

Exploring MiR-484 Regulation by Polyalthia longifolia: A Promising Biomarker and Therapeutic Target in Cervical Cancer through Integrated Bioinformatics and an In Vitro Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. MiR-484 Expression in Pan-Cancers
2.3. Clinical Value of miR-484 in CC
2.4. Differential Expression Analysis and Functional Implications in CC
2.5. Relationship between miR-484 Expression and Immune Landscape in Patients with CC
2.6. Tumor Stemness
2.7. Selection of m6A, m5C, and m1A Genes
2.8. Prediction of the Sensitivity Response to Therapeutic Agents
2.9. Construction and Verification of the Risk Model
2.10. Construction of an miRNA-mRNA-TF Network and Processing of scRNA-Seq Data
2.11. Relationship between the Risk Model and Immune Background
2.12. Cell Culture and qRT-PCR
2.13. Plant Collection, Extraction, and IC50 Determination
2.14. Transfection of miRNA Mimics
2.15. MTT Cell Proliferation Assay of Transfected HeLa Cells
2.16. Statistical Analyses
3. Results
3.1. MiR-484 Expression in Normal and Tumor Tissues in TCGA
3.2. MiR-484 Correlated Positively with TNM·T Stage
3.3. DEGs between High- and Low-miR-484 Groups in CC and Functional Enrichment Analysis
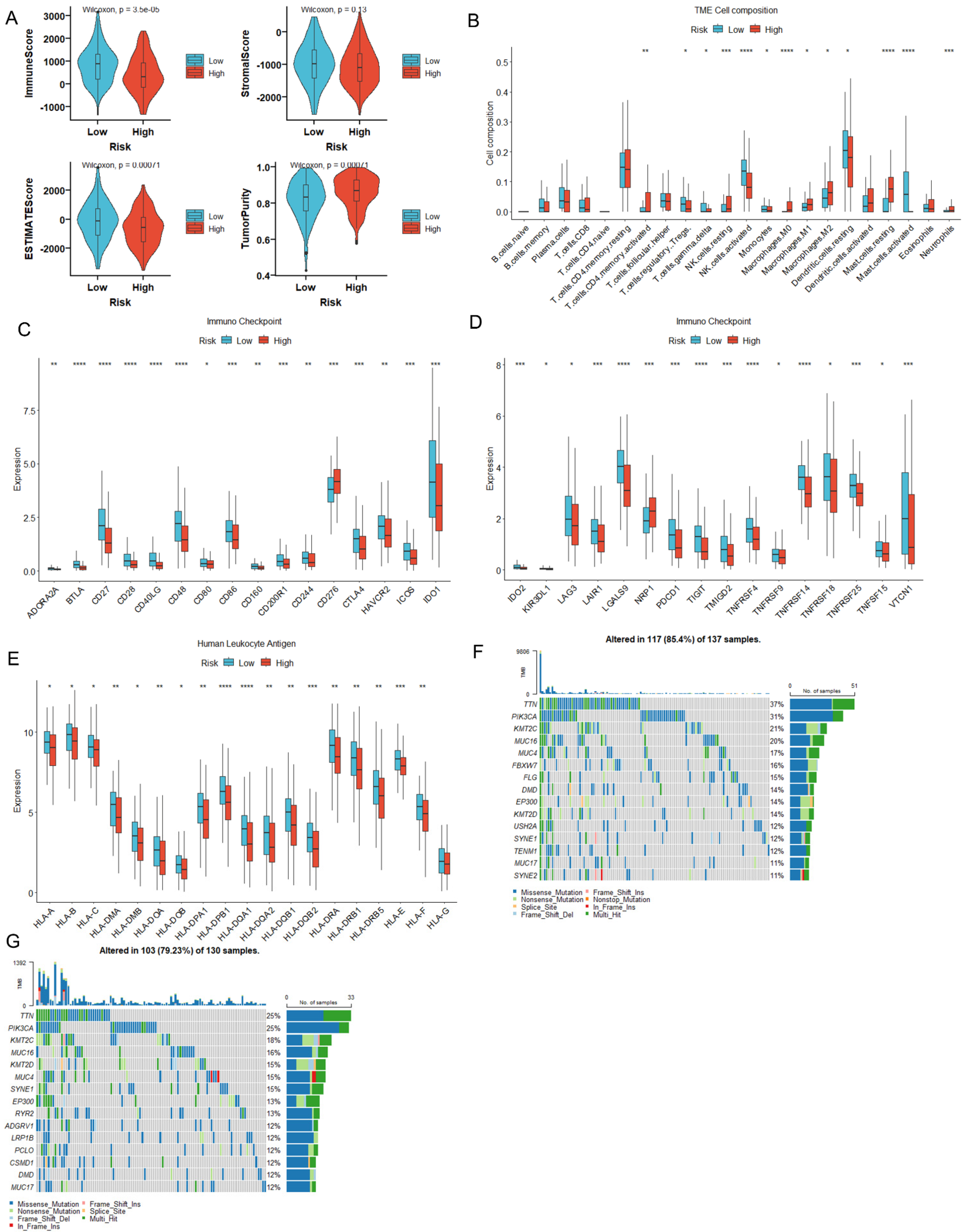
3.4. MiR-484 Expression Correlated with the Infiltration of Immune Cells in CC Tissues
3.5. MiR-484 Expression Correlated with ICPs, Mutation Landscape, and HLA Genes in CC
3.6. MiR-484 Expression Correlated with Tumor Stemness, Hypoxia, and RNA Modification
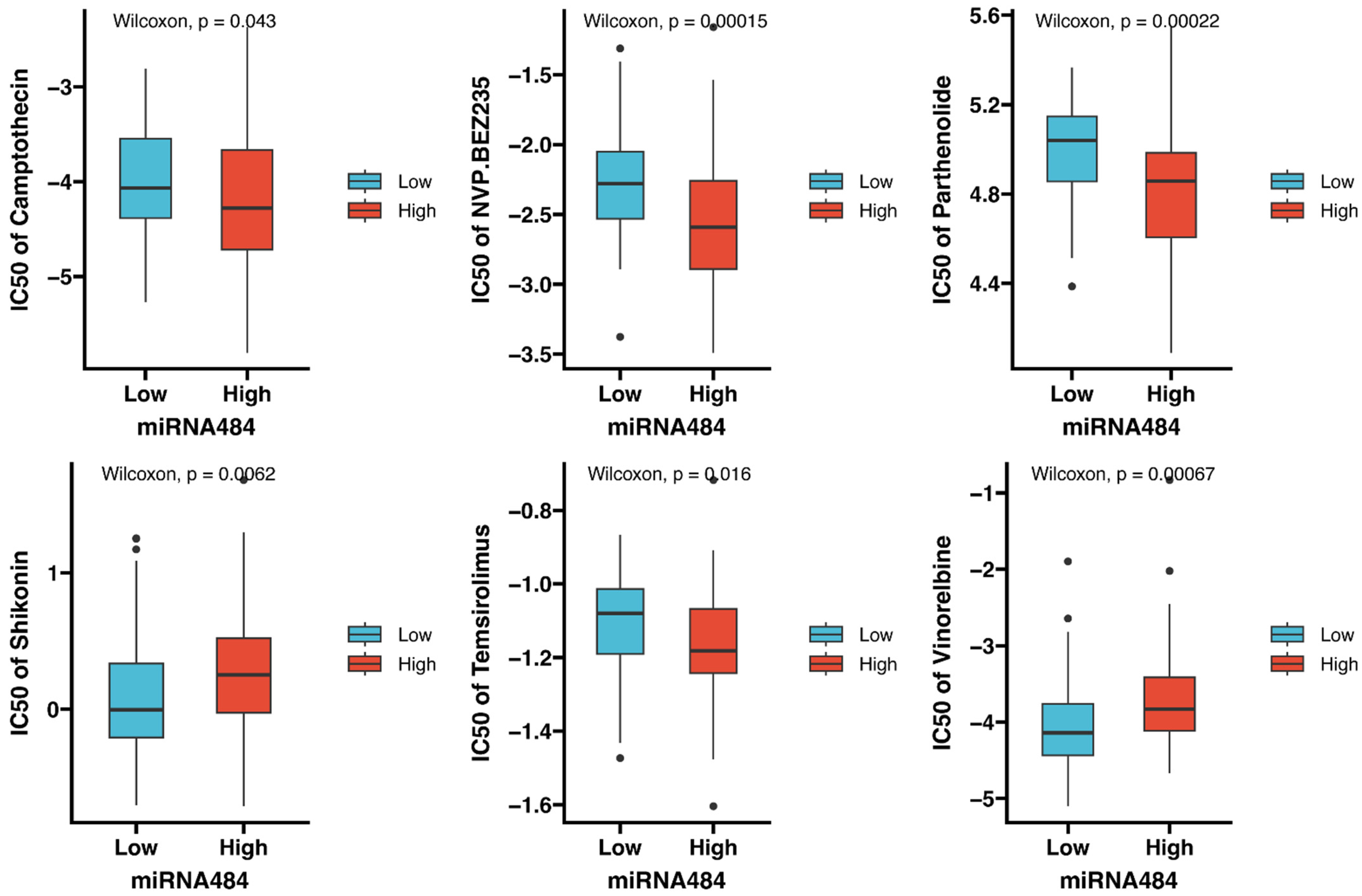
3.7. Identification of Potential Drugs Targeting miR-484
3.8. Construction and Verification of the Risk Model
3.9. MiRNA-mRNA-TF Network and Cell Localization in scRNA-Seq Dataset for Prognostic Genes
3.10. GSEA Analysis for DEGs between High- and Low-Risk Subgroups
3.11. The Immune Landscape and Immunotherapeutic Response of Prognostic Model
3.12. qRT-PCR Validation for Key Prognostic Genes
3.13. MiR-484 Increases the Survival of PLME-Treated HeLa Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Charoentong, P.; Angelova, M.; Efremova, M.; Gallasch, R.; Hackl, H.; Galon, J.; Trajanoski, Z. Bioinformatics for cancer immunology and immunotherapy. Cancer Immunol. Immunother. 2012, 61, 1885–1903. [Google Scholar] [CrossRef]
- Revathidevi, S.; Murugan, A.K.; Nakaoka, H.; Inoue, I.; Munirajan, A.K. APOBEC: A molecular driver in cervical cancer pathogenesis. Cancer Lett. 2021, 496, 104–116. [Google Scholar] [CrossRef]
- Reddy, K.B. MicroRNA (miRNA) in cancer. Cancer Cell Int. 2015, 15, 38. [Google Scholar] [CrossRef]
