
c-Met and Other Cell Surface Molecules: Interaction, Activation and Functional Consequences

Abstract
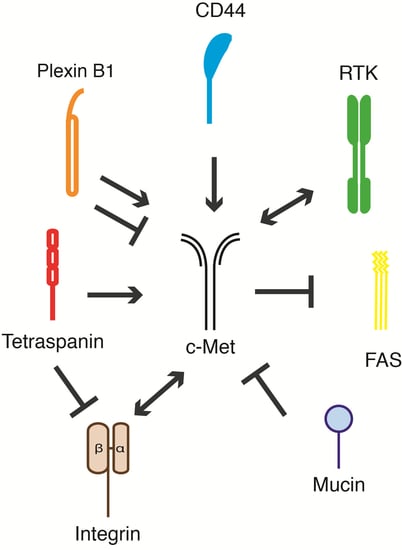
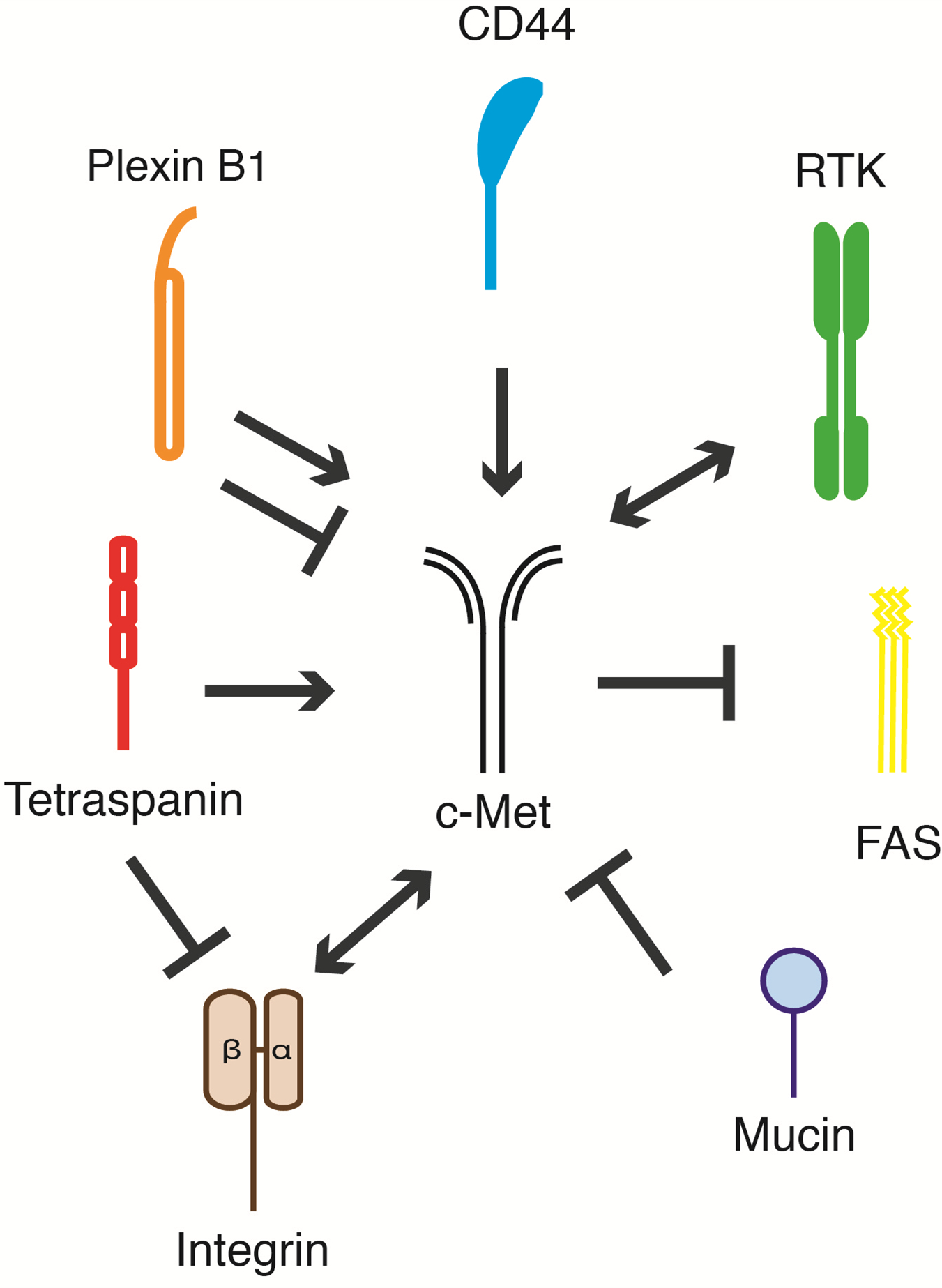
:
1. Introduction
2. c-Met Structure and Activation

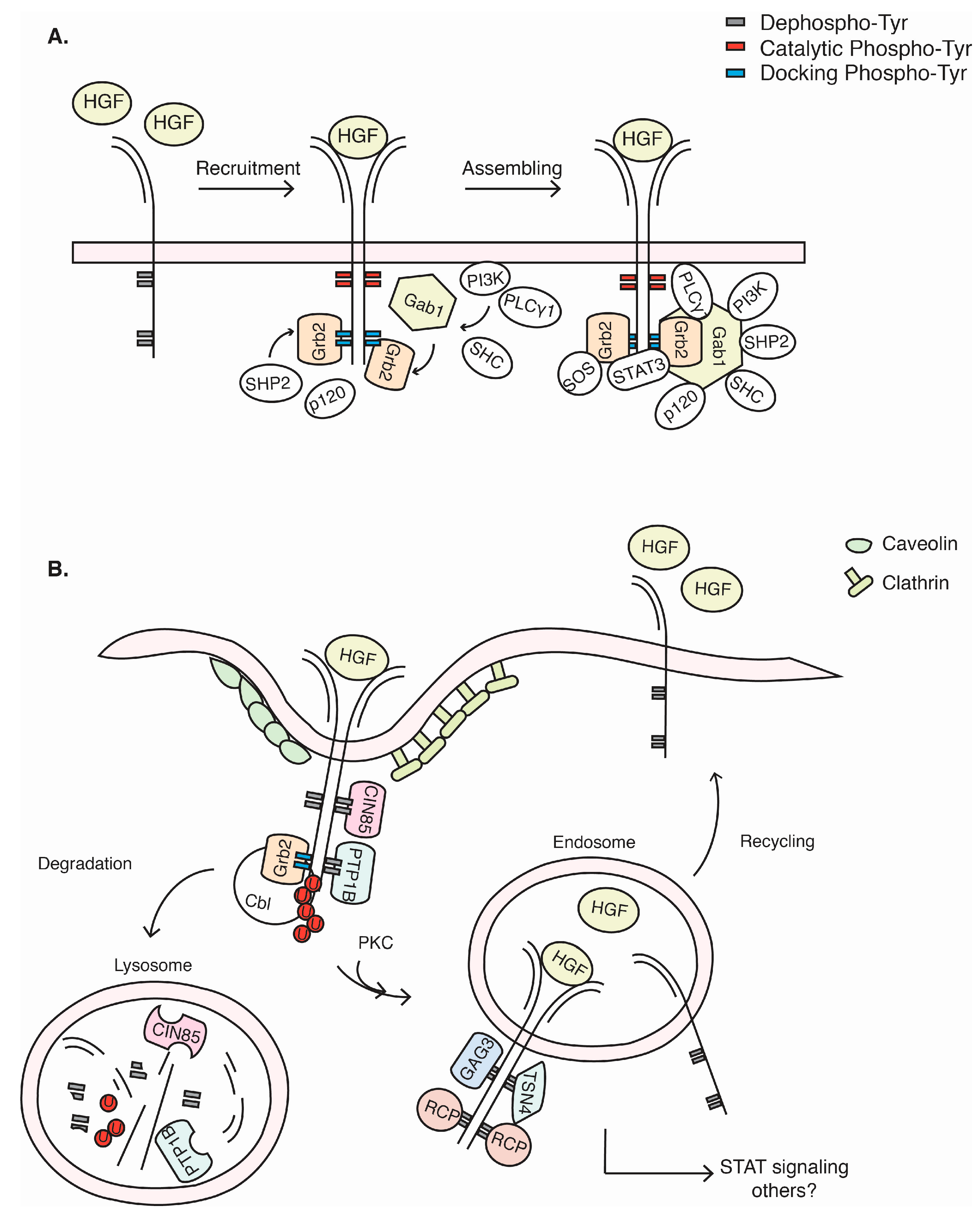
3. c-Met Signalling Cascade
4. c-Met Endocytosis and Recycling
5. c-Met and Its Membrane-Spanning Partner Molecules

{kind=link}
{kind=link}
{kind=link}
| Receptor | Cell System | Effect on c-Met | Biological Response | Reference | |
|---|---|---|---|---|---|
| Plexins | Plexin B1 | HUVEC | Inhibition | ↓ Angiogenesis | [54] |
| HT-29 | Activation | ↑ Invasion | [55] | ||
| SK-BR3, MLP29 | Activation | ↑ Migration ↑ Colony formation, ↑ Invasive growth | [56] | ||
| YUSIK, MDA-MB 468, MCF-7 | Inhibition | ↓ Migration | [57,58,59,60] | ||
| Plexin B3 | HUVECs | Activation | ↑ Migration | [61] | |
| CD44 | CD44v9 | C4-2, LNCap | Activation | ↑ Resistance, invasion | [62] |
| CD44v6 | WM9, WM164, 1205Lu | Activation | ↑ Migration | [63] | |
| HeLa, HT29, HepG2 | Activation | ↑ c-Met internalization, signalling, scattering | [40,64,65] | ||
| fibroblasts | Activation | ↑ Proliferation | [66] | ||
| CD44v10 | Human pulmonary microvascular EC, B-cells | Activation | ↑ EC barrier enhancement, B-cell survival | [67,68] | |
| Tetraspanin | CD151 | AccM, Acc2 | Activation | ↑ Migration, proliferation | [69] |
| MDA-MB-231 | Activation | ↑ Branching morphogenesis | [70] | ||
| GTL-16 | Activation | ↑ Proliferation, anchorage-independent growth | [71] | ||
| CD82 | PC3, Hepa1-6 | Inhibition | ↓ Migration, invasion | [72,73] | |
| Oligodendrocytes (O4+ cells) | Inhibition | ↓ Differentiation | [74] | ||
| HCV29/YTS1 | Inhibition | ↓ Invasion | [75] | ||
| H1299 | Inhibition | ↓ Migration, lamellipodia formation | [76] | ||
| Integrin | α6β4 | GTL-16, A431, MDA-MB-435 | Activation | ↑ Invasive growth | [71,77] |
| MEFs | Activation | ↑ Colony formation, tumour growth | [78] | ||
| DU145 | Activation | ↑ Self-renewal, invasion | [79] | ||
| HLMVEC, HPAEC | Activation | EC barrier integrity | [80] | ||
| α5β1 | HMVEC | Activation | ↑ Migration, proliferation | [81] | |
| SKOV3ip1, HeyA8 | Activation | ↑ Metastasis | [82] | ||
| α3β1 | Mouse papillary cells | Activation | Kidney morphogenesis | [83] | |
| αxβ1 | PC9 | Activation | ↑ Proliferation | [84] | |
| α2β1 | PMCs | Activation | ↑ PMC activation | [85] | |
| RTKs | Ron | NIH3T3 | Reciprocal Activation | ↑ Colony formation | [86] |
| EGFR | A431, HepG2, AKN-1, HuH6, MRC5 | Activation | ↑ c-Met signalling | [87] | |
| PyVmT, MDA-MB231, 4T1, NCl H596, DLD1, HT29 | Activation | ↑ Motility, proliferation | [88,89,90] | ||
| PC-9, HCC827, SNU-16, MKN45, BT474, SKBR3 | Activation | ↑ Drug resistance | [91,92,93] | ||
| 5637 tumour bladder cell line | Activation | ↑ Survival, cell growth | [94] | ||
| ARPE-19 | Activation, Ecto-domain shedding | ↑ Wound healing | [95] | ||
| A549 | Ecto-domain shedding | NA | [96] | ||
| H1993, EBC1 | Activation | ↑ Survival, proliferation | [97] | ||
| H1975, H520, A549 | Activation | ↑ Tumour growth and survival | [98] | ||
| GEO-CR, SW48-CR | Activation | ↑ c-Met phosphorylation, ↑ Survival | [99] | ||
| 32D, PC9 | Activation | ↑ c-Met phosphorylation, metastasis, invasion and colony formation | [100] | ||
| 201T, A549 | Activation | ↑ c-Met phosphorylation, xenograft growth | [101] | ||
| Her2 | SK-BR3, BT474 | Activation | ↑ Drug resistance | [102] | |
| H1993, EBC1 | Activation | ↑ Survival, proliferation, ↑ Migration | [97] | ||
| MDCK | Activation | ↑ EMT | [103] | ||
| Her3 | H1993, EBC1 | Activation | ↑ Survival, proliferation | [97] | |
| HCC827, | NA (HER3 activation) * | ↑ Drug resistance | [104] | ||
| MKN45, GTL16 | Activation | ↑ Drug resistance | [91,105] | ||
| IGFR | L3.6pl | Activation | ↑ Migration, invasion | [106] | |
| RET | H1993, EBC1 | Activation | ↑ Migration | [97] | |
| Death receptors | Fas | HepG2, Hepa1-6 | No effect | ↓ Apoptosis | [107,108] |
| HUVECs | NA | ↓ Apoptosis | [109] | ||
| DR5 | Medulloblastoma/glioma cell lines | NA | ↓ Apoptosis | [110] | |
| Mucins | Muc1 | Panc-1, HPAF2, MDA-MB-435, Mahlavu, SNU-449 | Inhibition | ↓ Invasion, EMT | [111,112,113] |
| Muc20 | HEK293, CHO-K1 | Inhibition | ↓ Invasion, EMT | [114] | |
| NRP1 | PCa cells | Activation | ↑ Bone metastasis | [115] | |
| ICAM1 | HT29, HepG2 | Activation | ↑ Proliferation | [116] | |

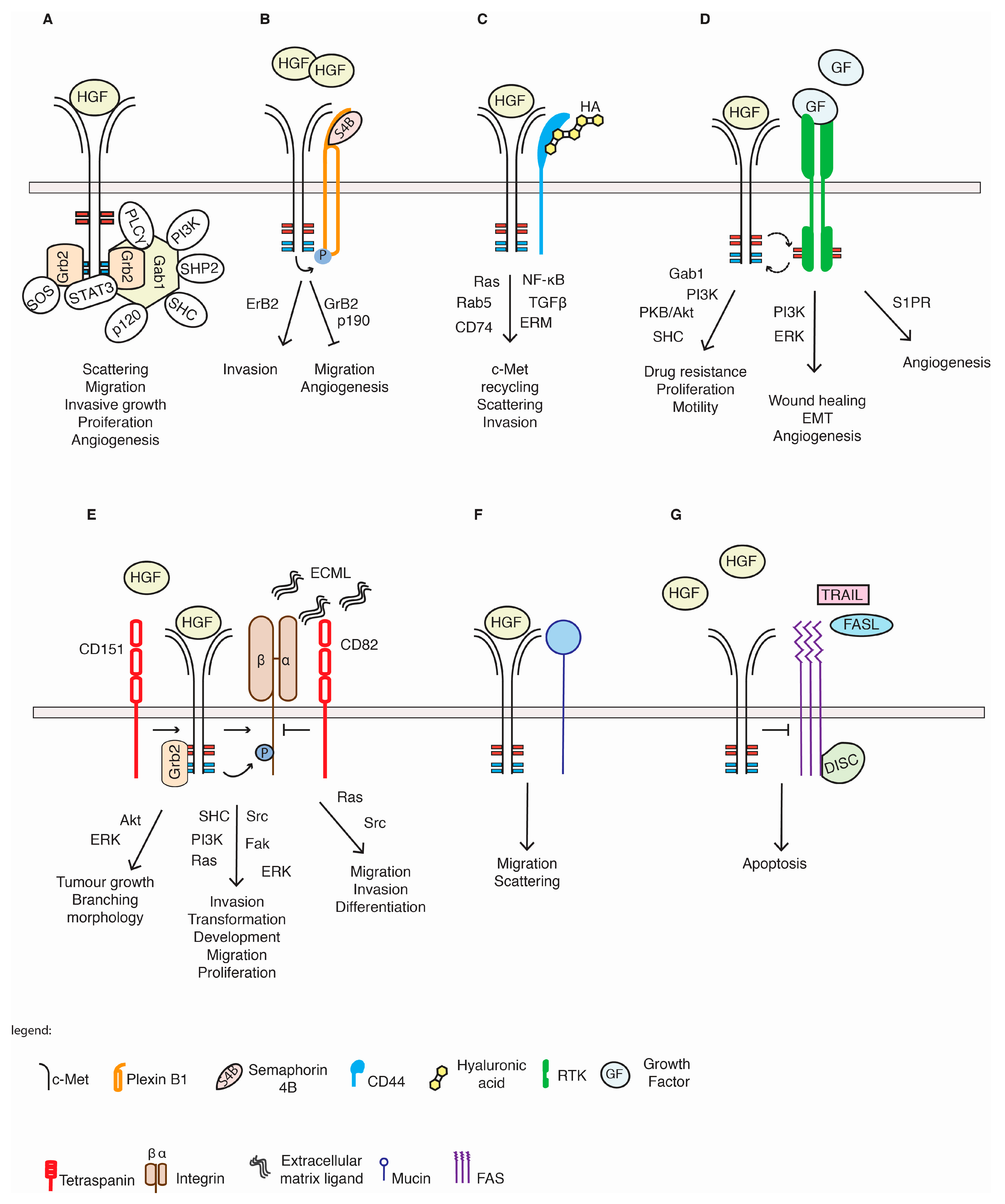
5.1. Plexin Proteins
5.2. CD44 Proteins
5.3. Integrins
5.4. Tetraspanins
5.5. Other RTKs
5.6. c-Met and Others
6. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Christofori, G. New signals from the invasive front. Nature 2006, 441, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Dean, M.; Kaul, K.; Braun, M.J.; Gonda, M.A.; vande Woude, G. Sequence of met protooncogene CDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc. Natl. Acad. Sci. USA 1987, 84, 6379–6383. [Google Scholar] [CrossRef] [PubMed]
