

Transcytosis-Driven Treatment of Neurodegenerative Disorders by mRNA-Expressed Antibody–Transferrin Conjugates



Abstract
:1. Introduction
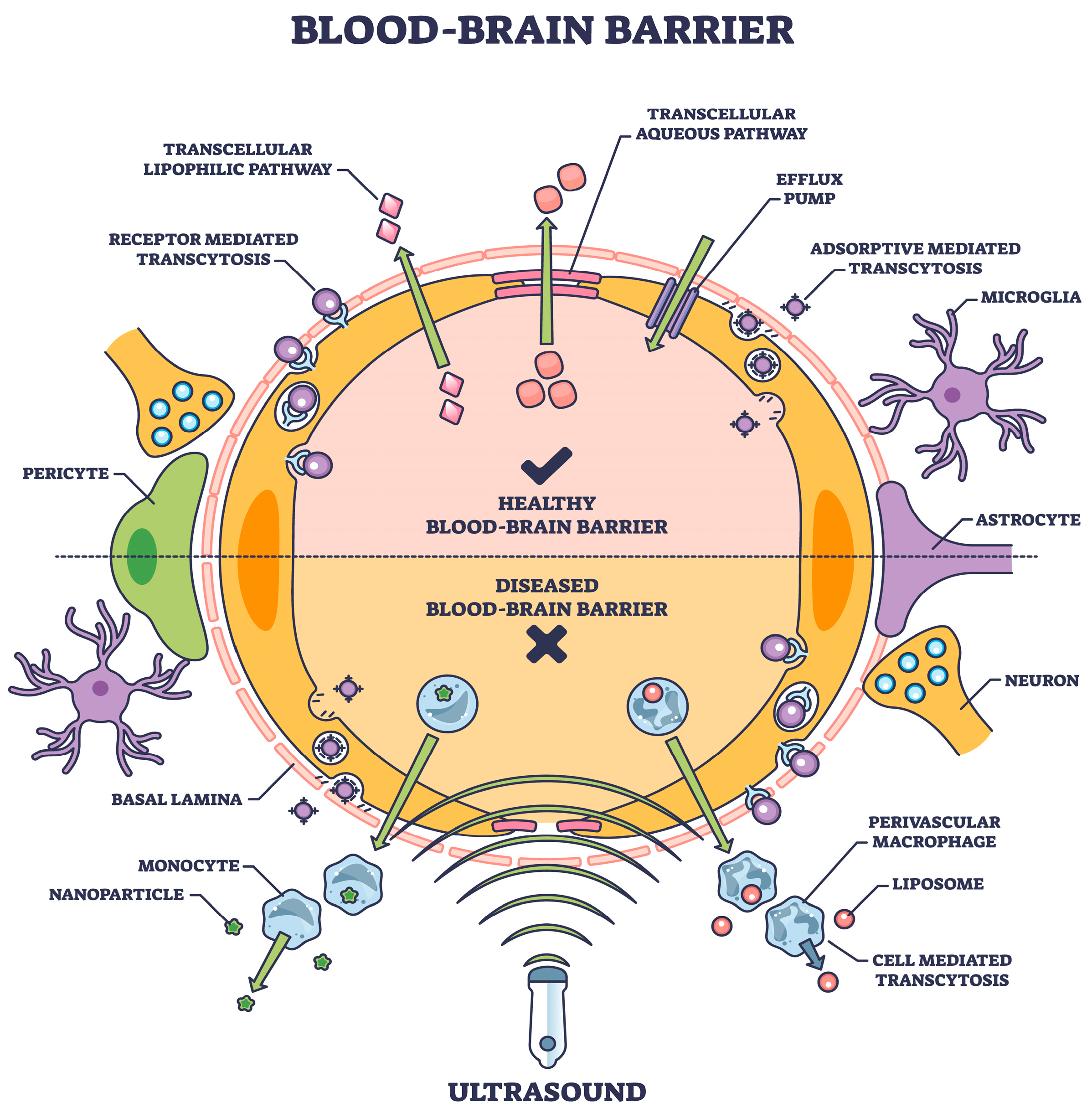
2. Transcytosis
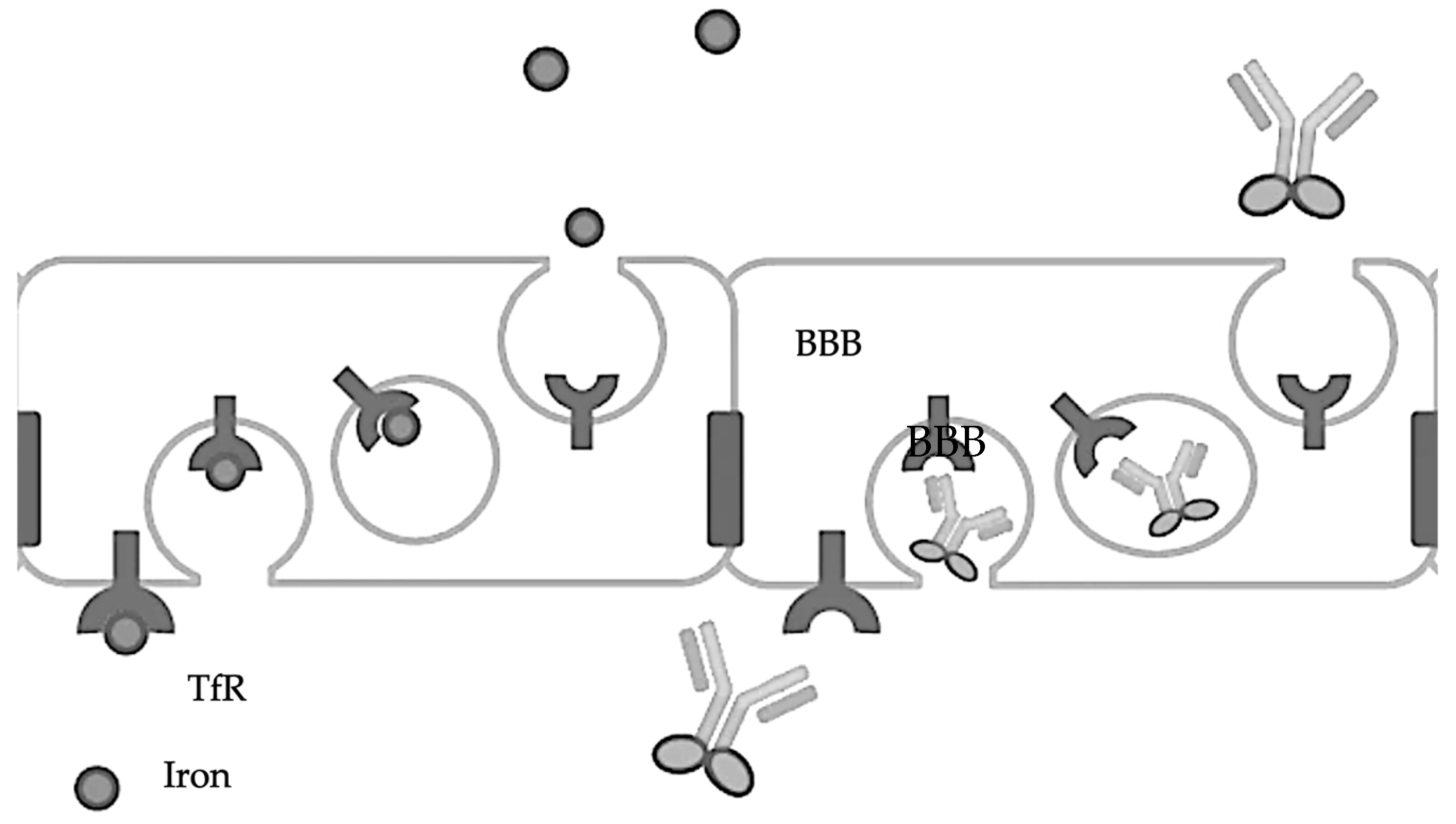
2.1. Transferrin
2.2. Insulin
2.3. Low-Density Lipoprotein (LDL) Receptor-Related Proteins (LRP)
3. Transcytosis Approach
4. Conjugation
5. Linkers
6. Molecular Modeling and Testing
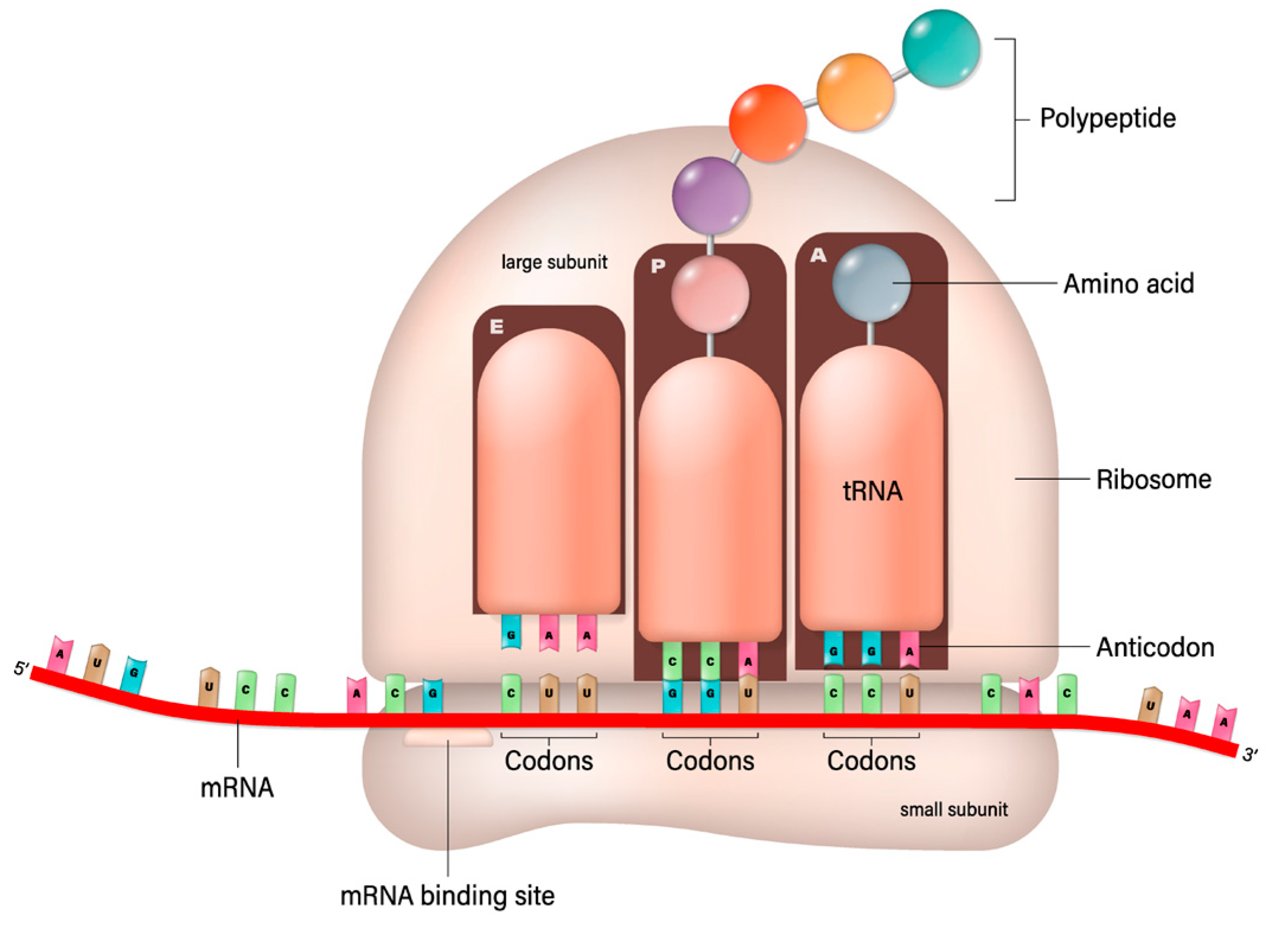
7. mRNA-Based Transcytosis
mRNA Design
8. Regulatory
9. Challenges
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Goedert, M. Neurodegenration: Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J. Clin. Invest. 2005, 115, 1449–1457. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat. Rev. Neurosci. 2016, 17, 251–260. [Google Scholar] [CrossRef]