- Doghish, A.S.; Ali, M.A.; Elyan, S.S.; Elrebehy, M.A.; Mohamed, H.H.; Mansour, R.M.; Elgohary, A.; Ghanem, A.; Faraag, A.H.I.; Abdelmaksoud, N.M.; et al. miRNAs role in cervical cancer pathogenesis and targeted therapy: Signaling pathways interplay. Pathol. Res. Pract. 2023, 244, 154386. [Google Scholar] [CrossRef]
- Wang, J.Y.; Chen, L.J. The role of miRNAs in the invasion and metastasis of cervical cancer. Biosci. Rep. 2019, 39, BSR20181377. [Google Scholar] [CrossRef]
- Jia, Y.Z.; Liu, J.; Wang, G.Q.; Song, Z.F. miR-484: A Potential Biomarker in Health and Disease. Front. Oncol. 2022, 12, 830420. [Google Scholar] [CrossRef]
- El-Maraghy, S.A.; Adel, O.; Zayed, N.; Yosry, A.; El-Nahaas, S.M.; Gibriel, A.A. Circulatory miRNA-484, 524, 615 and 628 expression profiling in HCV mediated HCC among Egyptian patients; implications for diagnosis and staging of hepatic cirrhosis and fibrosis. J. Adv. Res. 2020, 22, 57–66. [Google Scholar] [CrossRef]
- Lee, D.; Tang, W.; Dorsey, T.H.; Ambs, S. miR-484 is associated with disease recurrence and promotes migration in prostate cancer. Biosci. Rep. 2020, 40, BSR20191028. [Google Scholar] [CrossRef]
- Xue, X.; Wang, C.; Xue, Z.; Wen, J.; Han, J.; Ma, X.; Zang, X.; Deng, H.; Guo, R.; Asuquo, I.P. Exosomal miRNA profiling before and after surgery revealed potential diagnostic and prognostic markers for lung adenocarcinoma. Acta Biochim. Biophys. Sin. 2020, 52, 281–293. [Google Scholar] [CrossRef]
- Yi, R.; Feng, J.; Yang, S.; Huang, X.; Liao, Y.; Hu, Z.; Luo, M. miR-484/MAP2/c-Myc-positive regulatory loop in glioma promotes tumor-initiating properties through ERK1/2 signaling. J. Mol. Histol. 2018, 49, 209–218. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.M. MiR-484 suppressed proliferation, migration, invasion and induced apoptosis of gastric cancer via targeting CCL-18. Int. J. Exp. Pathol. 2020, 101, 203–214. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Yao, J.; Li, R.; Fan, X. Downregulation of miR-484 is associated with poor prognosis and tumor progression of gastric cancer. Diagn. Pathol. 2020, 15, 25. [Google Scholar] [CrossRef]
- Shen, Y.; Qi, L.; Li, Y.; Zhang, Y.; Gao, X.; Zhu, Y.; Wang, K. The downregulation of lncRNA PGM5-AS1 inhibits the proliferation and metastasis via increasing miR-484 expression in colorectal cancer. Cancer Biother. Radiopharm. 2021, 36, 220–229. [Google Scholar] [CrossRef]
- Luo, X.; Yue, M.; Li, C.; Sun, D.; Wang, L. Long Non-Coding RNA LINC00239 Functions as a Competitive Endogenous RNA by Sponging microRNA-484 and Enhancing KLF12 Expression to Promote the Oncogenicity of Colorectal Cancer [Retraction]. OncoTargets Ther. 2022, 15, 847–848. [Google Scholar] [CrossRef]