- Stoker, M.; Gherardi, E.; Perryman, M.; Gray, J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 1987, 327, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G.; Weidner, K.M.; Schwarz, H.; Birchmeier, W. The motility signal of scatter factor/hepatocyte growth factor mediated through the receptor tyrosine kinase met requires intracellular action of ras. J. Biol. Chem. 1994, 269, 21936–21939. [Google Scholar] [PubMed]
- Khwaja, A.; Lehmann, K.; Marte, B.M.; Downward, J. Phosphoinositide 3-kinase induces scattering and tubulogenesis in epithelial cells through a novel pathway. J. Biol. Chem. 1998, 273, 18793–18801. [Google Scholar] [CrossRef] [PubMed]
- Maroun, C.R.; Holgado-Madruga, M.; Royal, I.; Naujokas, M.A.; Fournier, T.M.; Wong, A.J.; Park, M. The gab1 ph domain is required for localization of gab1 at sites of cell–cell contact and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol. Cell. Biol. 1999, 19, 1784–1799. [Google Scholar] [PubMed]
- Schaeper, U.; Gehring, N.H.; Fuchs, K.P.; Sachs, M.; Kempkes, B.; Birchmeier, W. Coupling of gab1 to c-Met, Grb2, and Shp2 mediates biological responses. J. Cell Biol. 2000, 149, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- Weidner, K.M.; di Cesare, S.; Sachs, M.; Brinkmann, V.; Behrens, J.; Birchmeier, W. Interaction between gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 1996, 384, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Lamorte, L.; Kamikura, D.M.; Park, M. A switch from p130Cas/Crk to Gab1/Crk signaling correlates with anchorage independent growth and jnk activation in cells transformed by the Met receptor oncoprotein. Oncogene 2000, 19, 5973–5981. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Ponzetto, C.; di Renzo, M.F.; Cooper, C.S.; Comoglio, P.M. Tyrosine kinase receptor indistinguishable from the c-Met protein. Nature 1989, 339, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.; Lazarus, R.A.; Yao, X.; Kirchhofer, D.; Wiesmann, C. Crystal structure of the HGF beta-chain in complex with the sema domain of the met receptor. EMBO J. 2004, 23, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Gual, P.; Giordano, S.; Williams, T.A.; Rocchi, S.; van Obberghen, E.; Comoglio, P.M. Sustained recruitment of phospholipase C-gamma to Gab1 is required for hgf-induced branching tubulogenesis. Oncogene 2000, 19, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Maroun, C.R.; Naujokas, M.A.; Holgado-Madruga, M.; Wong, A.J.; Park, M. The tyrosine phosphatase Shp-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol. Cell. Biol. 2000, 20, 8513–8525. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Yart, A.; Dance, M.; Perret, B.; Salles, J.P.; Raynal, P. A novel role for Gab1 and Shp2 in epidermal growth factor-induced ras activation. J. Biol. Chem. 2005, 280, 5350–5360. [Google Scholar] [CrossRef] [PubMed]
- Sachs, M.; Brohmann, H.; Zechner, D.; Muller, T.; Hulsken, J.; Walther, I.; Schaeper, U.; Birchmeier, C.; Birchmeier, W. Essential role of Gab1 for signaling by the c-Met receptor in vivo. J. Cell Biol. 2000, 150, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Lock, L.S.; Royal, I.; Naujokas, M.A.; Park, M. Identification of an atypical Grb2 carboxyl-terminal Sh3 domain binding site in gab docking proteins reveals Grb2-dependent and -independent recruitment of Gab1 to receptor tyrosine kinases. J. Biol. Chem. 2000, 275, 31536–31545. [Google Scholar] [CrossRef] [PubMed]
- Machide, M.; Hashigasako, A.; Matsumoto, K.; Nakamura, T. Contact inhibition of hepatocyte growth regulated by functional association of the c-Met/hepatocyte growth factor receptor and lar protein–tyrosine phosphatase. J. Biol. Chem. 2006, 281, 8765–8772. [Google Scholar] [CrossRef] [PubMed]
- Palka, H.L.; Park, M.; Tonks, N.K. Hepatocyte growth factor receptor tyrosine kinase Met is a substrate of the receptor protein–tyrosine phosphatase Dep-1. J. Biol. Chem. 2003, 278, 5728–5735. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, V.; Paliouras, G.N.; Abella, J.V.; Dube, N.; Monast, A.; Tremblay, M.L.; Park, M. Regulation of the met receptor–tyrosine kinase by the protein-tyrosine phosphatase 1B and T-cell phosphatase. J. Biol. Chem. 2008, 283, 34374–34383. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Z.; Abella, J.V.; Park, M. Crosstalk in met receptor oncogenesis. Trends Cell Biol. 2009, 19, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Comoglio, P.M. Scatter-factor and semaphorin receptors: Cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. Met signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Pelicci, G.; Giordano, S.; Zhen, Z.; Salcini, A.E.; Lanfrancone, L.; Bardelli, A.; Panayotou, G.; Waterfield, M.D.; Ponzetto, C.; Pelicci, P.G.; et al. The motogenic and mitogenic responses to hgf are amplified by the shc adaptor protein. Oncogene 1995, 10, 1631–1638. [Google Scholar] [PubMed]
