
Myocardial Ischemia–Reperfusion Injury: Unraveling Pathophysiology, Clinical Manifestations, and Emerging Prevention Strategies

 ,
,  , , ,
, , ,  , ,
, , 

Abstract
:1. Introduction
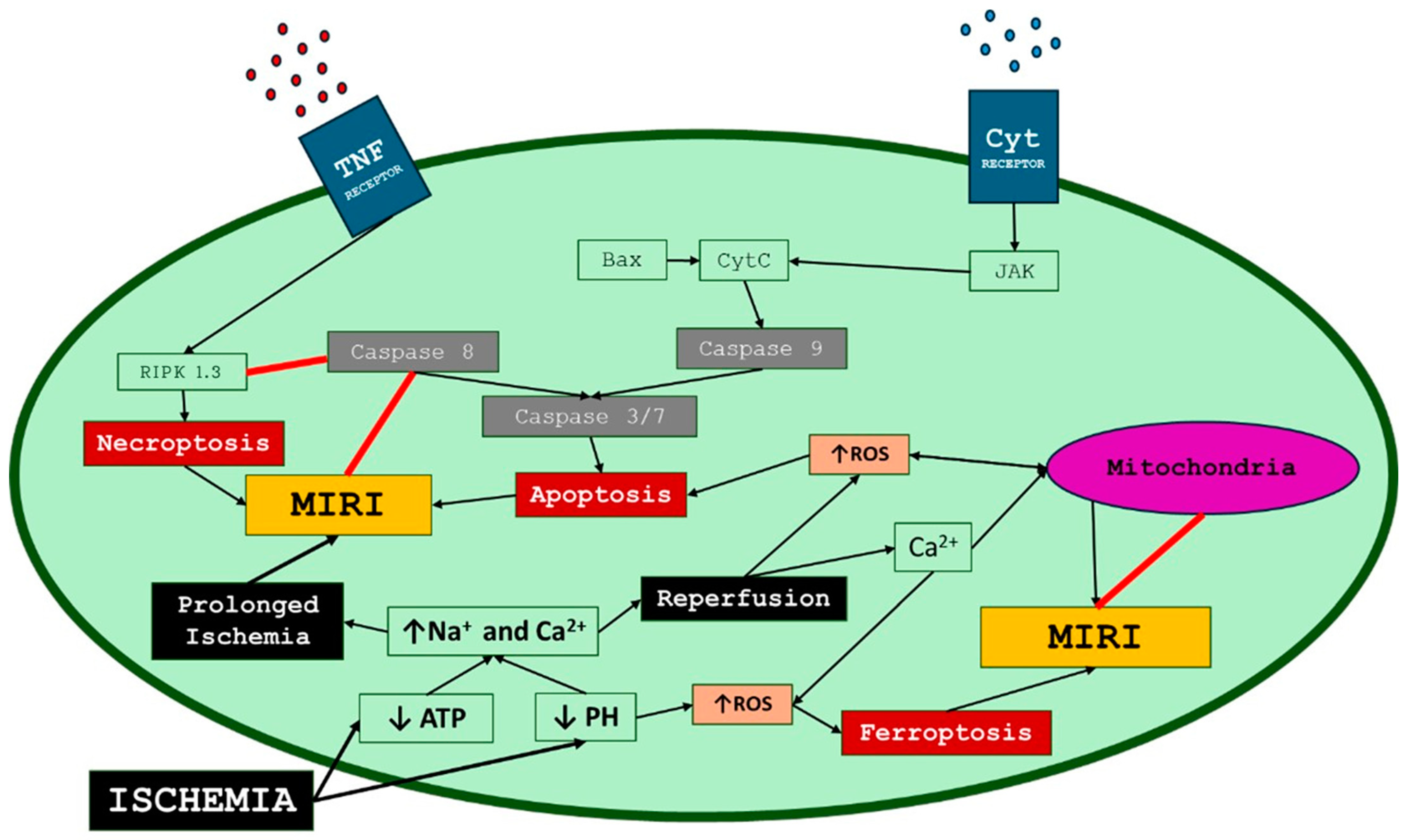
2. Myocardial Ischemia–Reperfusion Injury Mechanisms
2.1. Myocardial Stunning
2.2. No-Reflow Phenomenon
2.3. Reperfusion Arrhythmia
2.4. Lethal Reperfusion Injury
3. The Role of Endothelium in MIRI
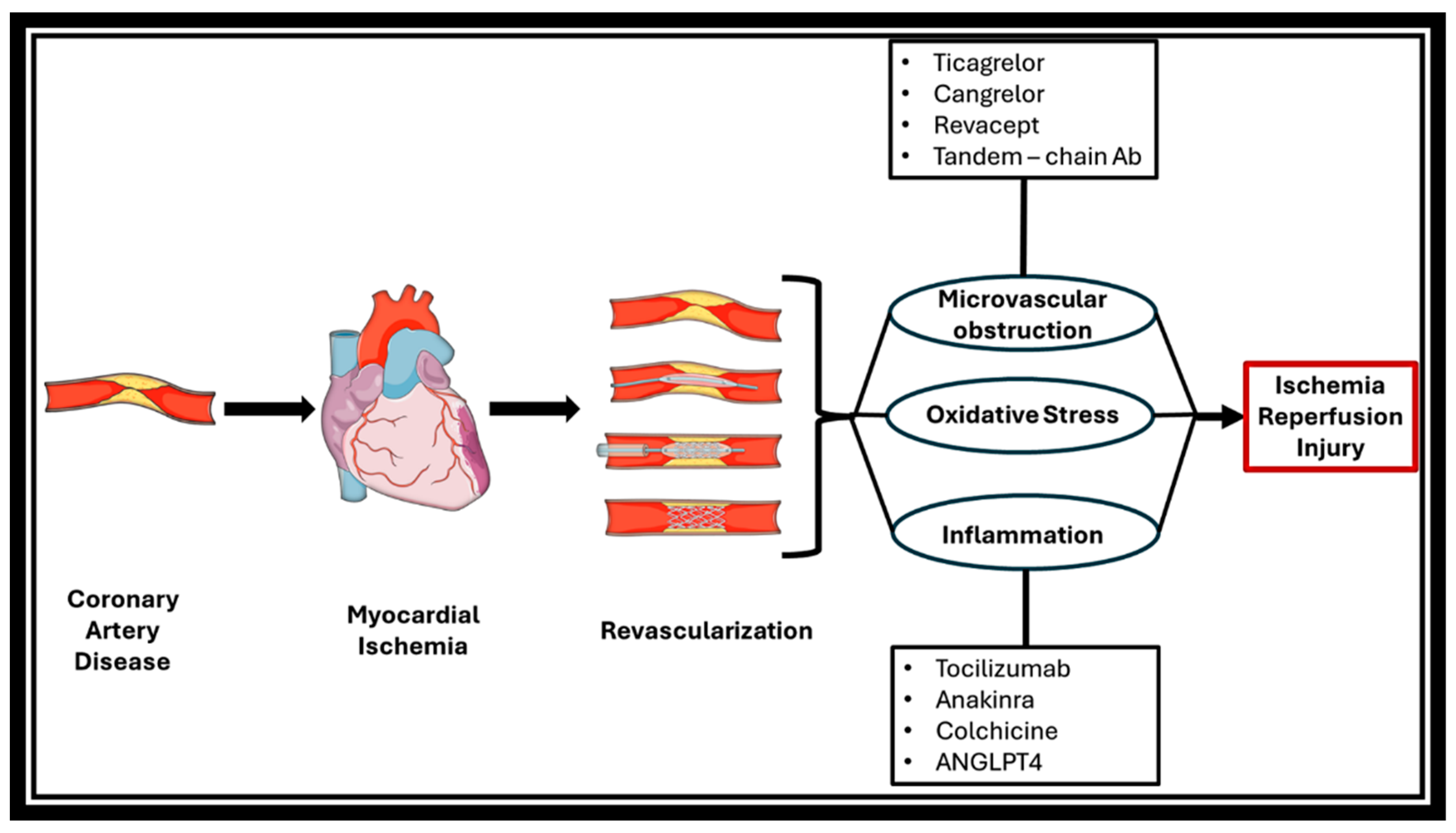
4. Preventive and Therapeutic Approaches
4.1. Ischemic Preconditioning
4.2. Ischemic Postconditioning
4.3. Remote Ischemic Conditioning
4.4. Hypothermia and Vagal Stimulation
4.5. Pharmaceutical Treatment
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jennings, R.B.; Sommers, H.M.; Smyth, G.A.; Flack, H.A.; Linn, H. Myocardial Necrosis Induced by Temporary Occlusion of a Coronary Artery in the Dog. Arch. Pathol. 1960, 70, 68–78. [Google Scholar] [PubMed]
- Levine, G.N.; Bates, E.R.; Blankenship, J.C.; Bailey, S.R.; Bittl, J.A.; Cercek, B.; Chambers, C.E.; Ellis, S.G.; Guyton, R.A.; Hollenberg, S.M.; et al. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. Catheter. Cardiovasc. Interv. 2013, 82, E266–E355. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial Reperfusion Injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, F.; Wang, Y.; Li, W.; Wu, J.; Hu, X.; Tang, T.; Liu, X. Multiple Delivery Strategies of Nanocarriers for Myocardial Ischemia-Reperfusion Injury: Current Strategies and Future Prospective. Drug Deliv. 2024, 31, 2298514. [Google Scholar] [CrossRef]
- Jennings, R.B.; Reimer, K.A. The Cell Biology of Acute Myocardial Ischemia. Annu. Rev. Med. 1991, 42, 225–246. [Google Scholar] [CrossRef]
- Bolli, R.; Marbán, E. Molecular and Cellular Mechanisms of Myocardial Stunning. Physiol. Rev. 1999, 79, 609–634. [Google Scholar] [CrossRef]
- Bolli, R. Myocardial “stunning” in Man. Circulation 1992, 86, 1671–1691. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Pasdois, P. The Role of the Mitochondrial Permeability Transition Pore in Heart Disease. Biochim. Biophys. Acta 2009, 1787, 1402–1415. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R. Mechanism of Myocardial “Stunning”. Circulation 1990, 82, 723–738. [Google Scholar] [CrossRef]
- Kim, S.J.; Kudej, R.K.; Yatani, A.; Kim, Y.K.; Takagi, G.; Honda, R.; Colantonio, D.A.; Van Eyk, J.E.; Vatner, D.E.; Rasmusson, R.L.; et al. A Novel Mechanism for Myocardial Stunning Involving Impaired Ca(2+) Handling. Circ. Res. 2001, 89, 831–837. [Google Scholar] [CrossRef]
- Ito, H. No-Reflow Phenomenon and Prognosis in Patients with Acute Myocardial Infarction. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Ramjane, K.; Han, L.; Jin, C. The Diagnosis and Treatment of the No-Reflow Phenomenon in Patients with Myocardial Infarction Undergoing Percutaneous Coronary Intervention. Exp. Clin. Cardiol. 2008, 13, 121–128. [Google Scholar] [PubMed]
- Resnic, F.S.; Wainstein, M.; Lee, M.K.Y.; Behrendt, D.; Wainstein, R.V.; Ohno-Machado, L.; Kirshenbaum, J.M.; Rogers, C.D.K.; Popma, J.J.; Piana, R. No-Reflow Is an Independent Predictor of Death and Myocardial Infarction after Percutaneous Coronary Intervention. Am. Heart J. 2003, 145, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S.; Aragonés, J.; Landázuri, M.O. Mitochondrial Reprogramming through Cardiac Oxygen Sensors in Ischaemic Heart Disease. Cardiovasc. Res. 2010, 88, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Reffelmann, T.; Kloner, R.A. The No-Reflow Phenomenon: A Basic Mechanism of Myocardial Ischemia and Reperfusion. Basic Res. Cardiol. 2006, 101, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Tatli, E.; Alicik, G.; Buturak, A.; Yilmaztepe, M.; Aktoz, M. Arrhythmias Following Revascularization Procedures in the Course of Acute Myocardial Infarction: Are They Indicators of Reperfusion or Ongoing Ischemia? Sci. World J. 2013, 2013, 160380. [Google Scholar] [CrossRef]
- Leventopoulos, G.; Koros, R.; Travlos, C.; Perperis, A.; Chronopoulos, P.; Tsoni, E.; Koufou, E.-E.; Papageorgiou, A.; Apostolos, A.; Kaouris, P.; et al. Mechanisms of Atrial Fibrillation: How Our Knowledge Affects Clinical Practice. Life 2023, 13, 1260. [Google Scholar] [CrossRef] [PubMed]
- Gorenek, B.; Blomström Lundqvist, C.; Brugada Terradellas, J.; Camm, A.J.; Hindricks, G.; Huber, K.; Kirchhof, P.; Kuck, K.-H.; Kudaiberdieva, G.; Lin, T.; et al. Cardiac Arrhythmias in Acute Coronary Syndromes: Position Paper from the Joint EHRA, ACCA, and EAPCI Task Force. EuroIntervention 2015, 10, 1095–1108. [Google Scholar] [CrossRef]
