 Deficiency Promotes Cellular Senescence and Aging Phenotypes in the Skin. Aging 2012, 4, 3–12. [Google Scholar] [CrossRef]
- Rübe, C.E.; Bäumert, C.; Schuler, N.; Isermann, A.; Schmal, Z.; Glanemann, M.; Mann, C.; Scherthan, H. Human Skin Aging Is Associated with Increased Expression of the Histone Variant H2A.J in the Epidermis. NPJ Aging Mech. Dis. 2021, 7, 7. [Google Scholar] [CrossRef]
- Jenkins, N.C.; Grossman, D. Role of Melanin in Melanocyte Dysregulation of Reactive Oxygen Species. BioMed Res. Int. 2013, 2013, 908797. [Google Scholar] [CrossRef]
- Jenkins, N.C.; Liu, T.; Cassidy, P.; Leachman, S.A.; Boucher, K.M.; Goodson, A.G.; Samadashwily, G.; Grossman, D. The p16INK4A Tumor Suppressor Regulates Cellular Oxidative Stress. Oncogene 2011, 30, 265–274. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, M.; Lemma, T.; Calcagnile, A.; De Santis, L.P.; Dogliotti, E. Cell Type and DNA Damage Specific Response of Human Skin Cells to Environmental Agents. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2007, 614, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Tigges, J.; Krutmann, J.; Fritsche, E.; Haendeler, J.; Schaal, H.; Fischer, J.W.; Kalfalah, F.; Reinke, H.; Reifenberger, G.; Stühler, K.; et al. The Hallmarks of Fibroblast Ageing. Mech. Ageing Dev. 2014, 138, 26–44. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Gruber, F.; Kremslehner, C.; Eckhart, L.; Tschachler, E. Cell Aging and Cellular Senescence in Skin Aging—Recent Advances in Fibroblast and Keratinocyte Biology. Exp. Gerontol. 2020, 130, 110780. [Google Scholar] [CrossRef]
- Chung, C.L.; Lawrence, I.; Hoffman, M.; Elgindi, D.; Nadhan, K.; Potnis, M.; Jin, A.; Sershon, C.; Binnebose, R.; Lorenzini, A.; et al. Topical Rapamycin Reduces Markers of Senescence and Aging in Human Skin: An Exploratory, Prospective, Randomized Trial. GeroScience 2019, 41, 861–869. [Google Scholar] [CrossRef]
- McDaniel, D.; Farris, P.; Valacchi, G. Atmospheric Skin Aging—Contributors and Inhibitors. J. Cosmet. Dermatol. 2018, 17, 124–137. [Google Scholar] [CrossRef]
- Idda, M.L.; McClusky, W.G.; Lodde, V.; Munk, R.; Abdelmohsen, K.; Rossi, M.; Gorospe, M. Survey of Senescent Cell Markers with Age in Human Tissues. Aging 2020, 12, 4052–4066. [Google Scholar] [CrossRef]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Characteristics of the Aging Skin. Adv. Wound Care 2013, 2, 5–10. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in Vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Ressler, S.; Bartkova, J.; Niederegger, H.; Bartek, J.; Scharffetter-Kochanek, K.; Jansen-Dürr, P.; Wlaschek, M. P16 INK4A Is a Robust in Vivo Biomarker of Cellular Aging in Human Skin. Aging Cell 2006, 5, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, K.A.U.; Fuchs, E. Skin and Its Regenerative Powers: An Alliance between Stem Cells and Their Niche. Dev. Cell 2017, 43, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Kim, M. Skin Barrier Function and the Microbiome. Int. J. Mol. Sci. 2022, 23, 13071. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-Y.; Chang, K.-W.; Liu, C.-J.; Tseng, Y.-H.; Lu, H.-H.; Lee, S.-Y.; Lin, S.-C. Ripe Areca Nut Extract Induces G 1 Phase Arrests and Senescence-Associated Phenotypes in Normal Human Oral Keratinocyte. Carcinogenesis 2006, 27, 1273–1284. [Google Scholar] [CrossRef]
- Suzuki, Y.; Takaya, K.; Watanabe, S.; Otaki, M.; Kono, H.; Kishi, K. Evaluation of the Effect of Age of the Younger Mice on the Rejuvenation of the Older Mice by Heterochronic Parabiosis. Aging 2022, 14, 2507–2512. [Google Scholar] [CrossRef]
- Kim, J.C.; Park, T.J.; Kang, H.Y. Skin-Aging Pigmentation: Who Is the Real Enemy? Cells 2022, 11, 2541. [Google Scholar] [CrossRef]