- Paumelle, R.; Tulasne, D.; Kherrouche, Z.; Plaza, S.; Leroy, C.; Reveneau, S.; vanden Bunder, B.; Fafeur, V. Hepatocyte growth factor/scatter factor activates the ets1 transcription factor by a ras-raf-mek-erk signaling pathway. Oncogene 2002, 21, 2309–2319. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Guzman, M.; Dolfi, F.; Zeh, K.; Vuori, K. Met-induced jnk activation is mediated by the adapter protein crk and correlates with the Gab1–Crk signaling complex formation. Oncogene 1999, 18, 7775–7786. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Ma, Y.X.; Gao, M.; Yuan, R.Q.; Meng, Q.; Goldberg, I.D.; Rosen, E.M. The multisubstrate adapter gab1 regulates hepatocyte growth factor (scatter factor)–c-Met signaling for cell survival and DNA repair. Mol. Cell. Biol. 2001, 21, 4968–4984. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.H.; Jeffers, M.; Bellacosa, A.; Mitsuuchi, Y.; vande Woude, G.F.; Testa, J.R. Anti-apoptotic signaling by hepatocyte growth factor/met via the phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways. Proc. Natl. Acad. Sci. USA 2001, 98, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Sipeki, S.; Bander, E.; Buday, L.; Farkas, G.; Bacsy, E.; Ways, D.K.; Farago, A. Phosphatidylinositol 3-kinase contributes to Erk1/Erk2 map kinase activation associated with hepatocyte growth factor-induced cell scattering. Cell. Signal. 1999, 11, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Boccaccio, C.; Ando, M.; Tamagnone, L.; Bardelli, A.; Michieli, P.; Battistini, C.; Comoglio, P.M. Induction of epithelial tubules by growth factor hgf depends on the stat pathway. Nature 1998, 391, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Wang, L.M.; Jove, R.; vande Woude, G.F. Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene 2002, 21, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, P.P.; Gill, G.N. Endocytosis and mitogenic signaling. Curr. Opin. Cell Biol. 1999, 11, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Hammond, D.E.; Urbe, S.; vande Woude, G.F.; Clague, M.J. Down-regulation of Met, the receptor for hepatocyte growth factor. Oncogene 2001, 20, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, A.; Gilestro, G.F.; Lanzardo, S.; Comoglio, P.M.; Migone, N.; Giordano, S. The endophilin–CIN85–CBL complex mediates ligand-dependent downregulation of c-Met. Nature 2002, 416, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Korhan, P.; Erdal, E.; Kandemis, E.; Cokakli, M.; Nart, D.; Yilmaz, F.; Can, A.; Atabey, N. Reciprocal activating crosstalk between c-Met and caveolin 1 promotes invasive phenotype in hepatocellular carcinoma. PLoS One 2014, 9, e105278. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lorinczi, M.; Ireton, K.; Elferink, L.A. Specific Grb2-mediated interactions regulate clathrin-dependent endocytosis of the c-Met-tyrosine kinase. J. Biol. Chem. 2007, 282, 16764–16775. [Google Scholar] [CrossRef] [PubMed]
- Abella, J.V.; Parachoniak, C.A.; Sangwan, V.; Park, M. Dorsal ruffle microdomains potentiate met receptor tyrosine kinase signaling and down-regulation. J. Biol. Chem. 2010, 285, 24956–24967. [Google Scholar] [CrossRef] [PubMed]
- Ogi, S.; Fujita, H.; Kashihara, M.; Yamamoto, C.; Sonoda, K.; Okamoto, I.; Nakagawa, K.; Ohdo, S.; Tanaka, Y.; Kuwano, M.; et al. Sorting nexin 2-mediated membrane trafficking of c-Met contributes to sensitivity of molecular-targeted drugs. Cancer Sci. 2013, 104, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Hasenauer, S.; Malinger, D.; Koschut, D.; Pace, G.; Matzke, A.; von Au, A.; Orian-Rousseau, V. Internalization of met requires the co-receptor CD44V6 and its link to erm proteins. PLoS One 2013, 8, e62357. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, M.; Taylor, G.A.; Weidner, K.M.; Omura, S.; vande Woude, G.F. Degradation of the Met tyrosine kinase receptor by the ubiquitin-proteasome pathway. Mol. Cell. Biol. 1997, 17, 799–808. [Google Scholar] [PubMed]
- Sangwan, V.; Abella, J.; Lai, A.; Bertos, N.; Stuible, M.; Tremblay, M.L.; Park, M. Protein–tyrosine phosphatase 1B modulates early endosome fusion and trafficking of met and epidermal growth factor receptors. J. Biol. Chem. 2011, 286, 45000–45013. [Google Scholar] [CrossRef] [PubMed]
- Miaczynska, M.; Pelkmans, L.; Zerial, M. Not just a sink: Endosomes in control of signal transduction. Curr. Opin. Cell Biol. 2004, 16, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Kermorgant, S.; Parker, P.J. Receptor trafficking controls weak signal delivery: A strategy used by c-met for stat3 nuclear accumulation. J. Cell Biol. 2008, 182, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Menard, L.; Parker, P.J.; Kermorgant, S. Receptor tyrosine kinase c-Met controls the cytoskeleton from different endosomes via different pathways. Nat. Commun. 2014, 5, 3907. [Google Scholar] [PubMed]
- Kermorgant, S.; Zicha, D.; Parker, P.J. Pkc controls hgf-dependent c-Met traffic, signalling and cell migration. EMBO J. 2004, 23, 3721–3734. [Google Scholar] [CrossRef] [PubMed]
