
Recombinant Poxvirus and the Tumor Microenvironment: Oncolysis, Immune Regulation and Immunization

Abstract
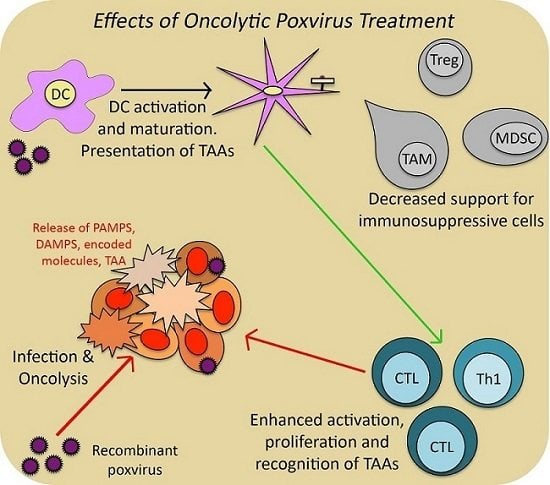
:
1. Introduction
2. Poxviridae
2.1. Background
2.2. Mechanism of Action
3. Recombinant Viruses
3.1. Virally Encoded Immune Stimulatory Molecules
3.2. Virally-Encoded Tumor-Associated Antigens and TME-Based Immunization
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| OV | oncolytic virus |
| TAA | tumor-associated antigen |
| TRICOM | triad of costimulatory molecules |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| VV | vaccinia virus |
| MVA | modified vaccinia Ankara |
| FV | fowlpox virus |
| CV | canarypox virus |
| DC | dendritic cells |
| TME | tumor microenvironment |
| APC | antigen-presenting cell |
| Tregs | T-regulatory cells |
| MDSCs | myeloid derived suppressor cells |
| TAMs | tumor-associated macrophages |
| TANs | tumor-associated neutrophils |
References
- Martuza, R.; Malick, A.; Markert, J.; Ruffner, K.; Coen, D. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Timiryasova, T.; Li, J.; Chen, B.; Chong, D.; Langridge, W.; Gridley, D.; Fodor, I. Antitumor effect of vaccinia virus in glioma model. Oncol. Res. 1999, 11, 133–144. [Google Scholar] [PubMed]
- Fulci, G.; Breymann, L.; Gianni, D.; Kurozomi, K.; Rhee, S.; Yu, J.; Kaur, B.; Louis, D.; Weissleder, R.; Caligiuri, M.; et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12873–12878. [Google Scholar] [CrossRef] [PubMed]
- Toda, M.; Rabkin, S.; Kojima, H.; Martuza, R. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum. Gene Ther. 1999, 10, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Poxviridae. In Fields Virology; Fields, B., Knipe, D., Howley, P., Eds.; Lippincott Williams & Wilkins: Philidelphia, PA, USA, 2013; pp. 2129–2159. [Google Scholar]
- McFadden, G. Poxvirus tropism. Nat. Rev. Microbiol. 2005, 3, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Bartee, E.; Chan, W.; Moreb, J.; Cogle, C.; McFadden, G. Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol. Blood Marrow Transplant. 2012, 18, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Rahman, M.; McFadden, G. Oncolytic myxoma virus: The path to clinic. Vaccine 2013, 31, 4252–4258. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Essani, K. Differential susceptibility of human cancer cell lines to wild-type tanapoxvirus infection. Open Virol. J. 2010, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Conrad, S.; El-Aswad, M.; Kurban, E.; Jeng, D.; Tripp, B.; Nutting, C.; Eversole, R.; Mackenzie, C.; Essani, K. Oncolytic tanapoxvirus expressing flic causes regression of human colorectal cancer xenografts in nude mice. J. Exp. Clin. Cancer Res. 2015, 34. [Google Scholar] [CrossRef] [PubMed]
- Rintoul, J.; Lemay, C.; Tai, L.; Stanford, M.; Falls, T.; de Souza, C.; Bridle, B.; Daneshmand, M.; Ohashi, P.; Wan, Y.; et al. ORFV: A novel oncolytic and immune stimulating parapoxvirus therapeutic. Mol. Ther. 2012, 20, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Weber, O.; Mercer, A.; Friebe, A.; Knolle, P.; Volk, H. Therapeutic immunomodulation using a virus—The potential of inactivated orf virus. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Cook, G. The smallpox saga and the origin(s) of vaccination. J. R. Soc. Health 1996, 116, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Hunter-Craig, I.; Newton, K.; Westbury, G.; Lacey, B. Use of vaccinia virus in the treatment of metastatic malignant melanoma. Br. Med. J. 1970, 2, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Burdick, K.; Hawk, W. Vitiligo in a case of vaccinia virus-treated melanoma. Cancer 1964, 17, 708–712. [Google Scholar] [CrossRef]