- Martic, I.; Wedel, S.; Jansen-Dürr, P.; Cavinato, M. A New Model to Investigate UVB-Induced Cellular Senescence and Pigmentation in Melanocytes. Mech. Ageing Dev. 2020, 190, 111322. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.M. Melanin Accumulation Accelerates Melanocyte Senescence by a Mechanism Involving P16 INK4a /CDK4/PRB and E2F1. Ann. N. Y. Acad. Sci. 2000, 908, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.S.; Dreesen, O. Biomarkers of Cellular Senescence and Skin Aging. Front. Genet. 2018, 9, 247. [Google Scholar] [CrossRef]
- Singh, K.; Maity, P.; Krug, L.; Meyer, P.; Treiber, N.; Lucas, T.; Basu, A.; Kochanek, S.; Wlaschek, M.; Geiger, H.; et al. Superoxide Anion Radicals Induce IGF-1 Resistance through Concomitant Activation of PTP1B and PTEN. EMBO Mol. Med. 2015, 7, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, D.; Bastonini, E.; Ottaviani, M.; Cota, C.; Migliano, E.; Dell’Anna, M.L.; Picardo, M. Vitiligo Skin: Exploring the Dermal Compartment. J. Investig. Dermatol. 2018, 138, 394–404. [Google Scholar] [CrossRef]
- Ciuciulete, A.R.; Stepan, A.E.; Badiu, A.M.; Andreiana, B.C.; Florescu, M.M.; Simionescu, C.E.; Vîlcea, A.M. E-Cadherin, Fibronectin and Slug Immunoexpression in Non-Melanoma Skin Cancers. Rom. J. Morphol. Embryol. 2021, 62, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Malaquin, N.; Vercamer, C.; Bouali, F.; Martien, S.; Deruy, E.; Wernert, N.; Chwastyniak, M.; Pinet, F.; Abbadie, C.; Pourtier, A. Senescent Fibroblasts Enhance Early Skin Carcinogenic Events via a Paracrine MMP-PAR-1 Axis. PLoS ONE 2013, 8, e63607. [Google Scholar] [CrossRef]
- Quan, T.; Fisher, G.J. Role of Age-Associated Alterations of the Dermal Extracellular Matrix Microenvironment in Human Skin Aging: A Mini-Review. Gerontology 2015, 61, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Fuks, K.B.; Woodby, B.; Valacchi, G. Skin Damage by Tropospheric Ozone. Der Hautarzt 2019, 70, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Souyoul, S.A.; Saussy, K.P.; Lupo, M.P. Nutraceuticals: A Review. Dermatol. Ther. 2018, 8, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Proksch, E. PH in Nature, Humans and Skin. J. Dermatol. 2018, 45, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Petracca, B.; Rothen-Rutishauser, B.; Valacchi, G.; Eeman, M. Bench Approaches to Study the Detrimental Cutaneous Impact of Tropospheric Ozone. J. Exp. Sci. Environ. Epidemiol. 2021, 31, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Holden, K.A.; Lee, A.R.; Hawcutt, D.B.; Sinha, I.P. The Impact of Poor Housing and Indoor Air Quality on Respiratory Health in Children. Breathe 2023, 19, 230058. [Google Scholar] [CrossRef] [PubMed]
- Farris, P.K.; Valacchi, G. Ultraviolet Light Protection: Is It Really Enough? Antioxidants 2022, 11, 1484. [Google Scholar] [CrossRef]
- Valacchi, G.; Sticozzi, C.; Pecorelli, A.; Cervellati, F.; Cervellati, C.; Maioli, E. Cutaneous Responses to Environmental Stressors. Ann. N. Y. Acad. Sci. 2012, 1271, 75–81. [Google Scholar] [CrossRef]
- Gerasymchuk, M.; Cherkasova, V.; Kovalchuk, O.; Kovalchuk, I. The Role of MicroRNAs in Organismal and Skin Aging. Int. J. Mol. Sci. 2020, 21, 5281. [Google Scholar] [CrossRef]
- Bauwens, E.; Parée, T.; Meurant, S.; Bouriez, I.; Hannart, C.; Wéra, A.-C.; Khelfi, A.; Fattaccioli, A.; Burteau, S.; Demazy, C.; et al. Senescence Induced by UVB in Keratinocytes Impairs Amino Acids Balance. J. Investig. Dermatol. 2023, 143, 554–565.e9. [Google Scholar] [CrossRef]
- Miyazawa, T.; Abe, C.; Burdeos, G.C.; Matsumoto, A.; Toda, M. Food Antioxidants and Aging: Theory, Current Evidence and Perspectives. Nutraceuticals 2022, 2, 181–204. [Google Scholar] [CrossRef]
- Cao, C.; Xiao, Z.; Wu, Y.; Ge, C. Diet and Skin Aging—From the Perspective of Food Nutrition. Nutrients 2020, 12, 870. [Google Scholar] [CrossRef]
- McMillan, T.J.; Leatherman, E.; Ridley, A.; Shorrocks, J.; Tobi, S.E.; Whiteside, J.R. Cellular Effects of Long Wavelength UV Light (UVA) in Mammalian Cells. J. Pharm. Pharmacol. 2010, 60, 969–976. [Google Scholar] [CrossRef]