- Masui, H.; Castro, L.; Mendelsohn, J. Consumption of EGF by A431 cells: Evidence for receptor recycling. J. Cell Biol. 1993, 120, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Joffre, C.; Barrow, R.; Menard, L.; Calleja, V.; Hart, I.R.; Kermorgant, S. A direct role for Met endocytosis in tumorigenesis. Nat. Cell Biol. 2011, 13, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Hammond, D.E.; Carter, S.; McCullough, J.; Urbe, S.; vande Woude, G.; Clague, M.J. Endosomal dynamics of met determine signaling output. Mol. Biol. Cell 2003, 14, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Muharram, G.; Sahgal, P.; Korpela, T.; de Franceschi, N.; Kaukonen, R.; Clark, K.; Tulasne, D.; Carpen, O.; Ivaska, J. Tensin-4-dependent met stabilization is essential for survival and proliferation in carcinoma cells. Dev. Cell 2014, 29, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Trinidad, A.G.; Timpson, P.; Morton, J.P.; Zanivan, S.; van den Berghe, P.V.; Nixon, C.; Karim, S.A.; Caswell, P.T.; Noll, J.E.; et al. Mutant p53 enhances met trafficking and signalling to drive cell scattering and invasion. Oncogene 2013, 32, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Parachoniak, C.A.; Luo, Y.; Abella, J.V.; Keen, J.H.; Park, M. Gga3 functions as a switch to promote met receptor recycling, essential for sustained erk and cell migration. Dev. Cell 2011, 20, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Conrotto, P.; Valdembri, D.; Corso, S.; Serini, G.; Tamagnone, L.; Comoglio, P.M.; Bussolino, F.; Giordano, S. Sema4d induces angiogenesis through met recruitment by plexin b1. Blood 2005, 105, 4321–4329. [Google Scholar] [CrossRef] [PubMed]
- Conrotto, P.; Corso, S.; Gamberini, S.; Comoglio, P.M.; Giordano, S. Interplay between scatter factor receptors and b plexins controls invasive growth. Oncogene 2004, 23, 5131–5137. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Corso, S.; Conrotto, P.; Artigiani, S.; Gilestro, G.; Barberis, D.; Tamagnone, L.; Comoglio, P.M. The semaphorin 4d receptor controls invasive growth by coupling with Met. Nat. Cell Biol. 2002, 4, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Soong, J.; Scott, G. Plexin B1 inhibits met through direct association and regulates SHP2 expression in melanocytes. J. Cell Sci. 2013, 126, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.; McClelland, L.; Fricke, A.; Williamson, M.; Kuo, I.; Scott, G. Plexin b1 suppresses c-Met in melanoma: A role for plexin b1 as a tumor-suppressor protein through regulation of c-Met. J. Investig. Dermatol. 2010, 130, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Krishnan, R.; Swiercz, J.M. Grb2 mediates semaphorin-4D-dependent rhoa inactivation. J. Cell Sci. 2012, 125, 3557–3567. [Google Scholar] [CrossRef] [PubMed]
- Swiercz, J.M.; Worzfeld, T.; Offermanns, S. Erbb-2 and met reciprocally regulate cellular signaling via plexin-b1. J. Biol. Chem. 2008, 283, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Artigiani, S.; Conrotto, P.; Fazzari, P.; Gilestro, G.F.; Barberis, D.; Giordano, S.; Comoglio, P.M.; Tamagnone, L. Plexin-B3 is a functional receptor for semaphorin 5A. EMBO Rep. 2004, 5, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Hascall, V.C.; Markwald, R.R.; Misra, S. Stromal hyaluronan interaction with epithelial cd44 variants promotes prostate cancer invasiveness by augmenting expression and function of hepatocyte growth factor and androgen receptor. J. Biol. Chem. 2010, 285, 19821–19832. [Google Scholar] [CrossRef] [PubMed]
- Damm, S.; Koefinger, P.; Stefan, M.; Wels, C.; Mehes, G.; Richtig, E.; Kerl, H.; Otte, M.; Schaider, H. HGF-promoted motility in primary human melanocytes depends on CD44V6 regulated via NF-kappaB, EGR-1, and C/EBP-beta. J. Investig. Dermatol. 2010, 130, 1893–1903. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V.; Chen, L.; Sleeman, J.P.; Herrlich, P.; Ponta, H. CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 2002, 16, 3074–3086. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V.; Morrison, H.; Matzke, A.; Kastilan, T.; Pace, G.; Herrlich, P.; Ponta, H. Hepatocyte growth factor-induced ras activation requires erm proteins linked to both CD44V6 and F-actin. Mol. Biol. Cell 2007, 18, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Bogatkevich, G.S.; Atnelishvili, I.; Akter, T.; Feghali-Bostwick, C.; Hoffman, S.; Fresco, V.M.; Fuchs, J.C.; Visconti, R.P.; Markwald, R.R.; et al. Overexpression of c-Met and CD44V6 receptors contributes to autocrine tgf-beta1 signaling in interstitial lung disease. J. Biol. Chem. 2014, 289, 7856–7872. [Google Scholar] [CrossRef] [PubMed]
- Gordin, M.; Tesio, M.; Cohen, S.; Gore, Y.; Lantner, F.; Leng, L.; Bucala, R.; Shachar, I. c-Met and its ligand hepatocyte growth factor/scatter factor regulate mature b cell survival in a pathway induced by cd74. J. Immunol. 2010, 185, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Singleton, P.A.; Salgia, R.; Moreno-Vinasco, L.; Moitra, J.; Sammani, S.; Mirzapoiazova, T.; Garcia, J.G. CD44 regulates hepatocyte growth factor-mediated vascular integrity. Role of c-Met, tiam1/rac1, dynamin 2, and cortactin. J. Biol. Chem. 2007, 282, 30643–30657. [Google Scholar] [CrossRef] [PubMed]