- Moss, B. Reflections on the early development of poxvirus vectors. Vaccine 2013, 31, 4220–4222. [Google Scholar] [CrossRef] [PubMed]
- Mackett, M.; Smith, G.; Moss, B. General method for production and selection of infectious vaccinia virus recombinants expressing foreign genes. J. Virol. 1984, 49, 857–864. [Google Scholar] [PubMed]
- Moss, B.; Smith, G.; Gerin, J.; Purcell, R. Live recombinant vaccinia virus protects chimpanzees against hepatitis B. Nature 1984, 311, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Murphy, B.; Moss, B. Construction and characterization of an infectious vaccinia virus recombinant that expresses the influenza hemagglutinin gene and induces resistance to influenza virus infection in hamsters. Proc. Natl. Acad. Sci. USA 1983, 80, 7155–7159. [Google Scholar] [CrossRef] [PubMed]
- Yilma, T.; Anderson, K.; Brechling, K.; Moss, B. Expression of an Adjuvant Gene (Interferon-Gamma) in Infectious Vaccinia Virus Recombinants. In Modern Approaches to Vaccines: Prevention of Aids and Other Viral, Bacterial, and Parasitic Diseases; Chanock, R., Lerner, R., Brown, F., Ginsberg, H., Eds.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1987; Volume 87, pp. 393–396. [Google Scholar]
- Joachim, A.; Nilsson, C.; Aboud, S.; Bakari, M.; Lyamuya, E.; Robb, M.; Marovich, M.; Earl, P.; Moss, B.; Ochsenbauer, C.; et al. Potent functional antibody responses elicited by HIV-I DNA priming and boosting with heterologous HIV-1 recombinant mva in healthy tanzanian adults. PLoS ONE 2015, 10, e0118486. [Google Scholar] [CrossRef] [PubMed]
- Antoine, G.; Scheiflinger, F.; Dorner, F.; Falkner, F. The complete genomic sequence of the modified vaccinia ankara strain: Comparison with other orthopoxviruses. Virology 1998, 244, 365–396. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Greenberg, R. Imvamune®: Modified vaccinia Ankara strain as an attenuated smallpox vaccine. Expert Rev. Vaccines 2009, 8, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Humrich, J.; Thuman, P.; Sauter, B.; Schuler, G.; Jenne, L. The highly attenuated vaccinia virus strain modified virus ankara induces apoptosis in melanoma cells and allows bystander dendritic cells to generate a potent anti-tumoral immunity. Clin. Exp. Immunol. 2006, 146, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Zeh, H.; Bartlett, D. Development of a replication-selective, oncolytic poxvirus for the treatment of human cancers. Cancer Gene Ther. 2002, 9, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- McCart, J.; Ward, J.; Lee, J.; Hu, Y.; Alexander, H.; Libutti, S.; Moss, B.; Bartlett, D. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001, 61, 8751–8757. [Google Scholar] [PubMed]