- Makin, S. The amyloid hypothesis on trial. Nature 2018, 559, S4–S7. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Orr, H.T.; Zoghbi, H.Y. SCA1 molecular genetics: A history of a 13 year collaboration against glutamines. Hum. Mol. Genet. 2001, 10, 2307–2311. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Foerster, B.R.; Welsh, R.C.; Feldman, E.L. 25 years of neuroimaging in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2013, 9, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Pappu, R.V.; Taylor, J.P. Beyond aggregation: Pathological phase transitions in neurodegenerative disease. Science 2020, 370, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.; Yau, W.M.; Mattson, M.P.; Tycko, R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D. Rogue antibodies involved in almost one-fifth of COVID deaths. Nature 2021, 597, 162. [Google Scholar] [CrossRef] [PubMed]
- Zakariya, S.M.; Zehra, A.; Khan, R.H. Biophysical Insight into Protein Folding, Aggregate Formation and its Inhibition Strategies. Protein Pept. Lett. 2022, 29, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Osse, A.M.L.; Cammann, D.; Powell, J.; Chen, J. Anti-Amyloid Monoclonal Antibodies for the Treatment of Alzheimer’s Disease. BioDrugs 2024, 38, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J. Anti-Amyloid Monoclonal Antibodies are Transformative Treatments that Redefine Alzheimer’s Disease Therapeutics. Drugs 2023, 83, 569–576. [Google Scholar] [CrossRef]
- Association, A.s. Aducanumab to Be Discontinued as an Alzheimer’s Treatment. Available online: https://www.alz.org/alzheimers-dementia/treatments/aducanumab (accessed on 14 March 2024).