- Scharffetter–Kochanek, K.; Brenneisen, P.; Wenk, J.; Herrmann, G.; Ma, W.; Kuhr, L.; Meewes, C.; Wlaschek, M. Photoaging of the Skin from Phenotype to Mechanisms. Exp. Gerontol. 2000, 35, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Fitsiou, E.; Pulido, T.; Campisi, J.; Alimirah, F.; Demaria, M. Cellular Senescence and the Senescence-Associated Secretory Phenotype as Drivers of Skin Photoaging. J. Investig. Dermatol. 2021, 141, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Kammeyer, A.; Luiten, R.M. Oxidation Events and Skin Aging. Ageing Res. Rev. 2015, 21, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Valacchi, G.; Fortino, V.; Bocci, V. The Dual Action of Ozone on the Skin. Br. J. Dermatol. 2005, 153, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Valacchi, G.; Sticozzi, C.; Belmonte, G.; Cervellati, F.; Demaude, J.; Chen, N.; Krol, Y.; Oresajo, C. Vitamin C Compound Mixtures Prevent Ozone-Induced Oxidative Damage in Human Keratinocytes as Initial Assessment of Pollution Protection. PLoS ONE 2015, 10, e0131097. [Google Scholar] [CrossRef]
- Kafoury, R.M.; Pryor, W.A.; Squadrito, G.L.; Salgo, M.G.; Zou, X.; Friedman, M. Lipid Ozonation Products Activate Phospholipases A2, C, and D. Toxicol. Appl. Pharmacol. 1998, 150, 338–349. [Google Scholar] [CrossRef]
- Reynolds, W.J.; Eje, N.; Christensen, P.; Li, W.; Daly, S.M.; Parsa, R.; Chavan, B.; Birch-Machin, M.A. Biological Effects of Air Pollution on the Function of Human Skin Equivalents. FASEB BioAdvances 2023, 5, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Zdanov, S.; Bernard, D.; Debacqchainiaux, F.; Martien, S.; Gosselin, K.; Vercamer, C.; Chelli, F.; Toussaint, O.; Abbadie, C. Normal or Stress-Induced Fibroblast Senescence Involves COX-2 Activity. Exp. Cell Res. 2007, 313, 3046–3056. [Google Scholar] [CrossRef] [PubMed]
- Fuller, B. Role of PGE-2 and Other Inflammatory Mediators in Skin Aging and Their Inhibition by Topical Natural Anti-Inflammatories. Cosmetics 2019, 6, 6. [Google Scholar] [CrossRef]
- Karen-Ng, L.P.; Ahmad, U.S.; Gomes, L.; Hunter, K.D.; Wan, H.; Hagi-Pavli, E.; Parkinson, E.K. Extracellular Prostaglandins E1 and E2 and Inflammatory Cytokines Are Regulated by the Senescence Program in Potentially Premalignant Oral Keratinocytes. Cancers 2022, 14, 2636. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Sharma, R.; Davis, S.S.; Lopez-Dominguez, J.A.; Mitchell, K.P.; Wiley, S.; Alimirah, F.; Kim, D.E.; Payne, T.; Rosko, A.; et al. Oxylipin Biosynthesis Reinforces Cellular Senescence and Allows Detection of Senolysis. Cell Metab. 2021, 33, 1124–1136.e5. [Google Scholar] [CrossRef]
- Packer, L.; Valacchi, G. Antioxidants and the Response of Skin to Oxidative Stress: Vitamin E as a Key Indicator. Ski. Pharmacol. Physiol. 2002, 15, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F.; Pambianchi, E.; Woodby, B.; Messano, N.; Therrien, J.-P.; Pecorelli, A.; Canella, R.; Valacchi, G. Evaluating the Effect of Ozone in UV Induced Skin Damage. Toxicol. Lett. 2021, 338, 40–50. [Google Scholar] [CrossRef]
- Xu, F.; Yan, S.; Wu, M.; Li, F.; Xu, X.; Song, W.; Zhao, J.; Xu, J.; Kan, H. Ambient Ozone Pollution as a Risk Factor for Skin Disorders. Br. J. Dermatol. 2011, 165, 224–225. [Google Scholar] [CrossRef]
- Valacchi, G.; Pecorelli, A.; Belmonte, G.; Pambianchi, E.; Cervellati, F.; Lynch, S.; Krol, Y.; Oresajo, C. Protective Effects of Topical Vitamin C Compound Mixtures against Ozone-Induced Damage in Human Skin. J. Investig. Dermatol. 2017, 137, 1373–1375. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Kang, K.A.; Piao, M.J.; Ahn, M.J.; Yi, J.M.; Bossis, G.; Hyun, Y.-M.; Park, C.O.; Hyun, J.W. Particulate Matter-Induced Senescence of Skin Keratinocytes Involves Oxidative Stress-Dependent Epigenetic Modifications. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Yang, G.; Zhang, C.; Liu, X.; Qian, G.; Deng, D. Effects of Cigarette Smoke Extracts on the Growth and Senescence of Skin Fibroblasts In Vitro. Int. J. Biol. Sci. 2013, 9, 613–623. [Google Scholar] [CrossRef]