- Klosek, S.K.; Nakashiro, K.; Hara, S.; Shintani, S.; Hasegawa, H.; Hamakawa, H. CD151 forms a functional complex with c-Met in human salivary gland cancer cells. Biochem. Biophys. Res. Commun. 2005, 336, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Klosek, S.K.; Nakashiro, K.; Hara, S.; Goda, H.; Hasegawa, H.; Hamakawa, H. CD151 regulates HGF-stimulated morphogenesis of human breast cancer cells. Biochem. Biophys. Res. Commun. 2009, 379, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.; Muratori, C.; Corso, S.; Tenaglia, E.; Bertotti, A.; Capparuccia, L.; Trusolino, L.; Comoglio, P.M.; Tamagnone, L. The tetraspanin CD151 is required for Met-dependent signaling and tumor cell growth. J. Biol. Chem. 2010, 285, 38756–38764. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.C.; Miranti, C.K. Tetraspanin KAI1/CD82 suppresses invasion by inhibiting integrin-dependent crosstalk with c-Met receptor and src kinases. Oncogene 2006, 25, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, X.; Zhang, J.; Li, Y.; Ma, K. Synergistic inhibition of cell migration by tetraspanin cd82 and gangliosides occurs via the egfr or cmet-activated pl3k/akt signalling pathway. Int. J. Biochem. Cell Biol. 2013, 45, 2349–2358. [Google Scholar] [CrossRef] [PubMed]
- Mela, A.; Goldman, J.E. Cd82 blocks cmet activation and overcomes hepatocyte growth factor effects on oligodendrocyte precursor differentiation. J. Neurosci. 2013, 33, 7952–7960. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, A.R.; dos Santos, J.N.; Handa, K.; Hakomori, S.I. Ganglioside GM2–tetraspanin CD82 complex inhibits Met and its cross-talk with integrins, providing a basis for control of cell motility through glycosynapse. J. Biol. Chem. 2007, 282, 8123–8133. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Sugiura, T.; Abe, M.; Ishii, K.; Shirasuna, K. Regulation of c-Met signaling by the tetraspanin KAI-1/CD82 affects cancer cell migration. Int. J. Cancer 2007, 121, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell 2001, 107, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Comoglio, P.M.; Trusolino, L. Beta4 integrin is a transforming molecule that unleashes met tyrosine kinase tumorigenesis. Cancer Res. 2005, 65, 10674–10679. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, T.; Otero, J.; Chen, Y.; Kim, Y.M.; Koutcher, J.A.; Satagopan, J.; Reuter, V.; Carver, B.; de Stanchina, E.; Enomoto, K.; et al. Beta4 integrin signaling induces expansion of prostate tumor progenitors. J. Clin. Investig. 2013, 123, 682–699. [Google Scholar] [PubMed]
- Ephstein, Y.; Singleton, P.A.; Chen, W.; Wang, L.; Salgia, R.; Kanteti, P.; Dudek, S.M.; Garcia, J.G.; Jacobson, J.R. Critical role of S1PR1 and integrin beta4 in HGF/c-Met-mediated increases in vascular integrity. J. Biol. Chem. 2013, 288, 2191–2200. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Patel, Y.; Murray, J.; Patel, K.V.; Sumathipala, R.; Sobel, M.; Wijelath, E.S. Novel hepatocyte growth factor (HGF) binding domains on fibronectin and vitronectin coordinate a distinct and amplified met-integrin induced signalling pathway in endothelial cells. BMC Cell Biol. 2005, 6, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.K.; Sawada, K.; Tiwari, P.; Mui, K.; Gwin, K.; Lengyel, E. Ligand-independent activation of c-Met by fibronectin and alpha5beta1-integrin regulates ovarian cancer invasion and metastasis. Oncogene 2011, 30, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chattopadhyay, N.; Qin, S.; Szekeres, C.; Vasylyeva, T.; Mahoney, Z.X.; Taglienti, M.; Bates, C.M.; Chapman, H.A.; Miner, J.H.; et al. Coordinate integrin and c-Met signaling regulate wnt gene expression during epithelial morphogenesis. Development 2009, 136, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.; Zhou, C. Association of integrin beta1 and c-Met in mediating EGFR TKI gefitinib resistance in non-small cell lung cancer. Cancer Cell Int. 2013, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- McCall-Culbreath, K.D.; Li, Z.; Zutter, M.M. Crosstalk between the alpha2beta1 integrin and c-Met/HGF-R regulates innate immunity. Blood 2008, 111, 3562–3570. [Google Scholar] [CrossRef] [PubMed]
- Follenzi, A.; Bakovic, S.; Gual, P.; Stella, M.C.; Longati, P.; Comoglio, P.M. Cross-talk between the proto-oncogenes met and ron. Oncogene 2000, 19, 3041–3049. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Stolz, D.B.; Esplen, J.E.; Dorko, K.; Michalopoulos, G.K.; Strom, S.C. Cross-talk between epidermal growth factor receptor and c-met signal pathways in transformed cells. J. Biol. Chem. 2000, 275, 8806–8811. [Google Scholar] [CrossRef] [PubMed]
- Bonine-Summers, A.R.; Aakre, M.E.; Brown, K.A.; Arteaga, C.L.; Pietenpol, J.A.; Moses, H.L.; Cheng, N. Epidermal growth factor receptor plays a significant role in hepatocyte growth factor mediated biological responses in mammary epithelial cells. Cancer Biol. Ther. 2007, 6, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Staal, B.; Essenburg, C.; Su, Y.; Kang, L.; West, R.; Kaufman, D.; Dekoning, T.; Eagleson, B.; Buchanan, S.G.; et al. Met kinase inhibitor SGX523 synergizes with epidermal growth factor receptor inhibitor erlotinib in a hepatocyte growth factor-dependent fashion to suppress carcinoma growth. Cancer Res. 2010, 70, 6880–6890. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Mukasa, A.; Bonavia, R.; Flynn, R.A.; Brewer, Z.E.; Cavenee, W.K.; Furnari, F.B.; White, F.M. Quantitative analysis of egfrviii cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl. Acad. Sci. USA 2007, 104, 12867–12872. [Google Scholar] [CrossRef] [PubMed]