- Li, N.; Yang, G.; Luo, L.; Ling, L.; Wang, X.; Shi, L.; Lan, J.; Jia, X.; Zhang, Q.; Long, Z. lncRNA THAP9-AS1 promotes pancreatic ductal adenocarcinoma growth and leads to a poor clinical outcome via sponging miR-484 and interacting with YAP. Clin. Cancer Res. 2020, 26, 1736–1748. [Google Scholar] [CrossRef]
- Vijayarathna, S.; Oon, C.E.; Al-Zahrani, M.; Abualreesh, M.H.; Chen, Y.; Kanwar, J.R.; Sahreen, S.; Ghazanfar, S.; Adnan, M.; Sasidharan, S. Standardized Polyalthia longifolia leaf extract induces the apoptotic HeLa cells death via microRNA regulation: Identification, validation, and therapeutic potential. Front. Pharmacol. 2023, 14, 1198425. [Google Scholar] [CrossRef]
- Wang, J.; Kang, Z.; Liu, Y.; Li, Z.; Liu, Y.; Liu, J. Identification of immune cell infiltration and diagnostic biomarkers in unstable atherosclerotic plaques by integrated bioinformatics analysis and machine learning. Front. Immunol. 2022, 13, 956078. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Pinto, J.P.; Kalathur, R.K.; Oliveira, D.V.; Barata, T.; Machado, R.S.; Machado, S.; Pacheco-Leyva, I.; Duarte, I.; Futschik, M.E. StemChecker: A web-based tool to discover and explore stemness signatures in gene sets. Nucleic Acids Res. 2015, 43, W72–W77. [Google Scholar] [CrossRef]
- Ru, Y.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.; Radcliffe, R.A.; Bowler, R.; Mahaffey, S.; Rossi, S.; Calin, G.A.; Bemis, L.; et al. The multiMiR R package and database: Integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res. 2014, 42, e133. [Google Scholar] [CrossRef]
- Yu, L.; Shen, N.; Shi, Y.; Shi, X.; Fu, X.; Li, S.; Zhu, B.; Yu, W.; Zhang, Y. Characterization of cancer-related fibroblasts (CAF) in hepatocellular carcinoma and construction of CAF-based risk signature based on single-cell RNA-seq and bulk RNA-seq data. Front. Immunol. 2022, 13, 1009789. [Google Scholar] [CrossRef] [PubMed]
- Vijayarathna, S.; Chen, Y.; Kanwar, J.R.; Sasidharan, S. Standardized Polyalthia longifolia leaf extract (PLME) inhibits cell proliferation and promotes apoptosis: The anti-cancer study with various microscopy methods. Biomed. Pharmacother. 2017, 91, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xiong, C.; Hsu, Y.-C.; Wang, X.; Chen, L. Human leukocyte antigen (HLA) and cancer immunotherapy: HLA-dependent and-independent adoptive immunotherapies. Ann. Blood 2020, 5, 14. [Google Scholar] [CrossRef]