- Arredondo, J.; Hall, L.L.; Ndoye, A.; Nguyen, V.T.; Chernyavsky, A.I.; Bercovich, D.; Orr-Urtreger, A.; Beaudet, A.L.; Grando, S.A. Central Role of Fibroblast A3 Nicotinic Acetylcholine Receptor in Mediating Cutaneous Effects of Nicotine. Lab. Investig. 2003, 83, 207–225. [Google Scholar] [CrossRef]
- Murakami, T.; Inagaki, N.; Kondoh, H. Cellular Senescence in Diabetes Mellitus: Distinct Senotherapeutic Strategies for Adipose Tissue and Pancreatic β Cells. Front. Endocrinol. 2022, 13, 869414. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Prattichizzo, F.; Grillari, J.; Balistreri, C.R. Cellular Senescence and Inflammaging in Age-Related Diseases. Mediat. Inflamm. 2018, 2018, 9076485. [Google Scholar] [CrossRef]
- Keaney, T.C. Aging in the Male Face: Intrinsic and Extrinsic Factors. Dermatol. Surg. 2016, 42, 797–803. [Google Scholar] [CrossRef]
- Bellei, B.; Picardo, M. Premature Cell Senescence in Human Skin: Dual Face in Chronic Acquired Pigmentary Disorders. Ageing Res. Rev. 2020, 57, 100981. [Google Scholar] [CrossRef] [PubMed]
- Reilly, D.M.; Lozano, J. Skin Collagen through the Lifestages: Importance for Skin Health and Beauty. Plast. Aesthetic Res. 2021, 2021, 2347–9264. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free Radicals and Antioxidants in Normal Physiological Functions and Human Disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Sörgel, C.A.; Cai, A.; Schmid, R.; Horch, R.E. Perspectives on the Current State of Bioprinted Skin Substitutes for Wound Healing. Biomedicines 2023, 11, 2678. [Google Scholar] [CrossRef]
- Smolina, N.; Bruton, J.; Kostareva, A.; Sejersen, T. Assaying Mitochondrial Respiration as an Indicator of Cellular Metabolism and Fitness. Cell Viability Assays Methods Protoc. 2017, 1601, 79–87. [Google Scholar]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Pitocco, D.; Zaccardi, F.; Di Stasio, E.; Romitelli, F.; Santini, S.A.; Zuppi, C.; Ghirlanda, G. Oxidative Stress, Nitric Oxide, and Diabetes. Rev. Diabet. Stud. RDS 2010, 7, 15–25. [Google Scholar] [CrossRef]
- Hebbar, S.; Knust, E. Reactive Oxygen Species (ROS) Constitute an Additional Player in Regulating Epithelial Development. BioEssays 2021, 43, 2100096. [Google Scholar] [CrossRef] [PubMed]
- Poljšak, B.; Dahmane, R. Free Radicals and Extrinsic Skin Aging. Dermatol. Res. Pract. 2012, 2012, 135206. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Azzi, A.; Zingg, J. Reduction of Senescence-associated Beta-galactosidase Activity by Vitamin E in Human Fibroblasts Depends on Subjects’ Age and Cell Passage Number. BioFactors 2020, 46, 665–674. [Google Scholar] [CrossRef]
- Younus, H. Therapeutic Potentials of Superoxide Dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Pérez-Cano, F.; Castell, M. Flavonoids, Inflammation and Immune System. Nutrients 2016, 8, 659. [Google Scholar] [CrossRef] [PubMed]
- Shammas, M.A. Telomeres, Lifestyle, Cancer, and Aging. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Karran, P.; Brem, R. Protein Oxidation, UVA and Human DNA Repair. DNA Repair 2016, 44, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Hamon, M.; Ahmed, E.K.; Baraibar, M.A.; Friguet, B. Proteome Oxidative Modifications and Impairment of Specific Metabolic Pathways During Cellular Senescence and Aging. Proteomics 2020, 20, 421. [Google Scholar] [CrossRef]
- Garcia-Peterson, L.M.; Wilking-Busch, M.J.; Ndiaye, M.A.; Philippe, C.G.A.; Setaluri, V.; Ahmad, N. Sirtuins in Skin and Skin Cancers. Ski. Pharmacol. Physiol. 2017, 30, 216–224. [Google Scholar] [CrossRef]