- Bachleitner-Hofmann, T.; Sun, M.Y.; Chen, C.T.; Tang, L.; Song, L.; Zeng, Z.; Shah, M.; Christensen, J.G.; Rosen, N.; Solit, D.B.; et al. Her kinase activation confers resistance to met tyrosine kinase inhibition in Met oncogene-addicted gastric cancer cells. Mol. Cancer Ther. 2008, 7, 3499–3508. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of met amplification in EGFR mutant nsclc. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008, 68, 9479–9487. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Mammadova, G.; Song, R.X.; Fukami, Y.; Sato, K. Tyrosine phosphorylation of p145Met mediated by egfr and src is required for serum-independent survival of human bladder carcinoma cells. J. Cell Sci. 2006, 119, 4623–4633. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.P.; Yu, F.S. Cross talk between c-met and epidermal growth factor receptor during retinal pigment epithelial wound healing. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2242–2248. [Google Scholar] [CrossRef]
- Nath, D.; Williamson, N.J.; Jarvis, R.; Murphy, G. Shedding of c-Met is regulated by crosstalk between a G-protein coupled receptor and the egf receptor and is mediated by a timp-3 sensitive metalloproteinase. J. Cell Sci. 2001, 114, 1213–1220. [Google Scholar] [PubMed]
- Tanizaki, J.; Okamoto, I.; Sakai, K.; Nakagawa, K. Differential roles of trans-phosphorylated EGFR, Her2, Her3, and Ret as heterodimerisation partners of met in lung cancer with Met amplification. Br. J. Cancer 2011, 105, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Du, R.; Jiang, S.; Wu, C.; Barkauskas, D.S.; Richey, J.; Molter, J.; Lam, M.; Flask, C.; Gerson, S.; et al. Dual Met–EGFR combinatorial inhibition against t790m–EGFR-mediated erlotinib-resistant lung cancer. Br. J. Cancer 2008, 99, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Troiani, T.; Martinelli, E.; Napolitano, S.; Vitagliano, D.; Ciuffreda, L.P.; Costantino, S.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; et al. Increased TGF-alpha as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR–Met interaction and activation of met signaling in colon cancer cells. Clin. Cancer Res. 2013, 19, 6751–6765. [Google Scholar] [CrossRef] [PubMed]
- Breindel, J.L.; Haskins, J.W.; Cowell, E.P.; Zhao, M.; Nguyen, D.X.; Stern, D.F. EGF receptor activates Met through mapk to enhance non-small cell lung carcinoma invasion and brain metastasis. Cancer Res. 2013, 73, 5053–5065. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Gubish, C.T.; Stabile, L.P.; Henry, C.; Siegfried, J.M. HGF-independent potentiation of EGFR action by c-Met. Oncogene 2011, 30, 3625–3635. [Google Scholar] [CrossRef] [PubMed]
- Shattuck, D.L.; Miller, J.K.; Carraway, K.L., 3rd; Sweeney, C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008, 68, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Khoury, H.; Naujokas, M.A.; Zuo, D.; Sangwan, V.; Frigault, M.M.; Petkiewicz, S.; Dankort, D.L.; Muller, W.J.; Park, M. HGF converts ERBB2/Neu epithelial morphogenesis to cell invasion. Mol. Biol. Cell 2005, 16, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. Met amplification leads to gefitinib resistance in lung cancer by activating erbb3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Villen, J.; Kornhauser, J.; Lee, K.A.; Stokes, M.P.; Rikova, K.; Possemato, A.; Nardone, J.; Innocenti, G.; Wetzel, R.; et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc. Natl. Acad. Sci. USA 2008, 105, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.W.; Somcio, R.J.; Fan, F.; Liu, W.; Johnson, M.; Lesslie, D.P.; Evans, D.B.; Gallick, G.E.; Ellis, L.M. Regulatory role of c-Met in insulin-like growth factor-I receptor-mediated migration and invasion of human pancreatic carcinoma cells. Mol. Cancer Ther. 2006, 5, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; DeFrances, M.C.; Dai, Y.; Pediaditakis, P.; Johnson, C.; Bell, A.; Michalopoulos, G.K.; Zarnegar, R. A mechanism of cell survival: Sequestration of fas by the HGF receptor Met. Mol. Cell 2002, 9, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Ma, J.; Wang, X.; Guo, L.; Zhu, Z.; Stoops, J.; Eaker, A.E.; Johnson, C.J.; Strom, S.; Michalopoulos, G.K.; et al. Lack of Fas antagonism by met in human fatty liver disease. Nat. Med. 2007, 13, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.A.; Brady, H.J. Cmet and Fas receptor interaction inhibits death-inducing signaling complex formation in endothelial cells. Hypertension 2005, 46, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Uslar, L.; Sevala, S.; Shah, K. Targeting c-Met receptor overcomes trail-resistance in brain tumors. PLoS One 2014, 9, e95490. [Google Scholar] [CrossRef] [PubMed]