- Guo, Z.; Naik, A.; O’Malley, M.; Popovic, P.; Demarco, R.; Hu, Y.; Yin, X.; Yang, S.; Zeh, H.; Moss, B.; et al. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2. Cancer Res. 2005, 65, 9991–9998. [Google Scholar] [CrossRef] [PubMed]
- Puhlmann, M.; Brown, C.; Gnant, M.; Huang, J.; Libutti, S.; Alexander, H.; Bartlett, D. Vaccinia as a vector for tumor-directed gene therapy: Biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther. 2000, 7, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, M.; Maguire, H.; Eisenlohr, L.; Laughlin, C.; Monken, C.; McCue, P.; Kovatich, A.; Lattime, E. Intratumoral recombinant gm-csf-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999, 6, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Monken, C.; Lattime, E. Intratumoral vaccination with vaccinia-expressed tumor antigen and granulocyte macrophage colony-stimulating factor overcomes immunological ignorance to tumor antigen. Cancer Res. 2003, 63, 6956–6961. [Google Scholar] [PubMed]
- Spagnoli, G.; Zajac, P.; Marti, W.; Oertli, D.; Padovan, E.; Noppen, C.; Kocher, T.; Adamina, M.; Heberer, M. Cytotoxic T-cell induction in metastatic melanoma patients undergoing recombinant vaccinia virus-based immuno-gene therapy. Recent Results Cancer Res. 2002, 160, 195–201. [Google Scholar] [PubMed]
- Taylor, J.; Paoletti, E. Fowlpox virus as a vector in non-avian species. Vaccine 1988, 6, 466–468. [Google Scholar] [CrossRef]
- Kundig, T.; Kalberer, C.; Hengartner, H.; Zinkernagel, R. Vaccination with two different vaccinia recombinant viruses: Long-term inhibition of secondary vaccination. Vaccine 1993, 11, 1154–1158. [Google Scholar] [CrossRef]
- Hodge, J.; McLaughlin, J.; Kantor, J.; Schlom, J. Diversified prime and boost protocols using recombinant vaccinia virus and recombinant non-replicating avian pox virus to enhance T-cell immunity and antitumor responses. Vaccine 1997, 15, 756–768. [Google Scholar] [CrossRef]
- Zanotto, C.; Pozzi, E.; Pacchioni, S.; Volonté, L.; De Giuli Morghen, C.; Radaelli, A. Canarypox and fowlpox viruses as recombinant vaccine vectors: A biological and immunological comparison. Antivir. Res. 2010, 88, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.; Moon, A.; Burke, J.; Ribas, A.; Stephenson, J.; Breitbach, C.; Daneshmand, M.; De Silva, N.; Parato, K.; Diallo, J.; et al. A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol. Ther. 2011, 19, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, M.; Maguire, H.J.; McCue, P.; Lee, S.; Alexander, A.; Nazarian, L.; Eisenlohr, L.; Nathan, F.; Berd, D.; Lattime, E. A pilot study demonstrating the feasibility of using intratumoral vaccinia injections as vector for gene transfer. Vaccine Res. 1995, 4, 55–69. [Google Scholar]
- Weibel, S.; Raab, V.; Yu, Y.; Worschech, A.; Wang, E.; Marincola, F.; Szalay, A. Viral-mediated oncolysis is the most critical factor in the late-phase of the tumor regression process upon vaccinia virus infection. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Whilding, L.; Archibald, K.; Kulbe, H.; Balkwill, F.; Öberg, D.; McNeish, I. Vaccinia virus induces programmed necrosis in ovarian cancer cells. Mol. Ther. 2013, 21, 2074–2086. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.; Paterson, J.; Lemay, C.; Falls, T.; McGuire, A.; Parato, K.; Stojdl, D.; Daneshmand, M.; Speth, K.; Kirn, D.; et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol. Ther. 2007, 15, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Donawho, C.; Muller, H.; Bucana, C.; Kripke, M. Enhanced growth of murine melanoma in ultraviolet-irradiated skin is associated with local inhibition of immune effector mechanisms. J. Immunol. 1996, 157, 781–786. [Google Scholar] [PubMed]