- Meerson, F.Z.; Belkina, L.M.; Sazontova, T.G.; Saltykova, V.A.; Arkhipenko, Y.V. The Role of Lipid Peroxidation in Pathogenesis of Arrhythmias and Prevention of Cardiac Fibrillation with Antioxidants. Basic Res. Cardiol. 1987, 82, 123–137. [Google Scholar] [CrossRef]
- Bernier, M.; Manning, A.S.; Hearse, D.J. Reperfusion Arrhythmias: Dose-Related Protection by Anti-Free Radical Interventions. Am. J. Physiol. 1989, 256, H1344–H1352. [Google Scholar] [CrossRef]
- Kaplinsky, E.; Ogawa, S.; Michelson, E.L.; Dreifus, L.S. Instantaneous and Delayed Ventricular Arrhythmias after Reperfusion of Acutely Ischemic Myocardium: Evidence for Multiple Mechanisms. Circulation 1981, 63, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R.A. Does Reperfusion Injury Exist in Humans? J. Am. Coll. Cardiol. 1993, 21, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Piper, H.M.; García-Dorado, D.; Ovize, M. A Fresh Look at Reperfusion Injury. Cardiovasc. Res. 1998, 38, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of Cell Death in Heart Disease. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell Death. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and Reperfusion--From Mechanism to Translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular Mechanisms of Necroptosis: An Ordered Cellular Explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Boengler, K.; Schulz, R. Inhibition of Mitochondrial Permeability Transition Pore Opening: The Holy Grail of Cardioprotection. Basic Res. Cardiol. 2010, 105, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Iliodromitis, E.K.; Farmakis, D.; Kremastinos, D.T. To Prevent, Protect and Save the Ischemic Heart: Antioxidants Revisited. Expert Opin. Ther. Targets 2009, 13, 945–956. [Google Scholar] [CrossRef]
- Carden, D.L.; Granger, D.N. Pathophysiology of Ischaemia-Reperfusion Injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Kumar, S.; Kasseckert, S.; Kostin, S.; Abdallah, Y.; Schafer, C.; Kaminski, A.; Reusch, H.; Piper, H.; Steinhoff, G.; Ladilov, Y. Ischemic Acidosis Causes Apoptosis in Coronary Endothelial Cells through Activation of Caspase-12. Cardiovasc. Res. 2007, 73, 172–180. [Google Scholar] [CrossRef]
- Kumar, S.; Reusch, H.P.; Ladilov, Y. Acidic Pre-conditioning Suppresses Apoptosis and Increases Expression of Bcl-xL in Coronary Endothelial Cells under Simulated Ischaemia. J. Cell. Mol. Med. 2008, 12, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Winn, R.K.; Ramamoorthy, C.; Vedder, N.B.; Sharar, S.R.; Harlan, J.M. Leukocyte—Endothelial Cell Interactions in Ischemia-Reperfusion Injury. Ann. N. Y. Acad. Sci. 1997, 832, 311–321. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial Dysfunction in Cardiovascular Diseases: The Role of Oxidant Stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef]
- Boyle, E.M.; Canty, T.G.; Morgan, E.N.; Yun, W.; Pohlman, T.H.; Verrier, E.D. Treating Myocardial Ischemia-Reperfusion Injury by Targeting Endothelial Cell Transcription. Ann. Thorac. Surg. 1999, 68, 1949–1953. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.K.; Brown, M.; Ren, X.; He, S.; McGuinness, M. NF-ΚB as an Integrator of Diverse Signaling Pathways: The Heart of Myocardial Signaling? Cardiovasc. Toxicol. 2003, 3, 229–254. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; He, G.-W.; Underwood, M.J.; Yu, C.-M. Cellular and Molecular Mechanisms of Endothelial Ischemia/Reperfusion Injury: Perspectives and Implications for Postischemic Myocardial Protection. Am. J. Transl. Res. 2016, 8, 765–777. [Google Scholar]
- Schafer, C.; Walther, S.; Schafer, M.; Dieterich, L.; Kasseckert, S.; Abdallah, Y.; Piper, H. Inhibition of Contractile Activation Reduces Reoxygenation-Induced Endothelial Gap Formation. Cardiovasc. Res. 2003, 58, 149–155. [Google Scholar] [CrossRef]
- MIURA, M. Regulation and Failure of Coronary Circulation. JPN Heart J. 1996, 37, 585–602. [Google Scholar] [CrossRef]
- Jorge, P.A.R.; Osaki, M.R.; de Almeida, E.; Dalva, M.; Credidio Neto, L. Endothelium-Dependent Coronary Flow in Ischemia Reperfusion. Exp. Toxicol. Pathol. 1997, 49, 147–151. [Google Scholar] [CrossRef]
- Qi, X.L.; Nguyen, T.L.; Andries, L.; Sys, S.U.; Rouleau, J.L. Vascular Endothelial Dysfunction Contributes to Myocardial Depression in Ischemia-Reperfusion in the Rat. Can. J. Physiol. Pharmacol. 1998, 76, 35–45. [Google Scholar] [CrossRef]
- Hashimoto, K.; Pearson, P.J.; Schaff, H.V.; Cartier, R. Endothelial Cell Dysfunction after Ischemic Arrest and Reperfusion: A Possible Mechanism of Myocardial Injury during Reflow. J. Thorac. Cardiovasc. Surg. 1991, 102, 688–694. [Google Scholar] [CrossRef]