- Morselli, E.; Mariño, G.; Bennetzen, M.V.; Eisenberg, T.; Megalou, E.; Schroeder, S.; Cabrera, S.; Bénit, P.; Rustin, P.; Criollo, A.; et al. Spermidine and Resveratrol Induce Autophagy by Distinct Pathways Converging on the Acetylproteome. J. Cell Biol. 2011, 192, 615–629. [Google Scholar] [CrossRef]
- Lin, J.; Epel, E. Stress and Telomere Shortening: Insights from Cellular Mechanisms. Ageing Res. Rev. 2022, 73, 101507. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-F.; Ou, C.-C.; Chien, P.-J.; Chang, H.-Y.; Ko, J.-L.; Wang, B.-Y. Chidamide-Induced ROS Accumulation and MiR-129-3p-Dependent Cell Cycle Arrest in Non-Small Lung Cancer Cells. Phytomedicine 2019, 56, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef]
- Akatli, A.N.; Ayva, E.S.; Bozdogan, O. P16 INK4a, and P14 ARF Expressions in Carcinogenesis of Squamous Cell Carcinoma of the Lip. Clin. Cancer Investig. J. 2022, 11, 1–8. [Google Scholar] [CrossRef]
- Ming, Z.; Lim, S.Y.; Rizos, H. Genetic Alterations in the INK4a/ARF Locus: Effects on Melanoma Development and Progression. Biomolecules 2020, 10, 1447. [Google Scholar] [CrossRef]
- Lagopati, N.; Belogiannis, K.; Angelopoulou, A.; Papaspyropoulos, A.; Gorgoulis, V. Non-Canonical Functions of the ARF Tumor Suppressor in Development and Tumorigenesis. Biomolecules 2021, 11, 86. [Google Scholar] [CrossRef]
- Young, A.R.; Narita, M. Connecting Autophagy to Senescence in Pathophysiology. Curr. Opin. Cell Biol. 2010, 22, 234–240. [Google Scholar] [CrossRef]
- Safwan-Zaiter, H.; Wagner, N.; Wagner, K.-D. p16INK4A—More Than a Senescence Marker. Life 2022, 12, 1332. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria Are Required for Pro-ageing Features of the Senescent Phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Kurz, E.U.; Lees-Miller, S.P. DNA Damage-Induced Activation of ATM and ATM-Dependent Signaling Pathways. DNA Repair 2004, 3, 889–900. [Google Scholar] [CrossRef]
- Song, S.B.; Hwang, E.S. High Levels of ROS Impair Lysosomal Acidity and Autophagy Flux in Glucose-Deprived Fibroblasts by Activating ATM and Erk Pathways. Biomolecules 2020, 10, 761. [Google Scholar] [CrossRef] [PubMed]
- Pezone, A.; Olivieri, F.; Napoli, M.V.; Procopio, A.; Avvedimento, E.V.; Gabrielli, A. Inflammation and DNA Damage: Cause, Effect or Both. Nat. Rev. Rheumatol. 2023, 19, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Vallese, A.; Cordone, V.; Pecorelli, A.; Valacchi, G. Ox-Inflammasome Involvement in Neuroinflammation. Free Radic. Biol. Med. 2023, 207, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef]
- Ferrara, F.; Prieux, R.; Woodby, B.; Valacchi, G. Inflammasome Activation in Pollution-Induced Skin Conditions. Plast. Reconstr. Surg. 2021, 147, 15S–24S. [Google Scholar] [CrossRef]
- Pecorelli, A.; Woodby, B.; Prieux, R.; Valacchi, G. Involvement of 4-hydroxy-2-nonenal in Pollution-induced Skin Damage. BioFactors 2019, 45, 536–547. [Google Scholar] [CrossRef]
- Martic, I.; Jansen-Dürr, P.; Cavinato, M. Effects of Air Pollution on Cellular Senescence and Skin Aging. Cells 2022, 11, 2220. [Google Scholar] [CrossRef]
- Gruber, F.; Kremslehner, C.; Narzt, M.-S. The Impact of Recent Advances in Lipidomics and Redox Lipidomics on Dermatological Research. Free Radic. Biol. Med. 2019, 144, 256–265. [Google Scholar] [CrossRef]
- Jia, Y.; Gan, Y.; He, C.; Chen, Z.; Zhou, C. The Mechanism of Skin Lipids Influencing Skin Status. J. Dermatol. Sci. 2018, 89, 112–119. [Google Scholar] [CrossRef]
- Feingold, K.R. The Importance of Lipids in Cutaneous Function. J. Lipid Res. 2007, 48, 2529–2530. [Google Scholar] [CrossRef] [PubMed]
- Nugteren, D.H.; Kivits, G.A.A. Conversion of Linoleic Acid and Arachidonic Acid by Skin Epidermal Lipoxygenases. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1987, 921, 135–141. [Google Scholar] [CrossRef]
- Nugteren, D.H.; Christ-Hazelhof, E.; van der Beek, A.; Houtsmuller, U.M.T. Metabolism of Linoleic Acid and Other Essential Fatty Acids in the Epidermis of the Rat. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1985, 834, 429–436. [Google Scholar] [CrossRef]