- Bozkaya, G.; Korhan, P.; Cokakli, M.; Erdal, E.; Sagol, O.; Karademir, S.; Korch, C.; Atabey, N. Cooperative interaction of muc1 with the HGF/c-Met pathway during hepatocarcinogenesis. Mol. Cancer 2012, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Horm, T.M.; Bitler, B.G.; Broka, D.M.; Louderbough, J.M.; Schroeder, J.A. Muc1 drives c-Met-dependent migration and scattering. Mol. Cancer Res. 2012, 10, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Behrens, M.E.; Eggers, J.P.; Cerny, R.L.; Bailey, J.M.; Shanmugam, K.; Gendler, S.J.; Bennett, E.P.; Hollingsworth, M.A. Phosphorylation of muc1 by met modulates interaction with p53 and mmp1 expression. J. Biol. Chem. 2008, 283, 26985–26995. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Orita, T.; Katsuya, K.; Yamasaki, Y.; Akiyama, K.; Li, H.; Yamamoto, T.; Saito, Y.; Nakamura, M. Muc20 suppresses the hepatocyte growth factor-induced GRB2–Ras pathway by binding to a multifunctional docking site of met. Mol. Cell. Biol. 2004, 24, 7456–7468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhau, H.E.; Osunkoya, A.O.; Iqbal, S.; Yang, X.; Fan, S.; Chen, Z.; Wang, R.; Marshall, F.F.; Chung, L.W.; et al. Vascular endothelial growth factor regulates myeloid cell leukemia-1 expression through neuropilin-1-dependent activation of c-Met signaling in human prostate cancer cells. Mol. Cancer 2010, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Olaku, V.; Matzke, A.; Mitchell, C.; Hasenauer, S.; Sakkaravarthi, A.; Pace, G.; Ponta, H.; Orian-Rousseau, V. c-Met recruits ICAM-1 as a coreceptor to compensate for the loss of CD44 in CD44 null mice. Mol. Biol. Cell 2011, 22, 2777–2786. [Google Scholar] [CrossRef] [PubMed]
- Tamagnone, L.; Artigiani, S.; Chen, H.; He, Z.; Ming, G.I.; Song, H.; Chedotal, A.; Winberg, M.L.; Goodman, C.S.; Poo, M.; et al. Plexins are a large family of receptors for transmembrane, secreted, and gpi-anchored semaphorins in vertebrates. Cell 1999, 99, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Driessens, M.H.; Hu, H.; Nobes, C.D.; Self, A.; Jordens, I.; Goodman, C.S.; Hall, A. Plexin-B semaphorin receptors interact directly with active rac and regulate the actin cytoskeleton by activating rho. Curr. Biol. 2001, 11, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Chugha, P.; Hota, P.K.; Alviani, R.S.; Li, M.; Tempel, W.; Shen, L.; Park, H.W.; Buck, M. Binding of Rac1, Rnd1, and Rhod to a novel Rho GTPase interaction motif destabilizes dimerization of the plexin-B1 effector domain. J. Biol. Chem. 2007, 282, 37215–37224. [Google Scholar] [CrossRef] [PubMed]
- Ch’ng, E.S.; Kumanogoh, A. Roles of SEMA4D and plexin-B1 in tumor progression. Mol. Cancer 2010, 9, 251. [Google Scholar] [CrossRef] [PubMed]
- Damola, A.; Legendre, A.; Ball, S.; Masters, J.R.; Williamson, M. Function of mutant and wild-type plexinb1 in prostate cancer cells. Prostate 2013, 73, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.F.; Ashkar, S.; Glimcher, M.J.; Cantor, H. Receptor-ligand interaction between CD44 and osteopontin (ETA-1). Science 1996, 271, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Van der Voort, R.; Taher, T.E.; Wielenga, V.J.; Spaargaren, M.; Prevo, R.; Smit, L.; David, G.; Hartmann, G.; Gherardi, E.; Pals, S.T. Heparan sulfate-modified CD44 promotes hepatocyte growth factor/scatter factor-induced signal transduction through the receptor tyrosine kinase c-Met. J. Biol. Chem. 1999, 274, 6499–6506. [Google Scholar] [CrossRef] [PubMed]
- Matzke, A.; Sargsyan, V.; Holtmann, B.; Aramuni, G.; Asan, E.; Sendtner, M.; Pace, G.; Howells, N.; Zhang, W.; Ponta, H.; et al. Haploinsufficiency of c-met in CD44−/− mice identifies a collaboration of CD44 and c-Met in vivo. Mol. Cell. Biol. 2007, 27, 8797–8806. [Google Scholar] [CrossRef] [PubMed]
- Crepaldi, T.; Gautreau, A.; Comoglio, P.M.; Louvard, D.; Arpin, M. Ezrin is an effector of hepatocyte growth factor-mediated migration and morphogenesis in epithelial cells. J. Cell Biol. 1997, 138, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Winograd-Katz, S.E.; Fassler, R.; Geiger, B.; Legate, K.R. The integrin adhesome: From genes and proteins to human disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.C.; Chen, S.Y.; Chen, C.H.; Chen, H.C. Crosstalk between hepatocyte growth factor and integrin signaling pathways. J. Biomed. Sci. 2006, 13, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.; Yoon, S.O.; Lipscomb, E.A.; Mercurio, A.M. The met receptor and alpha6beta4 integrin can function independently to promote carcinoma invasion. J. Biol. Chem. 2004, 279, 32287–32293. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Kobayashi, R.; Bishop, J.M. Cellular adherence elicits ligand-independent activation of the met cell-surface receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 8425–8430. [Google Scholar] [CrossRef] [PubMed]
- Ivaska, J.; Heino, J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu. Rev. Cell Dev. Biol. 2011, 27, 291–320. [Google Scholar] [CrossRef] [PubMed]