- Tratsiakovich, Y.; Thomas Gonon, A.; Krook, A.; Yang, J.; Shemyakin, A.; Sjöquist, P.-O.; Pernow, J. Arginase Inhibition Reduces Infarct Size via Nitric Oxide, Protein Kinase C Epsilon and Mitochondrial ATP-Dependent K+ Channels. Eur. J. Pharmacol. 2013, 712, 16–21. [Google Scholar] [CrossRef]
- Metais, C.; Li, J.; Li, J.; Simons, M.; Sellke, F.W. Serotonin-Induced Coronary Contraction Increases After Blood Cardioplegia-Reperfusion: Role of COX-2 Expression. Circulation 1999, 100, II-328–II-334. [Google Scholar] [CrossRef]
- Bonaventura, J.; Gow, A. NO and Superoxide: Opposite Ends of the Seesaw in Cardiac Contractility. Proc. Natl. Acad. Sci. USA 2004, 101, 16403–16404. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H.; Yasutake, H.; Fujii, K.; Owada, M.K.; Nakaike, R.; Fukumoto, Y.; Takayanagi, T.; Nagao, T.; Egashira, K.; Fujishima, M.; et al. The Importance of the Hyperpolarizing Mechanism Increases as the Vessel Size Decreases in Endothelium-Dependent Relaxations in Rat Mesenteric Circulation. J. Cardiovasc. Pharmacol. 1996, 28, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, T.; Ito, T.; Takahasi, R.; Tanaka, S. Influence of Normal to High Stroke Volume on Congestive Heart Failure Development after Transcatheter Aortic Valve Implantation: Case Series. J. Geriatr. Cardiol. 2021, 18, 83–88. [Google Scholar] [CrossRef]
- Edwards, G.; Félétou, M.; Weston, A.H. Endothelium-Derived Hyperpolarising Factors and Associated Pathways: A Synopsis. Pflugers Arch. 2010, 459, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.; Dora, K.A.; Gardener, M.J.; Garland, C.J.; Weston, A.H. K+ Is an Endothelium-Derived Hyperpolarizing Factor in Rat Arteries. Nature 1998, 396, 269–272. [Google Scholar] [CrossRef]
- Weston, A.H.; Félétou, M.; Vanhoutte, P.M.; Falck, J.R.; Campbell, W.B.; Edwards, G. Bradykinin-induced, Endothelium-dependent Responses in Porcine Coronary Arteries: Involvement of Potassium Channel Activation and Epoxyeicosatrienoic Acids. Br. J. Pharmacol. 2005, 145, 775–784. [Google Scholar] [CrossRef]
- Yang, Q.; Huang, J.-H.; Man, Y.-B.; Yao, X.-Q.; He, G.-W. Use of Intermediate/Small Conductance Calcium-Activated Potassium-Channel Activator for Endothelial Protection. J. Thorac. Cardiovasc. Surg. 2011, 141, 501–510.e1. [Google Scholar] [CrossRef] [PubMed]
- Ziberna, L.; Lunder, M.; Kužner, J.; Drevenšek, G. Normothermic and Hypothermic Models for Studying the Deleterious Effects of Hypoxia-Reoxygenation on EDHF-Mediated Relaxation in Isolated Porcine Coronary Arteries. J. Pharmacol. Toxicol. Methods 2009, 59, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.C.H.; Woodman, O.L. Enhanced Role for the Opening of Potassium Channels in Relaxant Responses to Acetylcholine after Myocardial Ischaemia and Reperfusion in Dog Coronary Arteries. Br. J. Pharmacol. 1999, 126, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Tomai, F. Warm up Phenomenon and Preconditioning in Clinical Practice. Heart 2002, 87, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with Ischemia: A Delay of Lethal Cell Injury in Ischemic Myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Richard, V.J.; Jennings, R.B.; Reimer, K.A. Myocardial Protection Is Lost before Contractile Function Recovers from Ischemic Preconditioning. Am. J. Physiol. 1991, 260, H796–H804. [Google Scholar] [CrossRef]
- Marber, M.S.; Latchman, D.S.; Walker, J.M.; Yellon, D.M. Cardiac Stress Protein Elevation 24 Hours after Brief Ischemia or Heat Stress Is Associated with Resistance to Myocardial Infarction. Circulation 1993, 88, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.P.; Pugsley, W.B.; Alkhulaifi, A.M.; Kemp, M.; Hooper, J.; Yellon, D.M. Ischaemic Preconditioning Reduces Troponin T Release in Patients Undergoing Coronary Artery Bypass Surgery. Heart 1997, 77, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Berger, M.; Kussmaul, W.G.; Hirshfeld, J.W.; Herrmann, H.C.; Laskey, W.K. Adaptation to Ischemia during Percutaneous Transluminal Coronary Angioplasty. Clinical, Hemodynamic, and Metabolic Features. Circulation 1990, 82, 2044–2051. [Google Scholar] [CrossRef]
- Zhao, Z.-Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.-P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of Myocardial Injury by Ischemic Postconditioning during Reperfusion: Comparison with Ischemic Preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef]