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Post, W. FDA delays Alzheimer’s Drug for Further Review in Surprise Move. Available online: https://www.washingtonpost.com/business/2024/03/08/eli-lilly-alzheimers-donanemab-fda/ (accessed on 14 March 2024).
- Times, N.A.L.S. Drug Relyvrio Fails Clinical Trial and May Be Withdrawn from the Market. Available online: https://www.nytimes.com/2024/03/08/health/als-drug-relyvrio.html (accessed on 14 March 2024).
- Today, P.s.N. Biogen Discontinues Development of Cinpanemab. Available online: https://parkinsonsnewstoday.com/news/biogen-announcement-discontinue-cinpanemab-parkinsons/ (accessed on 14 March 2024).
- Clinicaltrials.gov. Alzheimer’s Disease Antibody Response. Available online: https://clinicaltrials.gov/search?cond=Neurological%20Disorder&aggFilters=studyType:int&term=Antibody%20Response&intr=antibody2024 (accessed on 14 March 2024).
- Pardridge, W.M. Drug targeting to the brain. Pharm. Res. 2007, 24, 1733–1744. [Google Scholar] [CrossRef]
- Fortin, D.; Gendron, C.; Boudrias, M.; Garant, M.P. Enhanced chemotherapy delivery by intraarterial infusion and blood-brain barrier disruption in the treatment of cerebral metastasis. Cancer 2007, 109, 751–760. [Google Scholar] [CrossRef]
- Hynynen, K.; McDannold, N.; Vykhodtseva, N.; Jolesz, F.A. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology 2001, 220, 640–646. [Google Scholar] [CrossRef]
- Burgess, A.; Hynynen, K. Noninvasive and targeted drug delivery to the brain using focused ultrasound. ACS Chem. Neurosci. 2013, 4, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.O.; Swindell, C.S.; Anthony, F.H.; Witman, P.A.; Devanesan, P.; Webb, N.L.; Baker, S.D.; Wolff, A.C.; Donehower, R.C. Tumor targeting by conjugation of DHA to paclitaxel. J. Control Release 2001, 74, 233–236. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Szebeni, J. Stealth liposomes and long circulating nanoparticles: Critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog. Lipid Res. 2003, 42, 463–478. [Google Scholar] [CrossRef]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H., 2nd. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef]
- Jones, A.R.; Shusta, E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef]
- Niazi, S.K. Non-Invasive Drug Delivery across the Blood-Brain Barrier: A Prospective Analysis. Pharmaceutics 2023, 15, 2599. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Pardridge, W.M. Biopharmaceutical drug targeting to the brain. J. Drug Target. 2010, 18, 157–167. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Geerts, H.; Bergeler, S.; Walker, M.; van der Graaf, P.H.; Courade, J.P. Analysis of clinical failure of anti-tau and anti-synuclein antibodies in neurodegeneration using a quantitative systems pharmacology model. Sci. Rep. 2023, 13, 14342. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.T.; Ma, C.; Li, G.J.; Zheng, X.Y.; Hao, Y.T.; Yang, Y.; Wang, X. Application of Antibody Fragments Against Aβ With Emphasis on Combined Application With Nanoparticles in Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 654611. [Google Scholar] [CrossRef] [PubMed]
- Kniesel, U.; Wolburg, H. Tight Junctions of the Blood–Brain Barrier. Cell. Mol. Neurobiol. 2000, 20, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Moos, T.; Morgan, E.H. Transferrin and transferrin receptor function in brain barrier systems. Cell Mol. Neurobiol. 2000, 20, 77–95. [Google Scholar] [CrossRef]
- Gao, Y.; Zhu, J.; Lu, H. Single domain antibody-based vectors in the delivery of biologics across the blood-brain barrier: A review. Drug Deliv. Transl. Res. 2021, 11, 1818–1828. [Google Scholar] [CrossRef] [PubMed]
- Bien-Ly, N.; Yu, Y.J.; Bumbaca, D.; Elstrott, J.; Boswell, C.A.; Zhang, Y.; Luk, W.; Lu, Y.; Dennis, M.S.; Weimer, R.M.; et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 2014, 211, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow. Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pardridge, W.M. Blood-brain barrier targeting of BDNF improves motor function in rats with middle cerebral artery occlusion. Brain Res. 2006, 1111, 227–229. [Google Scholar] [CrossRef]