- Krieg, P.; Fürstenberger, G. The Role of Lipoxygenases in Epidermis. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 390–400. [Google Scholar] [CrossRef]
- Ni, Z.; Goracci, L.; Cruciani, G.; Fedorova, M. Computational Solutions in Redox Lipidomics—Current Strategies and Future Perspectives. Free Radic. Biol. Med. 2019, 144, 110–123. [Google Scholar] [CrossRef]
- Flor, A.C.; Kron, S.J. Lipid-Derived Reactive Aldehydes Link Oxidative Stress to Cell Senescence. Cell Death Dis. 2016, 7, e2366. [Google Scholar] [CrossRef]
- Wolf, G. Lipofuscin, the Age Pigment. Nutr. Rev. 2009, 51, 205–206. [Google Scholar] [CrossRef]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Cigarette Smoke Induces Senescence in Alveolar Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 643–649. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy Maintains Stemness by Preventing Senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Hamsanathan, S.; Gurkar, A.U. Lipids as Regulators of Cellular Senescence. Front. Physiol. 2022, 13, 796850. [Google Scholar] [CrossRef]
- Narzt, M.-S.; Pils, V.; Kremslehner, C.; Nagelreiter, I.-M.; Schosserer, M.; Bessonova, E.; Bayer, A.; Reifschneider, R.; Terlecki-Zaniewicz, L.; Waidhofer-Söllner, P.; et al. Epilipidomics of Senescent Dermal Fibroblasts Identify Lysophosphatidylcholines as Pleiotropic Senescence-Associated Secretory Phenotype (SASP) Factors. J. Investig. Dermatol. 2021, 141, 993–1006.e15. [Google Scholar] [CrossRef]
- Yang, H.H.; Kim, C.; Jung, B.; Kim, K.S.; Kim, J.-R. Involvement of IGF Binding Protein 5 in Prostaglandin E2-Induced Cellular Senescence in Human Fibroblasts. Biogerontology 2011, 12, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Brumwell, A.N.; Davis, S.S.; Jackson, J.R.; Valdovinos, A.; Calhoun, C.; Alimirah, F.; Castellanos, C.A.; Ruan, R.; Wei, Y.; et al. Secretion of Leukotrienes by Senescent Lung Fibroblasts Promotes Pulmonary Fibrosis. JCI Insight 2019, 4, e130056. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, Z. Fibroblast Senescence in Idiopathic Pulmonary Fibrosis. Front. Cell Dev. Biol. 2020, 8, 593283. [Google Scholar] [CrossRef]
- Flor, A.C.; Wolfgeher, D.; Wu, D.; Kron, S.J. A Signature of Enhanced Lipid Metabolism, Lipid Peroxidation and Aldehyde Stress in Therapy-Induced Senescence. Cell Death Discov. 2017, 3, 17075. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, R. Senotherapy: Growing Old and Staying Young? Pflügers Arch. Eur. J. Physiol. 2017, 469, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting Senescence for the Treatment of Cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Velarde, M.C.; Demaria, M. Targeting Senescent Cells: Possible Implications for Delaying Skin Aging: A Mini-Review. Gerontology 2016, 62, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Domaszewska-Szostek, A.; Puzianowska-Kuźnicka, M.; Kuryłowicz, A. Flavonoids in Skin Senescence Prevention and Treatment. Int. J. Mol. Sci. 2021, 22, 6814. [Google Scholar] [CrossRef]
- Lim, H.; Park, H.; Kim, H.P. Effects of Flavonoids on Senescence-Associated Secretory Phenotype Formation from Bleomycin-Induced Senescence in BJ Fibroblasts. Biochem. Pharmacol. 2015, 96, 337–348. [Google Scholar] [CrossRef]
- Laberge, R.; Zhou, L.; Sarantos, M.R.; Rodier, F.; Freund, A.; de Keizer, P.L.J.; Liu, S.; Demaria, M.; Cong, Y.; Kapahi, P.; et al. Glucocorticoids Suppress Selected Components of the Senescence-associated Secretory Phenotype. Aging Cell 2012, 11, 569–578. [Google Scholar] [CrossRef]
- Laberge, R.-M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR Regulates the Pro-Tumorigenic Senescence-Associated Secretory Phenotype by Promoting IL1A Translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Ido, Y.; Duranton, A.; Lan, F.; Weikel, K.A.; Breton, L.; Ruderman, N.B. Resveratrol Prevents Oxidative Stress-Induced Senescence and Proliferative Dysfunction by Activating the AMPK-FOXO3 Cascade in Cultured Primary Human Keratinocytes. PLoS ONE 2015, 10, e0115341. [Google Scholar] [CrossRef]