- Thuny, F.; Lairez, O.; Roubille, F.; Mewton, N.; Rioufol, G.; Sportouch, C.; Sanchez, I.; Bergerot, C.; Thibault, H.; Cung, T.T.; et al. Post-Conditioning Reduces Infarct Size and Edema in Patients with ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2012, 59, 2175–2181. [Google Scholar] [CrossRef] [PubMed]
- Traverse, J.H.; Swingen, C.M.; Henry, T.D.; Fox, J.; Wang, Y.L.; Chavez, I.J.; Lips, D.L.; Lesser, J.R.; Pedersen, W.R.; Burke, N.M.; et al. NHLBI-Sponsored Randomized Trial of Postconditioning During Primary Percutaneous Coronary Intervention for ST-Elevation Myocardial Infarction. Circ. Res. 2019, 124, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Laskey, W.K.; Yoon, S.; Calzada, N.; Ricciardi, M.J. Concordant Improvements in Coronary Flow Reserve and ST-Segment Resolution during Percutaneous Coronary Intervention for Acute Myocardial Infarction: A Benefit of Postconditioning. Catheter. Cardiovasc. Interv. 2008, 72, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Przyklenk, K.; Bauer, B.; Ovize, M.; Kloner, R.A.; Whittaker, P. Regional Ischemic “preconditioning” Protects Remote Virgin Myocardium from Subsequent Sustained Coronary Occlusion. Circulation 1993, 87, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Bøtker, H.E.; Przyklenk, K.; Redington, A.; Yellon, D. Remote Ischemic Conditioning. J. Am. Coll. Cardiol. 2015, 65, 177–195. [Google Scholar] [CrossRef] [PubMed]
- Crimi, G.; Pica, S.; Raineri, C.; Bramucci, E.; De Ferrari, G.M.; Klersy, C.; Ferlini, M.; Marinoni, B.; Repetto, A.; Romeo, M.; et al. Remote Ischemic Post-Conditioning of the Lower Limb during Primary Percutaneous Coronary Intervention Safely Reduces Enzymatic Infarct Size in Anterior Myocardial Infarction: A Randomized Controlled Trial. JACC Cardiovasc. Interv. 2013, 6, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, T.; Kawai, Y.; Miyoshi, T.; Mima, T.; Takagaki, K.; Tsukuda, S.; Kazatani, Y.; Nakamura, K.; Ito, H. Remote Ischemic Preconditioning Reduces Contrast-Induced Acute Kidney Injury in Patients with ST-Elevation Myocardial Infarction: A Randomized Controlled Trial. Int. J. Cardiol. 2015, 178, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Basalay, M.V.; Davidson, S.M.; Gourine, A.V.; Yellon, D.M. Neural Mechanisms in Remote Ischaemic Conditioning in the Heart and Brain: Mechanistic and Translational Aspects. Basic Res. Cardiol. 2018, 113, 25. [Google Scholar] [CrossRef] [PubMed]
- Tissier, R.; Ghaleh, B.; Cohen, M.V.; Downey, J.M.; Berdeaux, A. Myocardial Protection with Mild Hypothermia. Cardiovasc. Res. 2012, 94, 217–225. [Google Scholar] [CrossRef]
- Götberg, M.; van der Pals, J.; Götberg, M.; Olivecrona, G.K.; Kanski, M.; Koul, S.; Otto, A.; Engblom, H.; Ugander, M.; Arheden, H.; et al. Optimal Timing of Hypothermia in Relation to Myocardial Reperfusion. Basic Res. Cardiol. 2011, 106, 697–708. [Google Scholar] [CrossRef]
- Dixon, S.R.; Whitbourn, R.J.; Dae, M.W.; Grube, E.; Sherman, W.; Schaer, G.L.; Jenkins, J.S.; Baim, D.S.; Gibbons, R.J.; Kuntz, R.E.; et al. Induction of Mild Systemic Hypothermia with Endovascular Cooling during Primary Percutaneous Coronary Intervention for Acute Myocardial Infarction. J. Am. Coll. Cardiol. 2002, 40, 1928–1934. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Vagal Cardioprotection in Reperfused Acute Myocardial Infarction. JACC Cardiovasc. Interv. 2017, 10, 1521–1522. [Google Scholar] [CrossRef] [PubMed]
- Herzog, W.R.; Vogel, R.A.; Schlossberg, M.L.; Edenbaum, L.R.; Scott, H.J.; Serebruany, V.L. Short-Term Low Dose Intracoronary Diltiazem Administered at the Onset of Reperfusion Reduces Myocardial Infarct Size. Int. J. Cardiol. 1997, 59, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.-C.; Chen, H.-R.; Chiu, C.-C.; Liou, S.-F.; Chen, I.-J.; Yeh, J.-L. Protective Effect of Labedipinedilol-A, a Novel Dihydropyridine-Type Calcium Channel Blocker, on Myocardial Apoptosis in Ischemia–Reperfusion Injury. Life Sci. 2006, 79, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.H.; Pich, S.; Lindert, S.; Nebendahl, K.; Warneke, G.; Kreuzer, H. Treatment of Reperfusion Injury with Intracoronary Calcium Channel Antagonists and Reduced Coronary Free Calcium Concentration in Regionally Ischemic, Reperfused Porcine Hearts. J. Am. Coll. Cardiol. 1989, 13, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. Acute Methadone Treatment Reduces Myocardial Infarct Size via the δ-Opioid Receptor in Rats During Reperfusion. Anesth. Analg. 2009, 109, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.; Boulghobra, D.; Rochebloine, G.; Pallot, F.; Yehya, M.; Bornard, I.; Gayrard, S.; Coste, F.; Walther, G.; Meyer, G.; et al. Hyperglycemia triggers RyR2-dependent alterations of mitochondrial calcium homeostasis in response to cardiac ischemia-reperfusion: Key role of DRP1 activation. Redox Biol. 2024, 70, 103044. [Google Scholar] [CrossRef] [PubMed]