- Yang, Y.; Zuo, S.; Li, W.; Di, M.; Liu, J.; Chai, J.; Wang, J.; Yuan, Y.; Li, M.; Jia, Q. TRIM21 promotes tumor progression and cancer stemness in cervical squamous cell carcinoma. Pathol.-Res. Pract. 2023, 248, 154710. [Google Scholar] [CrossRef] [PubMed]
- Lyng, H.; Malinen, E. Hypoxia in cervical cancer: From biology to imaging. Clin. Transl. Imaging 2017, 5, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; West, C.; O’Connor, J.P.; Choudhury, A.; Hoskin, P. Impact of hypoxia on cervical cancer outcomes. Int. J. Gynecol. Cancer 2021, 31, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mao, Y.; Wang, C.; Jiang, X.; Tang, Q.; Wang, L.; Zhu, J.; Zhao, M. RNA methylation-related genes of m6A, m5C, and m1A predict prognosis and immunotherapy response in cervical cancer. Ann. Med. 2023, 55, 2190618. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Fang, Y.; Wu, K.; Liu, Y.; Zhang, W. Comprehensive gene and pathway analysis of cervical cancer progression. Oncol. Lett. 2020, 19, 3316–3332. [Google Scholar] [CrossRef]
- Wang, Q.; Song, R.; Zhao, C.; Liu, H.; Yang, Y.; Gu, S.; Feng, D.; He, J. HPV16 E6 promotes cervical cancer cell migration and invasion by downregulation of NHERF1. Int. J. Cancer 2019, 144, 1619–1632. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Wang, R.-C.; Cheng, K.; Ring, B.Z.; Su, L. Roles of Rap1 signaling in tumor cell migration and invasion. Cancer Biol. Med. 2017, 14, 90. [Google Scholar]
- Theocharis, A.D.; Karamanos, N.K. Proteoglycans remodeling in cancer: Underlying molecular mechanisms. Matrix Biol. 2019, 75, 220–259. [Google Scholar] [CrossRef]
- Sadri Nahand, J.; Moghoofei, M.; Salmaninejad, A.; Bahmanpour, Z.; Karimzadeh, M.; Nasiri, M.; Mirzaei, H.R.; Pourhanifeh, M.H.; Bokharaei-Salim, F.; Mirzaei, H. Pathogenic role of exosomes and microRNAs in HPV-mediated inflammation and cervical cancer: A review. Int. J. Cancer 2020, 146, 305–320. [Google Scholar] [CrossRef]
- Kurmyshkina, O.V.; Kovchur, P.I.; Schegoleva, L.V.; Volkova, T.O. T-and NK-cell populations with regulatory phenotype and markers of apoptosis in circulating lymphocytes of patients with CIN3 or microcarcinoma of the cervix: Evidence for potential mechanisms of immune suppression. Infect. Agents Cancer 2017, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mao, Z.; Lai, Y.; Wan, T.; Zhang, K.; Zhou, B. A review of the research progress in T-lymphocyte immunity and cervical cancer. Transl. Cancer Res. 2020, 9, 2026–2036. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Sharma, S.; Singhal, P.; Bansal, R.; Khare, A. Significance of Mast Cells in Uterine Cervical Lesions: A Cross-sectional Study. Natl. J. Lab. Med. 2021, 10, PO30–PO33. [Google Scholar] [CrossRef]