- Boado, R.J.; Zhang, Y.; Zhang, Y.; Pardridge, W.M. Humanization of anti-human insulin receptor antibody for drug targeting across the human blood-brain barrier. Biotechnol. Bioeng. 2007, 96, 381–391. [Google Scholar] [CrossRef]
- Banks, W.A. The source of cerebral insulin. Eur. J. Pharmacol. 2004, 490, 5–12. [Google Scholar] [CrossRef]
- Storck, S.E.; Meister, S.; Nahrath, J.; Meißner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.E.; Bouter, Y.; Prikulis, I.; et al. Endothelial LRP1 transports amyloid-β(1-42) across the blood-brain barrier. J. Clin. Invest. 2016, 126, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef]
- Liao, Y.; Du, X.; Li, J.; Lönnerdal, B. Human milk exosomes and their microRNAs survive digestion in vitro and are taken up by human intestinal cells. Mol. Nutr. Food Res. 2017, 61, 1700082. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain barrier drug delivery of IgG fusion proteins with a transferrin receptor monoclonal antibody. Expert. Opin. Drug Deliv. 2015, 12, 207–222. [Google Scholar] [CrossRef]
- Liao, G.S.; Li, X.B.; Zhang, C.Y.; Shu, Y.Y.; Tang, S.X. Pharmacological actions of nerve growth factor-transferrin conjugate on the central nervous system. J. Nat. Toxins 2001, 10, 291. [Google Scholar]
- Aloe, L.; Rocco, M.L.; Balzamino, B.O.; Micera, A. Nerve Growth Factor: A Focus on Neuroscience and Therapy. Curr. Neuropharmacol. 2015, 13, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Charles, V.; Bayer, R.; Bartus, R.T.; Putney, S.; Walus, L.R.; Friden, P.M. Intravenous administration of a transferrin receptor antibody-nerve growth factor conjugate prevents the degeneration of cholinergic striatal neurons in a model of Huntington disease. Proc. Natl. Acad. Sci. USA 1994, 91, 9077–9080. [Google Scholar] [CrossRef] [PubMed]
- Albeck, D.S.; Hoffer, B.J.; Quissell, D.O.; Sanders, L.A.; Zerbe, G.O.; Granholm, A.-C.E. A non-invasive transport system for GDNF across the blood-brain barrier. Neuroreport 1997, 8, 2293–2298. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.M.; Candy, J.M.; Court, J.A.; Whitford, C.A.; Edwardson, J.A. The role of transferrin in the uptake of aluminium and manganese by the IMR 32 neuroblastoma cell line. Biochem. Soc. Trans. 1987, 15, 498. [Google Scholar] [CrossRef]
- Harmatz, P.; Giugliani, R.; Martins, A.M.; Hamazaki, T.; Kubo, T.; Kira, R.; Minami, K.; Ikeda, T.; Moriuchi, H.; Kawashima, S.; et al. alpha-L-iduronidase fused with humanized anti-human transferrin receptor antibody (lepunafusp alfa) for mucopolysaccharidosis type I: A phase 1/2 trial. Mol. Ther. 2024, 32, 609–618. [Google Scholar] [CrossRef]
- Clarke, E.; Stocki, P.; Sinclair, E.H.; Gauhar, A.; Fletcher, E.J.R.; Krawczun-Rygmaczewska, A.; Duty, S.; Walsh, F.S.; Doherty, P.; Rutkowski, J.L. A Single Domain Shark Antibody Targeting the Transferrin Receptor 1 Delivers a TrkB Agonist Antibody to the Brain and Provides Full Neuroprotection in a Mouse Model of Parkinson’s Disease. Pharmaceutics 2022, 14, 1335. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, T.; Eto, Y.; Sakai, N.; Nakamura, K.; Yamamoto, T.; Yamaoka, M.; Ikeda, T.; So, S.; Tanizawa, K.; Sonoda, H.; et al. A Phase 2/3 Trial of Pabinafusp Alfa, IDS Fused with Anti-Human Transferrin Receptor Antibody, Targeting Neurodegeneration in MPS-II. Mol. Ther. 2021, 29, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sun, J.; Castellanos, D.M.; Pardridge, W.M.; Sumbria, R.K. Eliminating Fc N-Linked Glycosylation and Its Impact on Dosing Consideration for a Transferrin Receptor Antibody-Erythropoietin Fusion Protein in Mice. Mol. Pharm. 2020, 17, 2831–2839. [Google Scholar] [CrossRef]
- Ramalho, M.J.; Loureiro, J.A.; Coelho, M.A.N.; Pereira, M.C. Transferrin Receptor-Targeted Nanocarriers: Overcoming Barriers to Treat Glioblastoma. Pharmaceutics 2022, 14, 279. [Google Scholar] [CrossRef]