- Rout, A.; Tantry, U.S.; Novakovic, M.; Sukhi, A.; Gurbel, P.A. Targeted Pharmacotherapy for Ischemia Reperfusion Injury in Acute Myocardial Infarction. Expert Opin. Pharmacother. 2020, 21, 1851–1865. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Weil, M.H.; Tang, W.; Sun, S.; Wang, J. Comparison between Dobutamine and Levosimendan for Management of Postresuscitation Myocardial Dysfunction*. Crit. Care Med. 2005, 33, 487–491. [Google Scholar] [CrossRef]
- Kersten, J.R.; Montgomery, M.W.; Pagel, P.S.; Warltier, D.C. Levosimendan, a New Positive Inotropic Drug, Decreases Myocardial Infarct Size via Activation of KATP Channels. Anesth. Analg. 2000, 90, 5–11. [Google Scholar] [CrossRef]
- Kiraz, H.A.; Poyraz, F.; Kip, G.; Erdem, Ö.; Alkan, M.; Arslan, M.; Özer, A.; Şivgin, V.; Çomu, F.M. The Effect of Levosimendan on Myocardial Ischemia–Reperfusion Injury in Streptozotocin-Induced Diabetic Rats. Libyan J. Med. 2015, 10, 29269. [Google Scholar] [CrossRef]
- Leivaditis, V.; Koletsis, E.; Tsopanoglou, N.; Charokopos, N.; D’Alessandro, C.; Grapatsas, K.; Apostolakis, E.; Choleva, E.; Plota, M.; Emmanuil, A.; et al. The Coadministration of Levosimendan and Exenatide Offers a Significant Cardioprotective Effect to Isolated Rat Hearts against Ischemia/Reperfusion Injury. J. Cardiovasc. Dev. Dis. 2022, 9, 263. [Google Scholar] [CrossRef] [PubMed]
- Barrabés, J.A.; Garcia-Dorado, D.; Mirabet, M.; Inserte, J.; Agulló, L.; Soriano, B.; Massaguer, A.; Padilla, F.; Lidón, R.-M.; Soler-Soler, J. Antagonism of Selectin Function Attenuates Microvascular Platelet Deposition and Platelet-Mediated Myocardial Injury after Transient Ischemia. J. Am. Coll. Cardiol. 2005, 45, 293–299. [Google Scholar] [CrossRef]
- Xu, Y.; Huo, Y.; Toufektsian, M.-C.; Ramos, S.I.; Ma, Y.; Tejani, A.D.; French, B.A.; Yang, Z. Activated Platelets Contribute Importantly to Myocardial Reperfusion Injury. Am. J. Physiol.-Heart Circ. Physiol. 2006, 290, H692–H699. [Google Scholar] [CrossRef] [PubMed]
- Khandoga, A.; Biberthaler, P.; Enders, G.; Teupser, D.; Axmann, S.; Luchting, B.; Hutter, J.; Messmer, K.; Krombach, F. P-Selectin Mediates Platelet-Endothelial Cell Interactions and Reperfusion Injury in the Mouse Liver In Vivo. Shock 2002, 18, 529–535. [Google Scholar] [CrossRef]
- Mertens, P.; Maes, A.; Nuyts, J.; Belmans, A.; Desmet, W.; Esplugas, E.; Charlier, F.; Figueras, J.; Sambuceti, G.; Schwaiger, M.; et al. Recombinant P-Selectin Glycoprotein Ligand–Immunoglobulin, a P-Selectin Antagonist, as an Adjunct to Thrombolysis in Acute Myocardial Infarction. The P-Selectin Antagonist Limiting Myonecrosis (PSALM) Trial. Am. Heart. J. 2006, 152, 125.e1–125.e8. [Google Scholar] [CrossRef] [PubMed]
- Stähli, B.E.; Gebhard, C.; Duchatelle, V.; Cournoyer, D.; Petroni, T.; Tanguay, J.; Robb, S.; Mann, J.; Guertin, M.; Wright, R.S.; et al. Effects of the P-Selectin Antagonist Inclacumab on Myocardial Damage After Percutaneous Coronary Intervention According to Timing of Infusion: Insights From the SELECT-ACS Trial. J. Am. Hear. Assoc. 2016, 5, e004255. [Google Scholar] [CrossRef]
- Tardif, J.-C.; Tanguay, J.-F.; Wright, S.R.; Duchatelle, V.; Petroni, T.; Grégoire, J.C.; Ibrahim, R.; Heinonen, T.M.; Robb, S.; Bertrand, O.F.; et al. Effects of the P-Selectin Antagonist Inclacumab on Myocardial Damage After Percutaneous Coronary Intervention for Non–ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2013, 61, 2048–2055. [Google Scholar] [CrossRef]
- Wudexi, I.; Shokri, E.; Abo-Aly, M.; Shindo, K.; Abdel-Latif, A. Comparative Effectiveness of Anti-Inflammatory Drug Treatments in Coronary Heart Disease Patients: A Systematic Review and Network Meta-Analysis. Mediat. Inflamm. 2021, 2021, 5160728. [Google Scholar] [CrossRef]