- Sharma, S.; Stolina, M.; Yang, S.; Baratelli, F.; Lin, J.; Atianzar, K.; Luo, J.; Zhu, L.; Lin, Y.; Huang, M.; et al. Tumor cyclooxygenase 2-dependent suppression of dendritic cell function. Clin. Cancer Res. 2003, 9, 961–968. [Google Scholar] [PubMed]
- Halak, B.; Maguire, H.J.; Lattime, E. Tumor-induced interleukin-10 inhibits type 1 immune responses directed at a tumor antigen as well as a non-tumor antigen present at the tumor site. Cancer Res. 1999, 59, 911–917. [Google Scholar] [PubMed]
- Akasaki, Y.; Liu, G.; Chung, N.; Ehtesham, M.; Black, K.; Yu, J. Induction of a CD4+ T regulatory type 1 response by cyclooxygenase-2-overexpressing glioma. J. Immunol. 2004, 173, 4352–4359. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2011, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Strauss, L.; Zeidler, R.; Lang, S.; Whiteside, T. Expansion and characteristics of human T regulatory type 1 cells in co-cultures simulating tumor microenvironment. Cancer Immunol. Immunother. 2007, 56, 1429–1442. [Google Scholar] [CrossRef] [PubMed]
- Neeson, P.; Paterson, Y. Effects of the tumor microenvironment on the efficacy of tumor immunotherapy. Immunol. Investig. 2006, 35, 359–394. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A.; Koszinowski, U. Viral mechanisms of immune evasion. Immunol. Today 2000, 21, 447–455. [Google Scholar] [CrossRef]
- Alcami, A.; Smith, G. A soluble receptor for interleukin-1 beta encoded by vaccinia virus: A novel mechanism of virus modulation of the host response to infection. Cell 1992, 71, 153–167. [Google Scholar] [CrossRef]
- Tortorella, D.; Gewurz, B.; Furman, M.; Schust, D.; Ploegh, H. Viral subversion of the immune system. Annu. Rev. Immunol. 2000, 18, 861–926. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, M.; Hruby, D.; Maliszewski, C.; Pickup, D.; Sims, J.; Buller, R.; VanSlyke, J. Vaccinia and cowpox viruses encode a novel secreted interleukin-1-binding protein. Cell 1992, 71, 145–152. [Google Scholar] [CrossRef]
- McFadden, G.; Murphy, P. Host-related immunomodulators encoded by poxviruses and herpesviruses. Curr. Opin. Microbiol. 2000, 3, 371–378. [Google Scholar] [CrossRef]
- Fleming, S.; McCaughan, C.; Andrews, A.; Nash, A.; Mercer, A. A homolog of interleukin-10 is encoded by the poxvirus orf virus. J. Virol. 1997, 71, 4857–4861. [Google Scholar] [PubMed]