- Yao, H.; Jiang, X.; Fu, H.; Yang, Y.; Jin, Q.; Zhang, W.; Cao, W.; Gao, W.; Wang, S.; Zhu, Y.; et al. Exploration of the Immune-Related Long Noncoding RNA Prognostic Signature and Inflammatory Microenvironment for Cervical Cancer. Front. Pharmacol. 2022, 13, 870221. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Holub, K.; Biete, A. Impact of systemic inflammation biomarkers on the survival outcomes of cervical cancer patients. Clin. Transl. Oncol. 2019, 21, 836–844. [Google Scholar] [CrossRef]
- Ferrall, L.; Lin, K.Y.; Roden, R.B.S.; Hung, C.F.; Wu, T.C. Cervical Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2021, 27, 4953–4973. [Google Scholar] [CrossRef]
- Xu, Y.; Luo, H.; Hu, Q.; Zhu, H. Identification of potential driver genes based on multi-genomic data in cervical cancer. Front. Genet. 2021, 12, 598304. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Wang, B.; Zhang, T.; Cui, B. The roles of m6A methylation in cervical cancer: Functions, molecular mechanisms, and clinical applications. Cell Death Dis. 2023, 14, 734. [Google Scholar] [CrossRef]
- Choi, C.H.; Chung, J.-Y.; Kim, J.-H.; Kim, B.-G.; Hewitt, S.M. Expression of fibroblast growth factor receptor family members is associated with prognosis in early stage cervical cancer patients. J. Transl. Med. 2016, 14, 124. [Google Scholar] [CrossRef]
- Mahmood, H.T.N.A.; Tomas Bort, E.; Walker, A.J.; Grose, R.P.; Chioni, A.M. FGF signalling facilitates cervical cancer progression. FEBS J. 2022, 289, 3440–3456. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Lee, Y.; Kim, S. Bioinformatic analysis for the prognostic implication of genes encoding epithelial sodium channel in cervical cancer. Int. J. General. Med. 2022, 15, 1777–1787. [Google Scholar] [CrossRef]
- Ding, S.; Huang, X.; Zhu, J.; Xu, B.; Xu, L.; Gu, D.; Zhang, W. ADH7, miR-3065 and LINC01133 are associated with cervical cancer progression in different age groups. Oncol. Lett. 2020, 19, 2326–2338. [Google Scholar] [CrossRef]
- Yang, B.; Li, M.; Tang, W.; Liu, W.; Zhang, S.; Chen, L.; Xia, J. Dynamic network biomarker indicates pulmonary metastasis at the tipping point of hepatocellular carcinoma. Nat. Commun. 2018, 9, 678. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, S.; Takaoka, Y.; Ueda, Y.; Kitazawa, R. Identification of calmodulin-like protein 5 as tumor-suppressor gene silenced during early stage of carcinogenesis in squamous cell carcinoma of uterine cervix. Int. J. Cancer 2021, 149, 1358–1368. [Google Scholar] [CrossRef]