- Ritschel, V.N.; Seljeflot, I.; Arnesen, H.; Halvorsen, S.; Eritsland, J.; Fagerland, M.W.; Andersen, G.Ø. Circulating Levels of IL-6 Receptor and Gp130 and Long-Term Clinical Outcomes in ST-Elevation Myocardial Infarction. J. Am. Hear. Assoc. 2016, 5, e003014. [Google Scholar] [CrossRef]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a Single Dose of the Interleukin-6 Receptor Antagonist Tocilizumab on Inflammation and Troponin T Release in Patients with Non-ST-Elevation Myocardial Infarction: A Double-Blind, Randomized, Placebo-Controlled Phase 2 Trial. Eur. Hear. J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Broch, K.; Anstensrud, A.K.; Woxholt, S.; Sharma, K.; Tøllefsen, I.M.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen, B.H.; Damås, J.K.; et al. Randomized Trial of Interleukin-6 Receptor Inhibition in Patients with Acute ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2021, 77, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Van Tassell, B.W.; Biondi-Zoccai, G.; Kontos, M.C.; Grizzard, J.D.; Spillman, D.W.; Oddi, C.; Roberts, C.S.; Melchior, R.D.; Mueller, G.H.; et al. Effects of Interleukin-1 Blockade with Anakinra on Adverse Cardiac Remodeling and Heart Failure After Acute Myocardial Infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) Pilot Study]. Am. J. Cardiol. 2013, 111, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Trankle, C.R.; Buckley, L.F.; Lipinski, M.J.; Appleton, D.; Kadariya, D.; Canada, J.M.; Carbone, S.; Roberts, C.S.; Abouzaki, N.; et al. Interleukin-1 Blockade Inhibits the Acute Inflammatory Response in Patients With ST-Segment–Elevation Myocardial Infarction. J. Am. Hear. Assoc. 2020, 9, e014941. [Google Scholar] [CrossRef] [PubMed]
- Deftereos, S.; Giannopoulos, G.; Angelidis, C.; Alexopoulos, N.; Filippatos, G.; Papoutsidakis, N.; Sianos, G.; Goudevenos, J.; Alexopoulos, D.; Pyrgakis, V.; et al. Anti-Inflammatory Treatment with Colchicine in Acute Myocardial Infarction. Circulation 2015, 132, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Tsigkas, G.; Apostolos, A.; Trigka, A.; Chlorogiannis, D.; Katsanos, K.; Toutouzas, K.; Alexopoulos, D.; Brilakis, E.S.; Davlouros, P. Very Short Versus Longer Dual Antiplatelet Treatment After Coronary Interventions: A Systematic Review and Meta-Analysis. Am. J. Cardiovasc. Drugs 2023, 23, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Roubille, F.; Lairez, O.; Mewton, N.; Rioufol, G.; Ranc, S.; Sanchez, I.; Cung, T.T.; Elbaz, M.; Piot, C.; Ovize, M. Cardioprotection by Clopidogrel in Acute ST-Elevated Myocardial Infarction Patients: A Retrospective Analysis. Basic Res. Cardiol. 2012, 107, 275. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-M.; Liu, Y.; Cui, L.; Yang, X.; Liu, Y.; Tandon, N.; Kambayashi, J.; Downey, J.M.; Cohen, M.V. Platelet P2Y 12 Blockers Confer Direct Postconditioning-Like Protection in Reperfused Rabbit Hearts. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 251–262. [Google Scholar] [CrossRef]
- Bulluck, H.; Chan, M.H.H.; Bryant, J.A.; Chai, P.; Chawla, A.; Chua, T.S.; Chung, Y.; Fei, G.; Ho, H.H.; Ho, A.F.W.; et al. Platelet Inhibition to Target Reperfusion Injury Trial: Rationale and Study Design. Clin. Cardiol. 2019, 42, 5–12. [Google Scholar] [CrossRef]
- Cohen, M.V.; Yang, X.-M.; White, J.; Yellon, D.M.; Bell, R.M.; Downey, J.M. Cangrelor-Mediated Cardioprotection Requires Platelets and Sphingosine Phosphorylation. Cardiovasc. Drugs Ther. 2016, 30, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Aragno, M.; Cento, A.S.; Femminò, S.; Russo, I.; Bello, F.D.; Chiazza, F.; Collotta, D.; Alves, G.F.; Bertinaria, M.; et al. Ticagrelor Conditioning Effects Are Not Additive to Cardioprotection Induced by Direct NLRP3 Inflammasome Inhibition: Role of RISK, NLRP3, and Redox Cascades. Oxidative Med. Cell. Longev. 2020, 2020, 9219825. [Google Scholar] [CrossRef] [PubMed]
- Vilahur, G.; Gutiérrez, M.; Casani, L.; Varela, L.; Capdevila, A.; Pons-Lladó, G.; Carreras, F.; Carlsson, L.; Hidalgo, A.; Badimon, L. Protective Effects of Ticagrelor on Myocardial Injury After Infarction. Circulation 2016, 134, 1708–1719. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, L.; Becker, R.C.; Budaj, A.; Cannon, C.P.; Emanuelsson, H.; Held, C.; Horrow, J.; Husted, S.; James, S.; Katus, H. Ticagrelor versus Clopidogrel in Patients with Acute Coronary Syndromes. N. Engl. J. Med. 2009, 361, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Koh, J.S.; Lee, J.-H.; Park, J.-H.; Shin, E.-S.; Oh, J.H.; Chun, W.; Lee, S.Y.; Bae, J.-W.; Kim, J.S.; et al. Effect of Ticagrelor on Left Ventricular Remodeling in Patients With ST-Segment Elevation Myocardial Infarction (HEALING-AMI). JACC Cardiovasc. Interv. 2020, 13, 2220–2234. [Google Scholar] [CrossRef]
- Feliu, C.; Peyret, H.; Brassart-Pasco, S.; Oszust, F.; Poitevin, G.; Nguyen, P.; Millart, H.; Djerada, Z. Ticagrelor Prevents Endothelial Cell Apoptosis through the Adenosine Signalling Pathway in the Early Stages of Hypoxia. Biomolecules 2020, 10, 740. [Google Scholar] [CrossRef] [PubMed]