- Deane, D.; McInnes, C.; Percival, A.; Wood, A.; Thomson, J.; Lear, A.; Gilray, J.; Fleming, S.; Mercer, A.; Haig, D. Orf virus encodes a novel secreted protein inhibitor of granulocyte—Macrophage colony-stimulating factor and interleukin-2. J. Virol. 2000, 74, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; O’Malley, M.; Urban, J.; Sampath, P.; Guo, Z.; Kalinski, P.; Thorne, S.; Bartlett, D. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol. Ther. 2011, 19, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ravindranathan, R.; Li, J.; Kalinski, P.; Guo, Z.; Bartlett, D. CXCL11-armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Bhattacharya-Chatterjee, M.; O’Malley, B.J.; Chatterjee, S. Recombinant vaccinia virus expressing interleukin-2 invokes anti-tumor cellular immunity in an orthotopic murine model of head and neck squamous cell carcinoma. Mol. Ther. 2006, 13, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Jackaman, C.; Nelson, D. Cytokine-armed vaccinia virus infects the mesothelioma tumor microenvironment to overcome immune tolerance and mediate tumor resolution. Cancer Gene Ther. 2010, 17, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Chard, L.; Maniati, E.; Wang, P.; Zhang, Z.; Gao, D.; Wang, J.; Cao, F.; Ahmed, J.; El Khouri, M.; Hughes, J.; et al. A vaccinia virus armed with interleukin-10 is a promising therapeutic agent for treatment of murine pancreatic cancer. Clin. Cancer Res. 2015, 21, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lynn, R.; Cheng, G.; Alexander, E.; Kapoor, V.; Moon, E.; Sun, J.; Fridlender, Z.; Isaacs, S.; Thorne, S.; et al. Treating tumors with a vaccinia virus expressing ifnβ illustrates the complex relationships between oncolytic ability and immunogenicity. Mol. Ther. 2012, 20, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Su, Q.; Liang, Y.; Hu, J.; Yuan, S. Oncolytic vaccine virus harbouring the IL-24 gene suppresses the growth of lung cancer by inducing apoptosis. Biochem. Biophys. Res. Commun. 2016, 476, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.; Sabzevari, H.; Yafal, A.; Gritz, L.; Lorenz, M.; Schlom, J. A triad of costimulatory molecules synergize to amplify T-cell activation. Cancer Res. 1999, 59, 5800–5807. [Google Scholar] [PubMed]
- Kaufman, H.; Cheung, K.; Haskall, Z.; Horig, H.; Hesdorffer, C.; Panicali, D.; DeRaffele, G.; Spanknebel, K. Clinical protocol. Intra-Lesional rF-B7.1 versus rF-TRICOM vaccine in the treatment of metastatic cancer. Hum. Gene Ther. 2003, 14, 803–827. [Google Scholar] [PubMed]
- Kaufman, H.; DeRaffele, G.; Divito, J.; Hörig, H.; Lee, D.; Panicali, D.; Voulo, M. A phase I trial of intralesional rV-TRICOM vaccine in the treatment of malignant melanoma. Hum. Gene Ther. 2001, 12, 1459–1480. [Google Scholar] [PubMed]
- Kaufman, H.; Kim, D.; Kim-Schulze, S.; DeRaffele, G.; Jagoda, M.; Broucek, J.; Zloza, A. Results of a randomized phase I gene therapy clinical trial of nononcolytic fowlpox viruses encoding T cell costimulatory molecules. Hum. Gene Ther. 2014, 25, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Madan, R.; Bilusic, M.; Heery, C.; Schlom, J.; Gulley, J. Clinical evaluation of tricom vector therapeutic cancer vaccines. Semin. Oncol. 2012, 39, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.; Bernhard, H.; Shiota, F.; Hand, S.; Gralow, J.; Huseby, E.; Gillis, S.; Cheever, M. Granulocyte-macrophage colony-stimulating factor: An effective adjuvant for protein and peptide-based vaccines. Blood 1996, 88, 202–210. [Google Scholar] [PubMed]