- Liu, X.; Jia, Y.; Shi, C.; Kong, D.; Wu, Y.; Zhang, T.; Wei, A.; Wang, D. CYP4B1 is a prognostic biomarker and potential therapeutic target in lung adenocarcinoma. PLoS ONE 2021, 16, e0247020. [Google Scholar] [CrossRef]
- Yang, Y.; Yuan, S.; Yan, S.; Dong, K.; Yang, Y. Missense variants in CYP4B1 associated with increased risk of lung cancer among Chinese Han population. World J. Surg. Oncol. 2023, 21, 352. [Google Scholar] [CrossRef]
- Okino, K.; Nagai, H.; Nakayama, H.; Doi, D.; Yoneyama, K.; Konishi, H.; Takeshita, T. Inactivation of Crk SH3 domain-binding guanine nucleotide-releasing factor (C3G) in cervical squamous cell carcinoma. Int. J. Gynecol. Cancer 2006, 16, 763–771. [Google Scholar] [CrossRef]
- Xu, L.-P.; Qiu, H.-B.; Yuan, S.-Q.; Chen, Y.-M.; Zhou, Z.-W.; Chen, Y.-B. Downregulation of PSCA promotes gastric cancer proliferation and is related to poor prognosis. J. Cancer 2020, 11, 2708. [Google Scholar] [CrossRef] [PubMed]
- Ray, T.; Ryusaki, T.; Ray, P.S. Therapeutically targeting cancers that overexpress FOXC1: A transcriptional driver of cell plasticity, partial EMT, and cancer metastasis. Front. Oncol. 2021, 11, 721959. [Google Scholar] [CrossRef]
- Bai, L.; Mao, R.; Wang, J.; Ding, L.; Jiang, S.; Gao, C.; Kang, H.; Chen, X.; Sun, X.; Xu, J. ERK1/2 promoted proliferation and inhibited apoptosis of human cervical cancer cells and regulated the expression of c-Fos and c-Jun proteins. Med. Oncol. 2015, 32, 57. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, R.; Arora, H.; Rizvi, M. EMT in cervical cancer: Its role in tumour progression and response to therapy. Cancer Lett. 2015, 356, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Lu, J.; Ma, J.; Cai, W.; Wangpu, X.; Feng, H.; Zhao, J.; Guan, S.; Zong, Y.; Lu, A. CC motif chemokine ligand 19 suppressed colorectal cancer in vivo accompanied by an increase in IL-12 and IFN-γ. Biomed. Pharmacother. 2015, 69, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Sun, J.; He, C.; Huang, C.; Yu, H. CCL19 suppresses gastric cancer cell proliferation, migration, and invasion through the CCL19/CCR7/AIM2 pathway. Human. Cell 2020, 33, 1120–1132. [Google Scholar] [CrossRef]
- Liu, Q.; Qiao, M.; Lohinai, Z.; Mao, S.; Pan, Y.; Wang, Y.; Yang, S.; Zhou, F.; Jiang, T.; Yi, X. CCL19 associates with lymph node metastasis and inferior prognosis in patients with small cell lung cancer. Lung Cancer 2021, 162, 194–202. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, Y.; Zhang, W.; Alvarez, A.A.; Bai, Y.; Hu, B.; Cheng, S.-Y.; Yang, K.; Li, Y.; Feng, H. Lipolytic inhibitor G0S2 modulates glioma stem-like cell radiation response. J. Exp. Clin. Cancer Res. 2019, 38, 147. [Google Scholar] [CrossRef]