- Kilic Eren, M.; Kilincli, A.; Eren, Ö. Resveratrol Induced Premature Senescence Is Associated with DNA Damage Mediated SIRT1 and SIRT2 Down-Regulation. PLoS ONE 2015, 10, e0124837. [Google Scholar] [CrossRef] [PubMed]
- Faragher, R.G.A.; Burton, D.G.A.; Majecha, P.; Fong, N.S.Y.; Davis, T.; Sheerin, A.; Ostler, E.L. Resveratrol, but Not Dihydroresveratrol, Induces Premature Senescence in Primary Human Fibroblasts. AGE 2011, 33, 555–564. [Google Scholar] [CrossRef]
- Alimbetov, D.; Davis, T.; Brook, A.J.C.; Cox, L.S.; Faragher, R.G.A.; Nurgozhin, T.; Zhumadilov, Z.; Kipling, D. Suppression of the Senescence-Associated Secretory Phenotype (SASP) in Human Fibroblasts Using Small Molecule Inhibitors of P38 MAP Kinase and MK2. Biogerontology 2016, 17, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.J.; Kim, K.B.; Bae, S.; Choi, B.G.; An, S.; Ahn, K.J.; Kim, S.Y. Pretreatment of Ferulic Acid Protects Human Dermal Fibroblasts against Ultraviolet A Irradiation. Ann. Dermatol. 2016, 28, 740. [Google Scholar] [CrossRef]
- Sohn, E.-J.; Kim, J.M.; Kang, S.-H.; Kwon, J.; An, H.J.; Sung, J.-S.; Cho, K.A.; Jang, I.-S.; Choi, J.-S. Restoring Effects of Natural Anti-Oxidant Quercetin on Cellular Senescent Human Dermal Fibroblasts. Am. J. Chin. Med. 2018, 46, 853–873. [Google Scholar] [CrossRef]
- Jang, H.-J.; Yang, K.E.; Oh, W.K.; Lee, S.-I.; Hwang, I.-H.; Ban, K.-T.; Yoo, H.-S.; Choi, J.-S.; Yeo, E.-J.; Jang, I.-S. Nectandrin B-Mediated Activation of the AMPK Pathway Prevents Cellular Senescence in Human Diploid Fibroblasts by Reducing Intracellular ROS Levels. Aging 2019, 11, 3731–3749. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Jia, L.; Wang, X.; Liu, T.; Qin, W.; Ma, H.; Lv, Y.; Hu, J.; Guo, Q.; Tan, S.; et al. Anti-Aging Formula Protects Skin from Oxidative Stress-Induced Senescence through the Inhibition of CXCR2 Expression. J. Ethnopharmacol. 2024, 318, 116996. [Google Scholar] [CrossRef]
- Tian, L.-M.; Peng, Y.; Ke, D.; Li, H.; Chen, L.; Zhang, C.; Sen, L.; Tian, D.-Z.; Zhou, M.-S.; Ai, X.-S.; et al. The Effect of Yang Yan Qing E Wan on Senescent Phenotypes and the Expression of β-Catenin and p16INK4A in Human Skin Fibroblasts. J. Tissue Viability 2020, 29, 354–358. [Google Scholar] [CrossRef]
- Song, X.; Narzt, M.S.; Nagelreiter, I.M.; Hohensinner, P.; Terlecki-Zaniewicz, L.; Tschachler, E.; Grillari, J.; Gruber, F. Autophagy Deficient Keratinocytes Display Increased DNA Damage, Senescence and Aberrant Lipid Composition after Oxidative Stress In Vitro and In Vivo. Redox Biol. 2017, 11, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F.; Ouyang, S.-H.; Tu, L.-F.; Wang, X.; Yuan, W.-L.; Wang, G.-E.; Wu, Y.-P.; Duan, W.-J.; Yu, H.-M.; Fang, Z.-Z.; et al. Caffeine Protects Skin from Oxidative Stress-Induced Senescence through the Activation of Autophagy. Theranostics 2018, 8, 5713–5730. [Google Scholar] [CrossRef]
- Kvam, E.; Dahle, J. Pigmented Melanocytes Are Protected Against Ultraviolet-A-Induced Membrane Damage. J. Investig. Dermatol. 2003, 121, 564–569. [Google Scholar] [CrossRef]
- Handbook of Diet, Nutrition and the Skin; Preedy, V.R. (Ed.) Brill|Wageningen Academic: Wageningen, The Netherlands, 2012; ISBN 9789086867295. [Google Scholar]
- Evans, J.R.; Lawrenson, J.G. Antioxidant Vitamin and Mineral Supplements for Slowing the Progression of Age-Related Macular Degeneration. Cochrane Database Syst. Rev. 2017, 2017, CD000254. [Google Scholar] [CrossRef]
- Su, J.; Su, Q.; Hu, S.; Ruan, X.; Ouyang, S. Research Progress on the Anti-Aging Potential of the Active Components of Ginseng. Nutrients 2023, 15, 3286. [Google Scholar] [CrossRef] [PubMed]