- Johnsen, K.B.; Bak, M.; Melander, F.; Thomsen, M.S.; Burkhart, A.; Kempen, P.J.; Andresen, T.L.; Moos, T. Modulating the antibody density changes the uptake and transport at the blood-brain barrier of both transferrin receptor-targeted gold nanoparticles and liposomal cargo. J. Control. Release 2019, 295, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Bak, M.; Kempen, P.J.; Melander, F.; Burkhart, A.; Thomsen, M.S.; Nielsen, M.S.; Moos, T.; Andresen, T.L. Antibody affinity and valency impact brain uptake of transferrin receptor-targeted gold nanoparticles. Theranostics 2018, 8, 3416–3436. [Google Scholar] [CrossRef]
- Hultqvist, G.; Syvanen, S.; Fang, X.T.; Lannfelt, L.; Sehlin, D. Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor. Theranostics 2017, 7, 308–318. [Google Scholar] [CrossRef]
- Cabezon, I.; Manich, G.; Martin-Venegas, R.; Camins, A.; Pelegri, C.; Vilaplana, J. Trafficking of Gold Nanoparticles Coated with the 8D3 Anti-Transferrin Receptor Antibody at the Mouse Blood-Brain Barrier. Mol. Pharm. 2015, 12, 4137–4145. [Google Scholar] [CrossRef]
- Sumbria, R.K.; Hui, E.K.; Lu, J.Z.; Boado, R.J.; Pardridge, W.M. Disaggregation of amyloid plaque in brain of Alzheimer’s disease transgenic mice with daily subcutaneous administration of a tetravalent bispecific antibody that targets the transferrin receptor and the Abeta amyloid peptide. Mol. Pharm. 2013, 10, 3507–3513. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Delgado, T.; Rodriguez, J.A.; Helguera, G.; Penichet, M.L. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin. Immunol. 2006, 121, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Wilchek, M.; Bayer, E.A. The avidin-biotin complex in bioanalytical applications. Anal. Biochem. 1988, 171, 1–32. [Google Scholar] [CrossRef]
- Sasso, J.M.; Tenchov, R.; Bird, R.; Iyer, K.A.; Ralhan, K.; Rodriguez, Y.; Zhou, Q.A. The Evolving Landscape of Antibody–Drug Conjugates: In Depth Analysis of Recent Research Progress. Bioconjugate Chem. 2023, 34, 1951–2000. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Hervé, F.; Ghinea, N.; Scherrmann, J.M. CNS delivery via adsorptive transcytosis. AAPS J. 2008, 10, 455–472. [Google Scholar] [CrossRef]
- Salvati, E.; Re, F.; Sesana, S.; Cambianica, I.; Sancini, G.; Masserini, M.; Gregori, M. Liposomes functionalized to overcome the blood-brain barrier and to target amyloid-β peptide: The chemical design affects the permeability across an in vitro model. Int. J. Nanomedicine 2013, 8, 1749–1758. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- Pardridge, W.M. A Historical Review of Brain Drug Delivery. Pharmaceutics 2022, 14, 1283. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Sonawane, P.; Kumar, A.; Singh, H.; Naumovich, V.; Pathak, P.; Grishina, M.; Khalilullah, H.; Jaremko, M.; Emwas, A.H.; et al. Challenges and Opportunities in the Crusade of BRAF Inhibitors: From 2002 to 2022. ACS Omega 2023, 8, 27819–27844. [Google Scholar] [CrossRef]
- Limongi, T.; Susa, F.; Marini, M.; Allione, M.; Torre, B.; Pisano, R.; di Fabrizio, E. Lipid-Based Nanovesicular Drug Delivery Systems. Nanomaterials 2021, 11, 3391. [Google Scholar] [CrossRef]
- Tang, S.; Meng, Q.; Sun, H.; Su, J.; Yin, Q.; Zhang, Z.; Yu, H.; Chen, L.; Gu, W.; Li, Y. Dual pH-sensitive micelles with charge-switch for controlling cellular uptake and drug release to treat metastatic breast cancer. Biomaterials 2017, 114, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Beam, K.S.; Mixan, B.; Wahl, A.F. Identification and activities of human carboxylesterases for the activation of CPT-11, a clinically approved anticancer drug. Bioconjug Chem. 2001, 12, 1074–1080. [Google Scholar] [CrossRef]
- Zhuo, S.; Zhang, F.; Yu, J.; Zhang, X.; Yang, G.; Liu, X. pH-Sensitive Biomaterials for Drug Delivery. Molecules 2020, 25, 5649. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef]