- Fukunaga, T.; Fujita, Y.; Kishima, H.; Yamashita, T. Methylation dependent down-regulation of G0S2 leads to suppression of invasion and improved prognosis of IDH1-mutant glioma. PLoS ONE 2018, 13, e0206552. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Steger, A.; Mahner, S.; Jeschke, U.; Heidegger, H. The formation and therapeutic update of tumor-associated macrophages in cervical cancer. Int. J. Mol. Sci. 2019, 20, 3310. [Google Scholar] [CrossRef] [PubMed]
- Prete, G.D. Human Th1 and Th2 lymphocytes: Their role in the pathophysiology of atopy. Allergy 1992, 47, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Li, R.; Wang, G.; Yong, M.; Ran, W.; Liu, J.; Zhou, H. Expression and clinical significance of Th1/Th2 cells in different HPV types of cervical cancer. Int. J. Clin. Exp. Med. 2019, 12, 9953–9960. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Type | Primer Sequence (5′-3′) |
|---|---|---|
| FGFR3 | Forward | AGGAGCTCTTCAAGCTGCTG |
| Reverse | AGGTCCAGGTACTCGTCGG | |
| SCNN1B | Forward | TGCTACCCAGGCATTGACAG |
| Reverse | CAGGGCTCAGCTCCGAATAG | |
| CALML5 | Forward | GACGGCGAAATCAGCTTCCA |
| Reverse | AACTCCTCGTAGTTCACCCG | |
| GAPDH | Forward | CGAAGGTGGAGTCAACGGATTT |
| Reverse | ATGGGTGGAATCATATTGGAAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, J.; Chen, Y.; Chai, H.C.; Sasidharan, S. Exploring MiR-484 Regulation by Polyalthia longifolia: A Promising Biomarker and Therapeutic Target in Cervical Cancer through Integrated Bioinformatics and an In Vitro Analysis. Biomedicines 2024, 12, 909. https://doi.org/10.3390/biomedicines12040909
Niu J, Chen Y, Chai HC, Sasidharan S. Exploring MiR-484 Regulation by Polyalthia longifolia: A Promising Biomarker and Therapeutic Target in Cervical Cancer through Integrated Bioinformatics and an In Vitro Analysis. Biomedicines. 2024; 12(4):909. https://doi.org/10.3390/biomedicines12040909
Chicago/Turabian StyleNiu, Jiaojiao, Yeng Chen, Hwa Chia Chai, and Sreenivasan Sasidharan. 2024. "Exploring MiR-484 Regulation by Polyalthia longifolia: A Promising Biomarker and Therapeutic Target in Cervical Cancer through Integrated Bioinformatics and an In Vitro Analysis" Biomedicines 12, no. 4: 909. https://doi.org/10.3390/biomedicines12040909
APA StyleNiu, J., Chen, Y., Chai, H. C., & Sasidharan, S. (2024). Exploring MiR-484 Regulation by Polyalthia longifolia: A Promising Biomarker and Therapeutic Target in Cervical Cancer through Integrated Bioinformatics and an In Vitro Analysis. Biomedicines, 12(4), 909. https://doi.org/10.3390/biomedicines12040909