- Kass, E.; Parker, J.; Schlom, J.; Greiner, J. Comparative studies of the effects of recombinant GM-CSF and GM-CSF administered via a poxvirus to enhance the concentration of antigen-presenting cells in regional lymph nodes. Cytokine 2000, 12, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Qin, H.; Manna, S.; Tripathi, P. Recombinant vaccinia virus expressing cytokine GM-CSF as tumor vaccine. Anticancer Res. 1999, 19, 2869–2873. [Google Scholar] [PubMed]
- Reali, E.; Canter, D.; Zeytin, H.; Schlom, J.; Greiner, J. Comparative studies of avipox-GM-CSF versus recombinant GM-CSF protein as immune adjuvants with different vaccine platforms. Vaccine 2005, 23, 2909–2921. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Fan, J.; Guo, M.; Huang, B. Oncolytic and immunologic cancer therapy with GM-CSF-armed vaccinia virus of tian tan strain guang9. Cancer Lett. 2016, 372, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Oh, J.; Park, B.; Lee, D.; Kim, J.; Park, H.; Roh, M.; Je, J.; Yoon, J.; Thorne, S.; et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol. Ther. 2006, 14, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.; Chung, H.; Kim, C.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Cripe, T.; Ngo, M.; Geller, J.; Louis, C.; Currier, M.; Racadio, J.; Towbin, A.; Rooney, C.; Pelusio, A.; Moon, A.; et al. Phase 1 study of intratumoral pexa-vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol. Ther. 2015, 23, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Breitbach, C.; Lee, J.; Park, J.; Lim, H.; Kang, W.; Moon, A.; Mun, J.; Sommermann, E.; Maruri Avidal, L.; et al. Phase 1b trial of biweekly intravenous pexa-vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus in colorectal cancer. Mol. Ther. 2015, 23, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.; Burke, J.; Jonker, D.; Stephenson, J.; Haas, A.; Chow, L.; Nieva, J.; Hwang, T.; Moon, A.; Patt, R.; et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 2011, 477, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.; Parato, K.; Burke, J.; Hwang, T.; Bell, J.; Kirn, D. Pexa-vec double agent engineered vaccinia: Oncolytic and active immunotherapeutic. Curr. Opin. Virol. 2015, 13, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Hwang, T.; Park, B.; Bell, J.; Kirn, D. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-hbv activities in patients with hepatocellular carcinoma. Mol. Ther. 2008, 16, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Evgin, L.; Acuna, S.; Tanese de Souza, C.; Marguerie, M.; Lemay, C.; Ilkow, C.; Findlay, C.; Falls, T.; Parato, K.; Hanwell, D.; et al. Complement inhibition prevents oncolytic vaccinia virus neutralization in immune humans and cynomolgus macaques. Mol. Ther. 2015, 23, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.; Kaufman, H.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.; Spitler, L.; Puzanov, I.; Agarwala, S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.; Hodi, F. Talimogene laherparepvec for the treatment of advanced melanoma. Clin. Cancer Res. 2016, 22, 3127–3131. [Google Scholar] [CrossRef] [PubMed]
- Kohlhapp, F.; Zloza, A.; Kaufman, H. Talimogene laherparepvec (T-VEC) as cancer immunotherapy. Drugs Today 2015, 51, 549–558. [Google Scholar] [PubMed]