- Mattila, E.; Pellinen, T.; Nevo, J.; Vuoriluoto, K.; Arjonen, A.; Ivaska, J. Negative regulation of egfr signalling through integrin-alpha1beta1-mediated activation of protein tyrosine phosphatase tcptp. Nat. Cell Biol. 2005, 7, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Caswell, P.T.; Chan, M.; Lindsay, A.J.; McCaffrey, M.W.; Boettiger, D.; Norman, J.C. RAB-coupling protein coordinates recycling of alpha5beta1 integrin and egfr1 to promote cell migration in 3D microenvironments. J. Cell Biol. 2008, 183, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Bassani, S.; Cingolani, L.A. Tetraspanins: Interactions and interplay with integrins. Int. J. Biochem. Cell Biol. 2012, 44, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, Y.; Jin, Z.; Jing, X. Functional and biochemical studies of CD9 in fibrosarcoma cell line. Mol. Cell. Biochem. 2011, 350, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Murayama, Y.; Shinomura, Y.; Oritani, K.; Miyagawa, J.; Yoshida, H.; Nishida, M.; Katsube, F.; Shiraga, M.; Miyazaki, T.; Nakamoto, T.; et al. The tetraspanin CD9 modulates epidermal growth factor receptor signaling in cancer cells. J. Cell. Physiol. 2008, 216, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.L.; Hunter, L.A.; Ethier, S.P.; Boerner, J.L. Met and c-Src cooperate to compensate for loss of epidermal growth factor receptor kinase activity in breast cancer cells. Cancer Res. 2008, 68, 3314–3322. [Google Scholar] [CrossRef] [PubMed]
- Boccaccio, C.; Luraghi, P.; Comoglio, P.M. Met-mediated resistance to EGFR inhibitors: An old liaison rooted in colorectal cancer stem cells. Cancer Res. 2014, 74, 3647–3651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Staal, B.; Essenburg, C.; Lewis, S.; Kaufman, D.; vande Woude, G.F. Strengthening context-dependent anticancer effects on non-small cell lung carcinoma by inhibition of both Met and EGFR. Mol. Cancer Ther. 2013, 12, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Reznik, T.E.; Sang, Y.; Ma, Y.; Abounader, R.; Rosen, E.M.; Xia, S.; Laterra, J. Transcription-dependent epidermal growth factor receptor activation by hepatocyte growth factor. Mol. Cancer Res. 2008, 6, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Acunzo, M.; Romano, G.; Palmieri, D.; Lagana, A.; Garofalo, M.; Balatti, V.; Drusco, A.; Chiariello, M.; Nana-Sinkam, P.; Croce, C.M. Cross-talk between Met and EGFR in non-small cell lung cancer involves Mir-27a and sprouty2. Proc. Natl. Acad. Sci. USA 2013, 110, 8573–8578. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.T.; Jacobs, R.J.; Moravec, D.N.; Uppada, S.B.; Botting, G.M.; Nlend, M.; Puri, N. Alternative signaling pathways as potential therapeutic targets for overcoming EGFR and c-Met inhibitor resistance in non-small cell lung cancer. PLoS One 2013, 8, e78398. [Google Scholar] [CrossRef] [PubMed]
- Merlin, S.; Pietronave, S.; Locarno, D.; Valente, G.; Follenzi, A.; Prat, M. Deletion of the ectodomain unleashes the transforming, invasive, and tumorigenic potential of the met oncogene. Cancer Sci. 2009, 100, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Mukherjee, P. Muc1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Chappell, W.H.; Steelman, L.S.; Long, J.M.; Kempf, R.C.; Abrams, S.L.; Franklin, R.A.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; et al. Ras/Raf/Mek/ERK and pi3k/pten/Akt/MTOR inhibitors: Rationale and importance to inhibiting these pathways in human health. Oncotarget 2011, 2, 135–164. [Google Scholar] [PubMed]
- Accordi, B.; Pillozzi, S.; Dell’Orto, M.C.; Cazzaniga, G.; Arcangeli, A.; Kronnie, G.T.; Basso, G. Hepatocyte growth factor receptor c-met is associated with fas and when activated enhances drug-induced apoptosis in pediatric b acute lymphoblastic leukemia with tel-aml1 translocation. J. Biol. Chem. 2007, 282, 29384–29393. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; von Pawel, J.; Garmey, E.G.; Akerley, W.L.; Brugger, W.; Ferrari, D.; Chen, Y.; Costa, D.B.; Gerber, D.E.; Orlov, S.; et al. Randomized phase ii study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Ervin, T.J.; Ramlau, R.A.; Daniel, D.B.; Goldschmidt, J.H., Jr.; Blumenschein, G.R., Jr.; Krzakowski, M.J.; Robinet, G.; Godbert, B.; Barlesi, F.; et al. Randomized phase II trial of onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 4105–4114. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, T.E. Novel agents in development for advanced non-small cell lung cancer. Ther. Adv. Med. Oncol. 2014, 6, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Current status of targeted therapy in non-small cell lung cancer. Drugs Today 2014, 50, 503–525. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viticchiè, G.; Muller, P.A.J. c-Met and Other Cell Surface Molecules: Interaction, Activation and Functional Consequences. Biomedicines 2015, 3, 46-70. https://doi.org/10.3390/biomedicines3010046
Viticchiè G, Muller PAJ. c-Met and Other Cell Surface Molecules: Interaction, Activation and Functional Consequences. Biomedicines. 2015; 3(1):46-70. https://doi.org/10.3390/biomedicines3010046
Chicago/Turabian StyleViticchiè, Giuditta, and Patricia A. J. Muller. 2015. "c-Met and Other Cell Surface Molecules: Interaction, Activation and Functional Consequences" Biomedicines 3, no. 1: 46-70. https://doi.org/10.3390/biomedicines3010046
APA StyleViticchiè, G., & Muller, P. A. J. (2015). c-Met and Other Cell Surface Molecules: Interaction, Activation and Functional Consequences. Biomedicines, 3(1), 46-70. https://doi.org/10.3390/biomedicines3010046