- Yepes, M.; Sandkvist, M.; Moore, E.G.; Bugge, T.H.; Strickland, D.K.; Lawrence, D.A. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J. Clin. Invest. 2003, 112, 1533–1540. [Google Scholar] [CrossRef]
- Ferrari, E.; Soloviev, M.; Niranjan, D.; Arsenault, J.; Gu, C.; Vallis, Y.; O‘Brien, J.; Davletov, B. Assembly of protein building blocks using a short synthetic peptide. Bioconjug Chem. 2012, 23, 479–484. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed]
- Minibaeva, G.; Ivanova, A.; Polishchuk, P. EasyDock: Customizable and scalable docking tool. J. Cheminformatics 2023, 15, 102. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Repasky, M.P.; Shelley, M.; Friesner, R.A. Flexible ligand docking with Glide. Curr. Protoc. Bioinform. 2007, 18, 8.12.1–8.12.36. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.J.; Moughon, S.; Wang, C.; Schueler-Furman, O.; Kuhlman, B.; Rohl, C.A.; Baker, D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. [Google Scholar] [CrossRef]
- Johnson, K.A.; Fox, N.C.; Sperling, R.A.; Klunk, W.E. Brain imaging in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006213. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Argos, P. An investigation of oligopeptides linking domains in protein tertiary structures and possible candidates for general gene fusion. J. Mol. Biol. 1990, 211, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.; Wells, J.A. Dissecting the catalytic triad of a serine protease. Nature 1988, 332, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Urry, D.W. Physical Chemistry of Biological Free Energy Transduction As Demonstrated by Elastic Protein-Based Polymers. J. Phys. Chem. B 1997, 101, 11007–11028. [Google Scholar] [CrossRef]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001, 14, 529–532. [Google Scholar] [CrossRef]
- Huang, X.; Kong, N.; Zhang, X.; Cao, Y.; Langer, R.; Tao, W. The landscape of mRNA nanomedicine. Nat. Med. 2022, 28, 2273–2287. [Google Scholar] [CrossRef] [PubMed]
- Blakney, A.K.; Ip, S.; Geall, A.J. An Update on Self-Amplifying mRNA Vaccine Development. Vaccines 2021, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K. The FDA’s New Guideline “Generally Accepted Scientific Knowledge” (GASK): An Opportunity to Expedite the Approval of Biosimilars. Pharmaceuticals 2023, 16, 1517. [Google Scholar] [CrossRef] [PubMed]
- Ban, W.; You, Y.; Yang, Z. Imaging Technologies for Cerebral Pharmacokinetic Studies: Progress and Perspectives. Biomedicines 2022, 10, 2447. [Google Scholar] [CrossRef] [PubMed]
- Marathe, P.H.; Shyu, W.C.; Humphreys, W.G. The use of radiolabeled compounds for ADME studies in discovery and exploratory development. Curr. Pharm. Des. 2004, 10, 2991–3008. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ren, J.; Si, X.; Sun, Z.; Wang, P.; Zhang, X.; Liu, K.; Wei, B. The global mRNA vaccine patent landscape. Hum. Vaccin. Immunother. 2022, 18, 2095837. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K. RNA Therapeutics: A Healthcare Paradigm Shift. Biomedicines 2023, 11, 1275. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, J.; Xu, Q. Current Developments and Challenges of mRNA Vaccines. Annu. Rev. Biomed. Eng. 2022, 24, 85–109. [Google Scholar] [CrossRef]
- Brende, B.; Farrar, J.; Gashumba, D.; Moedas, C.; Mundel, T.; Shiozaki, Y.; Vardhan, H.; Wanka, J.; Røttingen, J.A. CEPI-a new global R&D organisation for epidemic preparedness and response. Lancet 2017, 389, 233–235. [Google Scholar] [CrossRef]
- Md Khairi, L.N.H.; Fahrni, M.L.; Lazzarino, A.I. The Race for Global Equitable Access to COVID-19 Vaccines. Vaccines 2022, 10, 1306. [Google Scholar] [CrossRef]
- Yoo, K.J.; Mehta, A.; Mak, J.; Bishai, D.; Chansa, C.; Patenaude, B. COVAX and equitable access to COVID-19 vaccines. Bull. World Health Organ. 2022, 100, 315–328. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Regional Office for Europe. The mRNA Vaccine Technology Transfer Hub. Available online: https://www.who.int/initiatives/the-mrna-vaccine-technology-transfer-hub (accessed on 14 March 2024).