- Dolcetti, L.; Peranzoni, E.; Ugel, S.; Marigo, I.; Fernandez Gomez, A.; Mesa, C.; Geilich, M.; Winkels, G.; Traggiai, E.; Casati, A.; et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur. J. Immunol. 2010, 40, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Thorn, M.; Guha, P.; Cunetta, M.; Espat, N.; Miller, G.; Junghans, R.; Katz, S. Tumor-associated GM-CSF overexpression induces immunoinhibitory molecules via stat3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene Ther. 2016, 23, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, T.; Zhao, C.; Pavicic, P.J.; Datta, S. Myeloid colony-stimulating factors as regulators of macrophage polarization. Front. Immunol. 2014, 5, 554. [Google Scholar] [CrossRef] [PubMed]
- Nebiker, C.; Han, J.; Eppenberger-Castori, S.; Iezzi, G.; Hirt, C.; Amicarella, F.; Cremonesi, E.; Huber, X.; Padovan, E.; Angrisani, B.; et al. GM-CSF production by tumor cells is associated with improved survival in colorectal cancer. Clin. Cancer Res. 2014, 20, 3094–3106. [Google Scholar] [CrossRef] [PubMed]
- Lattime, E.C.; Mastrangelo, M.J.; Bagasra, O.; Li, W.; Berd, D. Expression of cytokine mrna in human melanoma tissues. Cancer Immunol. Immunother. 1995, 41, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Lattime, E. Tumor-induced interleukin 10 suppresses the ability of splenic dendritic cells to stimulate CD4 and CD8 T-cell responses. Cancer Res. 2003, 63, 2150–2157. [Google Scholar] [PubMed]
- Strauss, L.; Bergmann, C.; Szczepanski, M.; Gooding, W.; Johnson, J.; Whiteside, T. A unique subset of CD4+CD25highFoxP3+ T cells secreting interleukin-10 and transforming growth factor-β1 mediates suppression in the tumor microenvironment. Clin. Cancer Res. 2007, 13, 4345–4354. [Google Scholar] [CrossRef] [PubMed]
- De Vries, C.; Poplin, E.; Weiss, R.; August, D.; Gabriel, E.; DiPaola, R.; Lattime, E. Poxvirus-Based Strategies for Combined Vaccine and Tumor Microenvironment Manipulation. In Gene Therapy of Cancer: Translational Approaches from Preclinical Studies to Clinical Implementation, 3rd ed.; Lattime, E., Gerson, S., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 241–257. [Google Scholar]
- De Vries, C.; Monken, C.; Lattime, E. The addition of recombinant vaccinia HER2/neu to oncolytic vaccinia-gmcsf given into the tumor microenvironment overcomes mdsc-mediated immune escape and systemic anergy. Cancer Gene Ther. 2015, 22, 154–162. [Google Scholar] [CrossRef] [PubMed]
- McNeel, D.; Chen, Y.; Gulley, J.; Dwyer, A.; Madan, R.; Carducci, M.; DiPaola, R. Randomized phase II trial of docetaxel with or without PSA-TRICOM vaccine in patients with castrate-resistant metastatic prostate cancer: A trial of the ECOG-ACRIN cancer research group (E1809). Hum. Vaccines Immunother. 2015, 11, 2469–2474. [Google Scholar] [CrossRef] [PubMed]
- Oudard, S.; Rixe, O.; Beuselinck, B.; Linassier, C.; Banu, E.; Machiels, J.; Baudard, M.; Ringeisen, F.; Velu, T.; Lefrere-Belda, M.; et al. A phase II study of the cancer vaccine TG4010 alone and in combination with cytokines in patients with metastatic renal clear-cell carcinoma: Clinical and immunological findings. Cancer Immunol. Immunother. 2011, 60, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Ottolino-Perry, K.; Acuna, S.; Angarita, F.; Sellers, C.; Zerhouni, S.; Tang, N.; McCart, J. Oncolytic vaccinia virus synergizes with irinotecan in colorectal cancer. Mol. Oncol. 