- Kunichika, H.; Ben-Yehuda, O.; Lafitte, S.; Kunichika, N.; Peters, B.; DeMaria, A.N. Effects of Glycoprotein Iib/Iiia Inhibition on Microvascular Flow after Coronary Reperfusion. J. Am. Coll. Cardiol. 2004, 43, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Kupatt, C.; Wichels, R.; Horstkotte, J.; Krombach, F.; Habazettl, H.; Boekstegers, P. Molecular Mechanisms of Platelet-Mediated Leukocyte Recruitment during Myocardial Reperfusion. J. Leukoc. Biol. 2002, 72, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.; Haigh, K.; Nguyen, T.; Wang, X.; Lim, B.; Yap, M.L.; Eddy, E.M.; Haigh, J.J.; Peter, K. The Pulmonary Microvasculature Entraps Induced Vascular Progenitor Cells (iVPCs) Systemically Delivered after Cardiac Ischemia-reperfusion Injury: Indication for Preservation of Heart Function via Paracrine Effects beyond Engraftment. Microcirculation 2019, 26, e12493. [Google Scholar] [CrossRef]
- Ziegler, M.; Wang, X.; Lim, B.; Leitner, E.; Klingberg, F.; Ching, V.; Yao, Y.; Huang, D.; Gao, X.-M.; Kiriazis, H.; et al. Platelet-Targeted Delivery of Peripheral Blood Mononuclear Cells to the Ischemic Heart Restores Cardiac Function after Ischemia-Reperfusion Injury. Theranostics 2017, 7, 3192–3206. [Google Scholar] [CrossRef]
- Schönberger, T.; Ziegler, M.; Borst, O.; Konrad, I.; Nieswandt, B.; Massberg, S.; Ochmann, C.; Jürgens, T.; Seizer, P.; Langer, H.; et al. The Dimeric Platelet Collagen Receptor GPVI-Fc Reduces Platelet Adhesion to Activated Endothelium and Preserves Myocardial Function after Transient Ischemia in Mice. Am. J. Physiol.-Cell Physiol. 2012, 303, C757–C766. [Google Scholar] [CrossRef] [PubMed]
- Pachel, C.; Mathes, D.; Arias-Loza, A.-P.; Heitzmann, W.; Nordbeck, P.; Deppermann, C.; Lorenz, V.; Hofmann, U.; Nieswandt, B.; Frantz, S. Inhibition of Platelet GPVI Protects Against Myocardial Ischemia–Reperfusion Injury. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.; Hein-Rothweiler, R.; Schüpke, S.; Janisch, M.; Bernlochner, I.; Ndrepepa, G.; Sibbing, D.; Gori, T.; Borst, O.; Holdenrieder, S.; et al. Efficacy and Safety of Revacept, a Novel Lesion-Directed Competitive Antagonist to Platelet Glycoprotein VI, in Patients Undergoing Elective Percutaneous Coronary Intervention for Stable Ischemic Heart Disease. JAMA Cardiol. 2021, 6, 753. [Google Scholar] [CrossRef]
- Do Carmo, H.; Arjun, S.; Petrucci, O.; Yellon, D.M.; Davidson, S.M. The Caspase 1 Inhibitor VX-765 Protects the Isolated Rat Heart via the RISK Pathway. Cardiovasc. Drugs Ther. 2018, 32, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-M.; Downey, J.M.; Cohen, M.V.; Housley, N.A.; Alvarez, D.F.; Audia, J.P. The Highly Selective Caspase-1 Inhibitor VX-765 Provides Additive Protection Against Myocardial Infarction in Rat Hearts When Combined with a Platelet Inhibitor. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 574–578. [Google Scholar] [CrossRef]
- Audia, J.P.; Yang, X.-M.; Crockett, E.S.; Housley, N.; Haq, E.U.; O’Donnell, K.; Cohen, M.V.; Downey, J.M.; Alvarez, D.F. Caspase-1 Inhibition by VX-765 Administered at Reperfusion in P2Y12 Receptor Antagonist-Treated Rats Provides Long-Term Reduction in Myocardial Infarct Size and Preservation of Ventricular Function. Basic Res. Cardiol. 2018, 113, 32. [Google Scholar] [CrossRef] [PubMed]
- Cung, T.-T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Lincoff, A.M.; Roe, M.; Aylward, P.; Galla, J.; Rynkiewicz, A.; Guetta, V.; Zelizko, M.; Kleiman, N.; White, H.; McErlean, E.; et al. Inhibition of Delta-Protein Kinase C by Delcasertib as an Adjunct to Primary Percutaneous Coronary Intervention for Acute Anterior ST-Segment Elevation Myocardial Infarction: Results of the PROTECTION AMI Randomized Controlled Trial. Eur. Hear. J. 2014, 35, 2516–2523. [Google Scholar] [CrossRef]
- Gibson, C.M.; Giugliano, R.P.; Kloner, R.A.; Bode, C.; Tendera, M.; Jánosi, A.; Merkely, B.; Godlewski, J.; Halaby, R.; Korjian, S.; et al. EMBRACE STEMI Study: A Phase 2a Trial to Evaluate the Safety, Tolerability, and Efficacy of Intravenous MTP-131 on Reperfusion Injury in Patients Undergoing Primary Percutaneous Coronary Intervention. Eur. Hear. J. 2016, 37, 1296–1303. [Google Scholar] [CrossRef]
- Dewey, F.E.; Gusarova, V.; O’Dushlaine, C.; Gottesman, O.; Trejos, J.; Hunt, C.; Van Hout, C.V.; Habegger, L.; Buckler, D.; Lai, K.-M.V.; et al. Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N. Engl. J. Med. 2016, 374, 1123–1133. [Google Scholar] [CrossRef]