- Son, H.-U.; Choi, H.-J.; Alam, M.B.; Jeong, C.G.; Lee, H.I.; Kim, S.L.; Zhao, P.; Kim, T.-H.; Lee, S.-H. Prunus Mume Seed Exhibits Inhibitory Effect on Skin Senescence via SIRT1 and MMP-1 Regulation. Oxidative Med. Cell. Longev. 2021, 2021, 5528795. [Google Scholar] [CrossRef] [PubMed]
- Boyajian, J.L.; Ghebretatios, M.; Schaly, S.; Islam, P.; Prakash, S. Microbiome and Human Aging: Probiotic and Prebiotic Potentials in Longevity, Skin Health and Cellular Senescence. Nutrients 2021, 13, 4550. [Google Scholar] [CrossRef]
- Schagen, S.K.; Zampeli, V.A.; Makrantonaki, E.; Zouboulis, C.C. Discovering the Link between Nutrition and Skin Aging. Derm. Endocrinol. 2012, 4, 298–307. [Google Scholar] [CrossRef]
- Gašperlin, M.; Gosenca, M. Main Approaches for Delivering Antioxidant Vitamins through the Skin to Prevent Skin Ageing. Expert Opin. Drug Deliv. 2011, 8, 905–919. [Google Scholar] [CrossRef]


{kind=link}
| References | Skin Cell | Main Senescence Markers | Senotherapeutic Strategy | Intervention Origin | Main Senotherapeutic Target | Mechanism of Action |
|---|---|---|---|---|---|---|
| [171] | Human fibroblast | BrdU and SA β-gal | Flavonoids (apigenin and kaempferol) | Synthetic | Reduced SASP inhibited NF-κB p65 activity | Reduced inflammation |
| [172] | Human fibroblast | BrdU and SA β-gal | Glucocorticoids cortisol and corticosterone | Synthetic | Reduced SASP inhibited NF-κB p65 activity and suppressed IL1A | Inhibited the ability of SASP to stimulate tumor cell invasion |
| [173] | Human fibroblast | SASP components | Rapamycin | Synthetic | Reduced SASP inhibited NF-κB | Inhibited cell proliferative activity |
| [174] | Human Keratinocytes | SA β-gal | Resveratrol | Synthetic | Reduced SASP increased Activation of AMPK-FOXO3 | Inhibited cell proliferative activity |
| [184] | Human Keratinocytes | ROS | Caffeine | Synthetic | A2AR/SIRT3/AMPK-mediated autophagy and ROS reduction | Reduced ROS increased proliferative activity |
| [177] | Human fibroblast | BrdU, SA β-gal and SASP components | UR-13756 and BIRB 796 | Synthetic | Reduced SASP inhibited p38 MAPK and MAPKAPK2 | Reduced inflammation |
| [178] | Human fibroblast | ROS and SA β-gal | Ferulic acid | Synthetic | Increased proliferation and cell cycle progression | Increased proliferative activity and antioxidant effects |
| [180] | Human diploid fibroblasts | ROS and SA β-gal | Nectandrin B | Natural | Cell cycle progression, reduction in p16, p21, p27, p53 | Increased proliferative activity and antioxidant effects |
| [181] | Human fibroblast | SAHF and SA β-gal | Rehmannia glutinosa, Panax Ginseng, Asparagus cochinchinensis, Ophiopogon japonicus, Cortex Lycii., Poria cocos | Natural | Cell cycle progression, reduction in p16, p21 | Improved skin morphological parameters |
| [182] | Human fibroblast | ROS, SA β-gal and p16INK4a | Yang Yan Qing E Wan (YYQEW) | Natural | Cell cycle progression, reduction in p16 INK4a, and reduced ROS | Increased antioxidant effects |
| [179] | Human fibroblast | ROS and SA β-gal | Quercetin | Synthetic | Cell cycle progression, reduction in p16, p53, and reduced ROS | Increased antioxidant effects |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarandy, M.M.; Gonçalves, R.V.; Valacchi, G. Cutaneous Redox Senescence. Biomedicines 2024, 12, 348. https://doi.org/10.3390/biomedicines12020348
Sarandy MM, Gonçalves RV, Valacchi G. Cutaneous Redox Senescence. Biomedicines. 2024; 12(2):348. https://doi.org/10.3390/biomedicines12020348
Chicago/Turabian StyleSarandy, Mariáurea Matias, Reggiani Vilela Gonçalves, and Giuseppe Valacchi. 2024. "Cutaneous Redox Senescence" Biomedicines 12, no. 2: 348. https://doi.org/10.3390/biomedicines12020348
APA StyleSarandy, M. M., Gonçalves, R. V., & Valacchi, G. (2024). Cutaneous Redox Senescence. Biomedicines, 12(2), 348. https://doi.org/10.3390/biomedicines12020348