2015, 9, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Quoix, E.; Lena, H.; Losonczy, G.; Forget, F.; Chouaid, C.; Papai, Z.; Gervais, R.; Ottensmeier, C.; Szczesna, A.; Kazarnowicz, A.; et al. TG4010 immunotherapy and first-line chemotherapy for advanced non-small-cell lung cancer (time): Results from the phase 2b part of a randomised, double-blind, placebo-controlled, phase 2B/3 trial. Lancet Oncol. 2016, 17, 212–223. [Google Scholar] [CrossRef]
- Foy, S.; Sennino, B.; dela Cruz, T.; Cote, J.; Gordon, E.; Kemp, F.; Xavier, V.; Franzusoff, A.; Rountree, R.; Mandl, S. Poxvirus-based active immunotherapy with PD-1 and LAG-3 dual immune checkpoint inhibition overcomes compensatory immune regulation, yielding complete tumor regression in mice. PLoS ONE 2016, 11, e0150084. [Google Scholar] [CrossRef] [PubMed]
- Foy, S.; Mandl, S.; Dela Cruz, T.; Cote, J.; Gordon, E.; Trent, E.; Delcayre, A.; Breitmeyer, J.; Franzusoff, A.; Rountree, R. Poxvirus-based active immunotherapy synergizes with CTLA-4 blockade to increase survival in a murine tumor model by improving the magnitude and quality of cytotoxic t cells. Cancer Immunol. Immunother. 2016, 65, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Cappuccini, F.; Stribbling, S.; Pollock, E.; Hill, A.; Redchenko, I. Immunogenicity and efficacy of the novel cancer vaccine based on simian adenovirus and MVA vectors alone and in combination with PD-1 mAb in a mouse model of prostate cancer. Cancer Immunol. Immunother. 2016, 65, 701–713. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
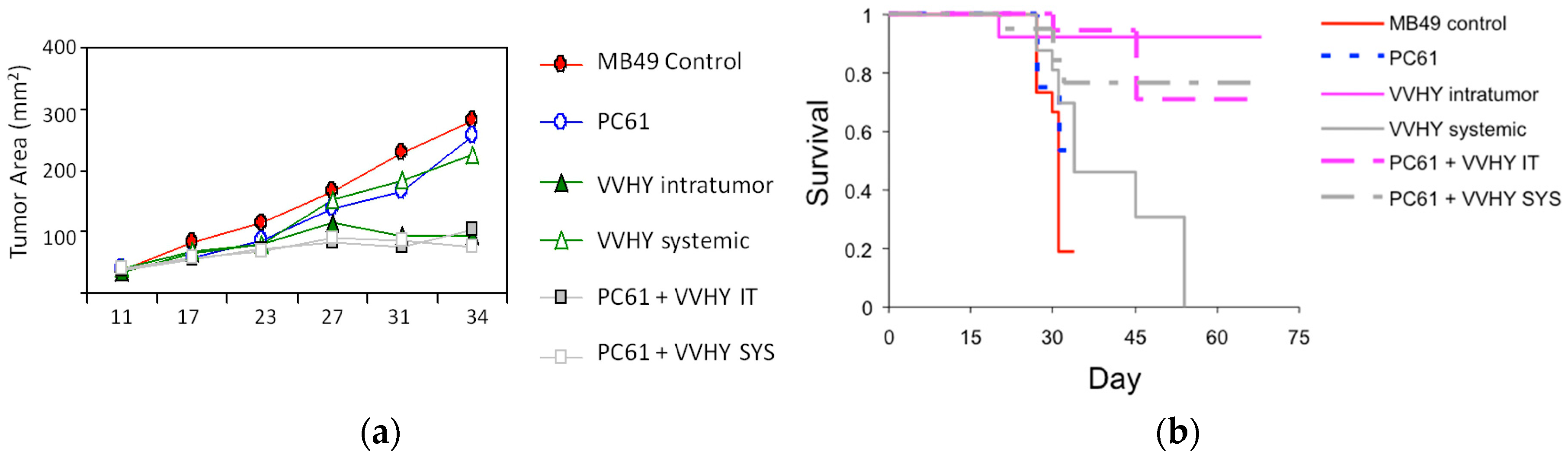{kind=link}
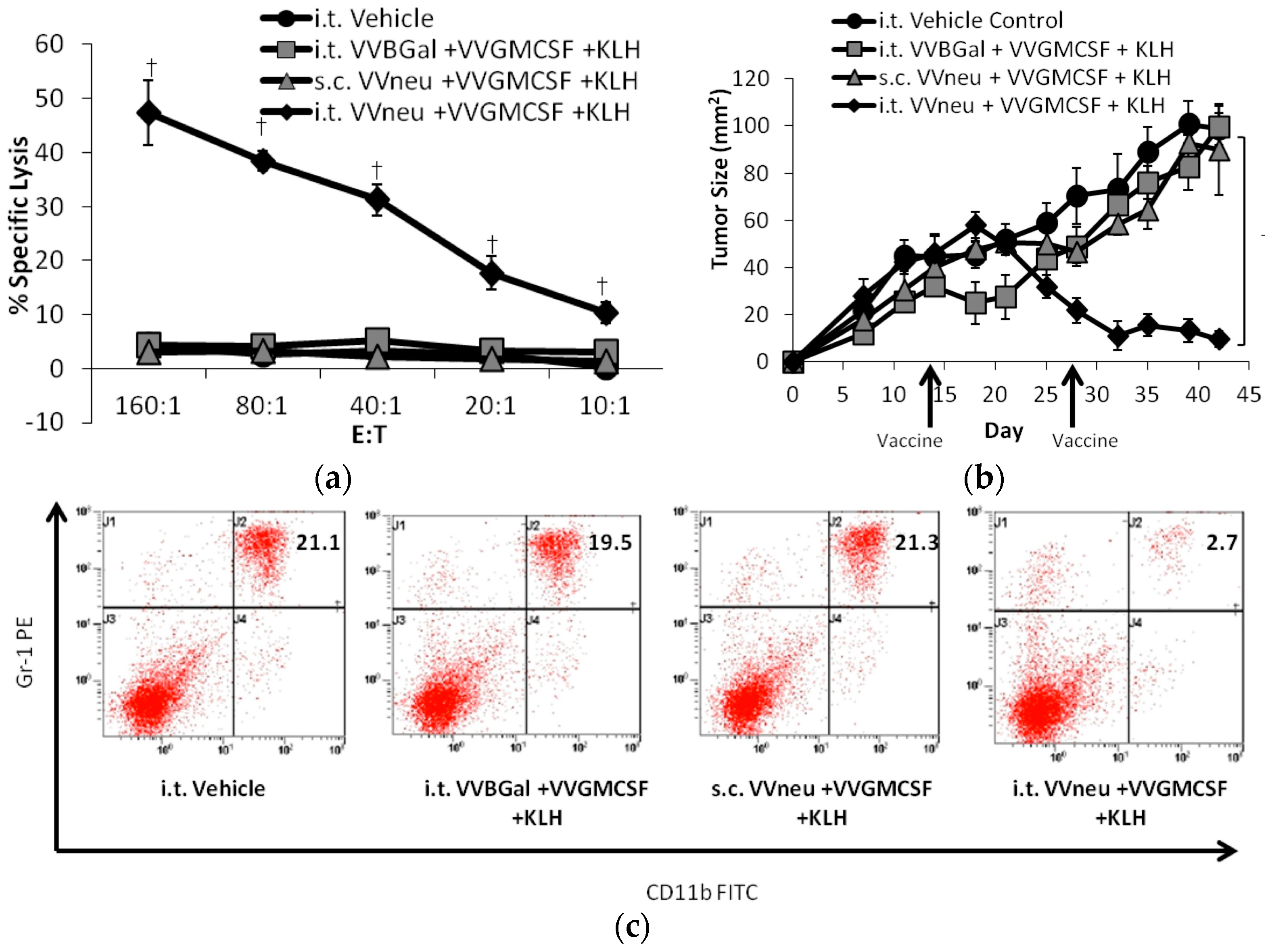{kind=link}
| Genera | Example Species | Primary Hosts | Human Infectivity | Use in Cancer Research and Therapy |
|---|---|---|---|---|
| Orthopoxvirus | Variola Vaccinia | Vertebrates and Arthropods | Yes | Extensive clinical trials |
| Avipoxvirus | Canarpox Fowlpox | Birds | Infects but does not replicate | Extensive clinical trials |
| Leporipoxvirus | Myxoma | Rabbits | Infects but does not replicate | Some preclinical models [7,8] |
| Yatapoxvirus | Tanapox | Monkeys and Baboons | Yes | Limited preclinical models [9,10] |
| Parapoxvirus | Orf | Sheep and Goats | Yes | Limited preclinical models [11,12] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharp, D.W.; Lattime, E.C. Recombinant Poxvirus and the Tumor Microenvironment: Oncolysis, Immune Regulation and Immunization. Biomedicines 2016, 4, 19. https://doi.org/10.3390/biomedicines4030019
Sharp DW, Lattime EC. Recombinant Poxvirus and the Tumor Microenvironment: Oncolysis, Immune Regulation and Immunization. Biomedicines. 2016; 4(3):19. https://doi.org/10.3390/biomedicines4030019
Chicago/Turabian StyleSharp, Daniel W., and Edmund C. Lattime. 2016. "Recombinant Poxvirus and the Tumor Microenvironment: Oncolysis, Immune Regulation and Immunization" Biomedicines 4, no. 3: 19. https://doi.org/10.3390/biomedicines4030019
APA StyleSharp, D. W., & Lattime, E. C. (2016). Recombinant Poxvirus and the Tumor Microenvironment: Oncolysis, Immune Regulation and Immunization. Biomedicines, 4(3), 19. https://doi.org/10.3390/biomedicines4030019