- Cho, D.I.; Kang, H.; Jeon, J.H.; Eom, G.H.; Cho, H.H.; Kim, M.R.; Cho, M.; Jeong, H.; Cho, H.C.; Hong, M.H.; et al. Antiinflammatory Activity of ANGPTL4 Facilitates Macrophage Polarization to Induce Cardiac Repair. JCI Insight 2019, 4, e125437. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Kim, Y.S.; Park, J.; Choe, G.; Lee, S.; Kang, B.G.; Jun, J.H.; Shin, Y.; Kim, M.; Ahn, Y.; et al. A Paintable and Adhesive Hydrogel Cardiac Patch with Sustained Release of ANGPTL4 for Infarcted Heart Repair. Bioact. Mater. 2024, 31, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Yao, W.; Yi, K.; Zheng, C.; Lv, S.; Tao, Y.; Hei, Z.; Li, M. Nanotheranostics for the Management of Hepatic Ischemia-Reperfusion Injury. Small 2021, 17, 2007727. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Gersh, B.J. The Pathophysiology of Acute Myocardial Infarction and Strategies of Protection beyond Reperfusion: A Continual Challenge. Eur. Hear. J. 2016, 38, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Weng, X.; Tan, H.; Huang, Z.; Chen, J.; Zhang, N.; Wang, Q.; Li, Q.; Gao, J.; Sun, D.; Yakufu, W.; et al. Targeted Delivery and ROS-Responsive Release of Resolvin D1 by Platelet Chimeric Liposome Ameliorates Myocardial Ischemia–Reperfusion Injury. J. Nanobiotechnol. 2022, 20, 454. [Google Scholar] [CrossRef] [PubMed]
- Papastamos, C.; Antonopoulos, A.S.; Simantiris, S.; Koumallos, N.; Theofilis, P.; Sagris, M.; Tsioufis, K.; Androulakis, E.; Tousoulis, D. Stem Cell-Based Therapies in Cardiovascular Diseases: From Pathophysiology to Clinical Outcomes. Curr. Pharm. Des. 2023, 29, 2795–2801. [Google Scholar] [CrossRef] [PubMed]
- Ottersbach, A.; Mykhaylyk, O.; Heidsieck, A.; Eberbeck, D.; Rieck, S.; Zimmermann, K.; Breitbach, M.; Engelbrecht, B.; Brügmann, T.; Hesse, M.; et al. Improved Heart Repair upon Myocardial Infarction: Combination of Magnetic Nanoparticles and Tailored Magnets Strongly Increases Engraftment of Myocytes. Biomaterials 2018, 155, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Oikonomou, E.; Tsioufis, K.; Tousoulis, D. Genetic Predisposition and Inflammatory Inhibitors in COVID-19: Where Do We Stand? Biomedicines 2022, 10, 242. [Google Scholar] [CrossRef]
- Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Tsioufis, K.; Tousoulis, D. Telomere Length: A Cardiovascular Biomarker and a Novel Therapeutic Target. Int. J. Mol. Sci. 2022, 23, 16010. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Before Coronary Occlusion | During Coronary Reperfusion | After Coronary Reperfusion |
|---|---|---|
| Mechanical Preconditioning | Mechanical Remote Conditioning | Mechanical Postconditioning |
| Mechanical Remote Conditioning | Hypothermia | Mechanical Remote Conditioning |
| Infusion of Nitrates | Vagal Stimulation | Regulation of Circadian Rhythm |
| TLR4 inhibitors | Infusion of TLR4 inhibitors | Erythropoietin |
| Infusion of Adenosine | Infusion of Adenosine | Epoetin Alpha |
| Etomoxir | Glucose–Insulin–Potassium (GIK) | |
| Trimetazidine | Acetaminophen | |
| Ranolazine | ||
| Dichloroacetate (DCA) | ||
| Levosimendan |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sagris, M.; Apostolos, A.; Theofilis, P.; Ktenopoulos, N.; Katsaros, O.; Tsalamandris, S.; Tsioufis, K.; Toutouzas, K.; Tousoulis, D. Myocardial Ischemia–Reperfusion Injury: Unraveling Pathophysiology, Clinical Manifestations, and Emerging Prevention Strategies. Biomedicines 2024, 12, 802. https://doi.org/10.3390/biomedicines12040802
Sagris M, Apostolos A, Theofilis P, Ktenopoulos N, Katsaros O, Tsalamandris S, Tsioufis K, Toutouzas K, Tousoulis D. Myocardial Ischemia–Reperfusion Injury: Unraveling Pathophysiology, Clinical Manifestations, and Emerging Prevention Strategies. Biomedicines. 2024; 12(4):802. https://doi.org/10.3390/biomedicines12040802
Chicago/Turabian StyleSagris, Marios, Anastasios Apostolos, Panagiotis Theofilis, Nikolaos Ktenopoulos, Odysseas Katsaros, Sotirios Tsalamandris, Konstantinos Tsioufis, Konstantinos Toutouzas, and Dimitris Tousoulis. 2024. "Myocardial Ischemia–Reperfusion Injury: Unraveling Pathophysiology, Clinical Manifestations, and Emerging Prevention Strategies" Biomedicines 12, no. 4: 802. https://doi.org/10.3390/biomedicines12040802
APA StyleSagris, M., Apostolos, A., Theofilis, P., Ktenopoulos, N., Katsaros, O., Tsalamandris, S., Tsioufis, K., Toutouzas, K., & Tousoulis, D. (2024). Myocardial Ischemia–Reperfusion Injury: Unraveling Pathophysiology, Clinical Manifestations, and Emerging Prevention Strategies. Biomedicines, 12(4), 802. https://doi.org/10.3390/biomedicines12040802