- Zuber, C.; Mitteregger, G.; Schuhmann, N.; Rey, C.; Knackmuss, S.; Rupprecht, W.; Reusch, U.; Pace, C.; Little, M.; Kretzschmar, H.A.; et al. Delivery of single-chain antibodies (scFvs) directed against the 37/67 kDa laminin receptor into mice via recombinant adeno-associated viral vectors for prion disease gene therapy. J. Gen. Virol. 2008, 89, 2055–2061. [Google Scholar] [CrossRef] [PubMed]
- Waraho-Zhmayev, D.; Meksiriporn, B.; Portnoff, A.D.; DeLisa, M.P. Optimizing recombinant antibodies for intracellular function using hitchhiker-mediated survival selection. Protein Eng. Des. Sel. 2014, 27, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K.; Magoola, M. mRNA and Synthesis-Based Therapeutic Proteins: A Non-Recombinant Affordable Option. Biologics 2023, 3, 355–379. [Google Scholar] [CrossRef]
- Klein, K.; Gencoglu, M.; Heisterberg, J.; Acha, V.; Stolk, P. The Global Landscape of Manufacturers of Follow-on Biologics: An Overview of Five Major Biosimilar Markets and 15 Countries. BioDrugs 2023, 37, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.L.; Weinberg, M.S.; Arnold, S.E. Cost-effectiveness of Aducanumab and Donanemab for Early Alzheimer Disease in the US. JAMA Neurol. 2022, 79, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K. Making COVID-19 mRNA vaccines accessible: Challenges resolved. Expert. Rev. Vaccines 2022, 21, 1163–1176. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
 | |
| Element | Description |
| Cap (2) | A modified 5′-cap1 structure (m7G+m3′-5′-ppp-5′-Am): GA |
| 5′-UTR (52) | The 5′-untranslated region derived from human alpha-globin RNA with an optimized Kozak sequence. GAATAAACTAGTATTCTTCTGGTCCCCACAGACTCAGAGAGAACCCGCCACC |
| Signal peptide (48) | S glycoprotein signal peptide (extended leader sequence) guides translocation of the nascent polypeptide chain into the endoplasmic reticulum: ATGTTCGTGTTCCTGGTGCTGCTGCCTCTGGTGTCCAGCCAGTGTGTG |
| Coding region (n) | Codon-optimized sequence (ORF). Replace U with Ψ, but mRNA sequencing projections require replacement with T for projections. |
| 3′-UTR (268) | The 3′ untranslated region comprises two sequence elements derived from the amino-terminal enhancer of split (AES) mRNA and the mitochondrial encoded 12S ribosomal RNA to confer RNA stability and high total protein expression: GCTAGCTGCCCCTTTCCCGTCCTGGGTACCCCGAGTCTCCCCCGACCTCGGGTCCCAGGTATGCTCCCACCTCCACCTGCCCCACTCACCACCTCTGCTAGTTCCAGACACCTCCCAAGCACGCAGCAATGCAGCTCAAAACGCTTAGCCTAGCCACACCCCCACGGGAAACAGCAGTGATTAACCTTTAGCAATAAACGAAAGTTTAACTAAGCTATACTAACCCCAGGGTTGGTCAATTTCGTGCCAGCCACACCCTGGAGCTAGC |
| poly(A) (110) | A 110-nucleotide poly(A)-tail consisting of a stretch of 30 adenosine residues, followed by a 10-nucleotide linker sequence and another 70 adenosine residues: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGCATATGACTAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, S.K.; Magoola, M. Transcytosis-Driven Treatment of Neurodegenerative Disorders by mRNA-Expressed Antibody–Transferrin Conjugates. Biomedicines 2024, 12, 851. https://doi.org/10.3390/biomedicines12040851
Niazi SK, Magoola M. Transcytosis-Driven Treatment of Neurodegenerative Disorders by mRNA-Expressed Antibody–Transferrin Conjugates. Biomedicines. 2024; 12(4):851. https://doi.org/10.3390/biomedicines12040851
Chicago/Turabian StyleNiazi, Sarfaraz K., and Matthias Magoola. 2024. "Transcytosis-Driven Treatment of Neurodegenerative Disorders by mRNA-Expressed Antibody–Transferrin Conjugates" Biomedicines 12, no. 4: 851. https://doi.org/10.3390/biomedicines12040851
APA StyleNiazi, S. K., & Magoola, M. (2024). Transcytosis-Driven Treatment of Neurodegenerative Disorders by mRNA-Expressed Antibody–Transferrin Conjugates. Biomedicines, 12(4), 851. https://doi.org/10.3390/biomedicines12040851