The NF-κB Activating Pathways in Multiple Myeloma


Abstract
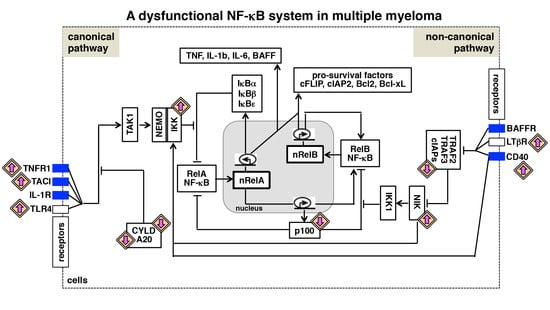
:
1. General Introduction
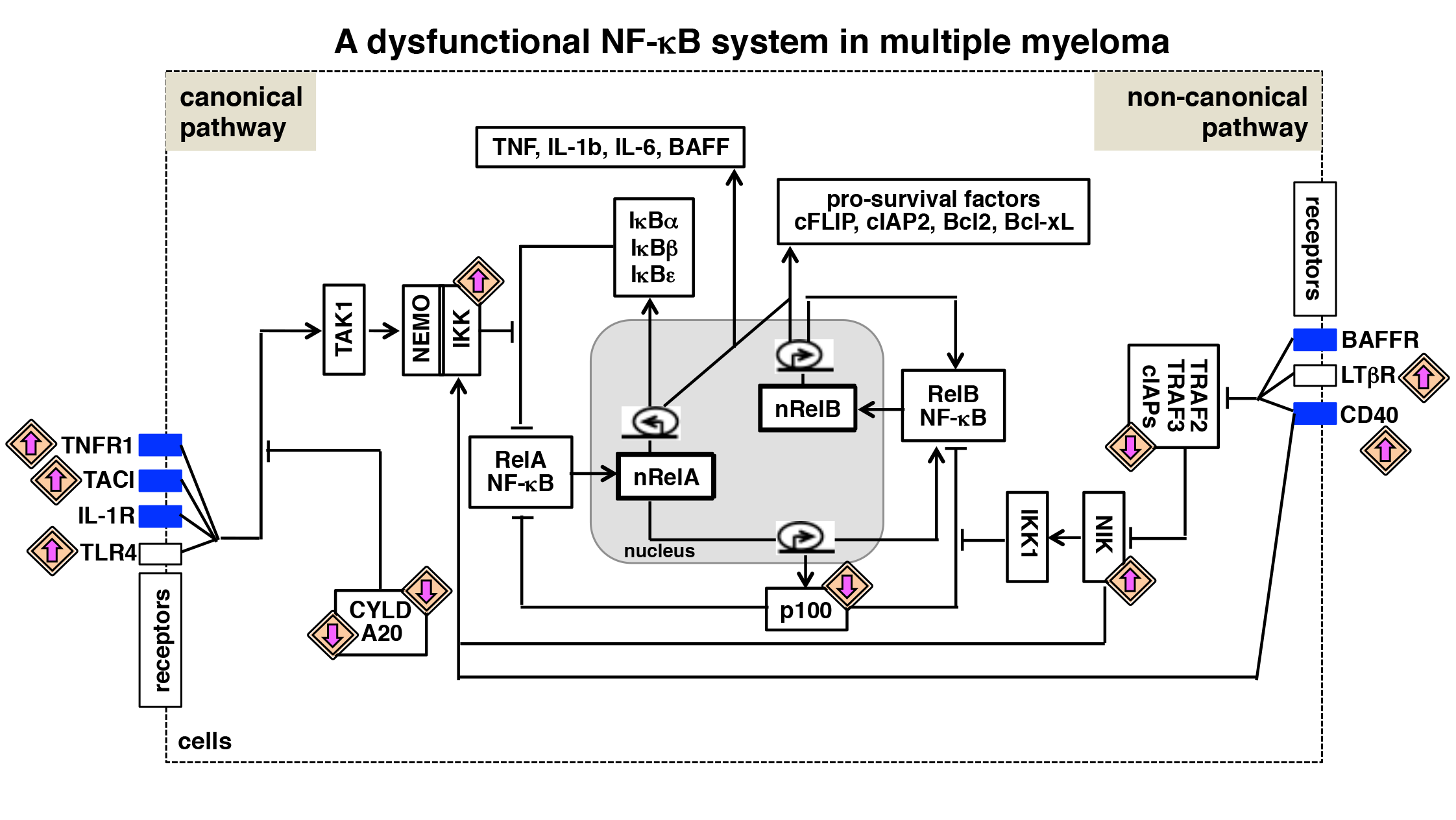
2. Multiple Myeloma—Epidemiology and Aetiology
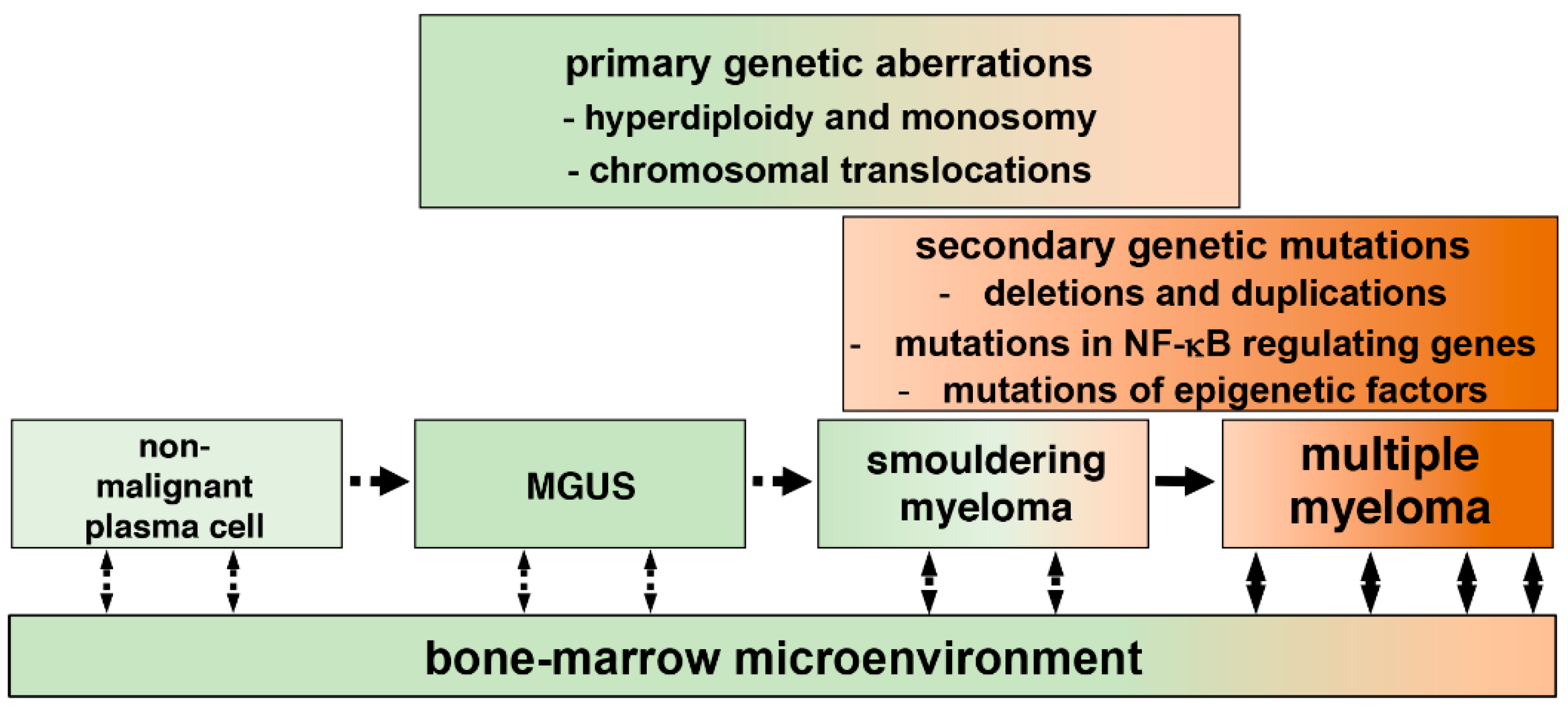
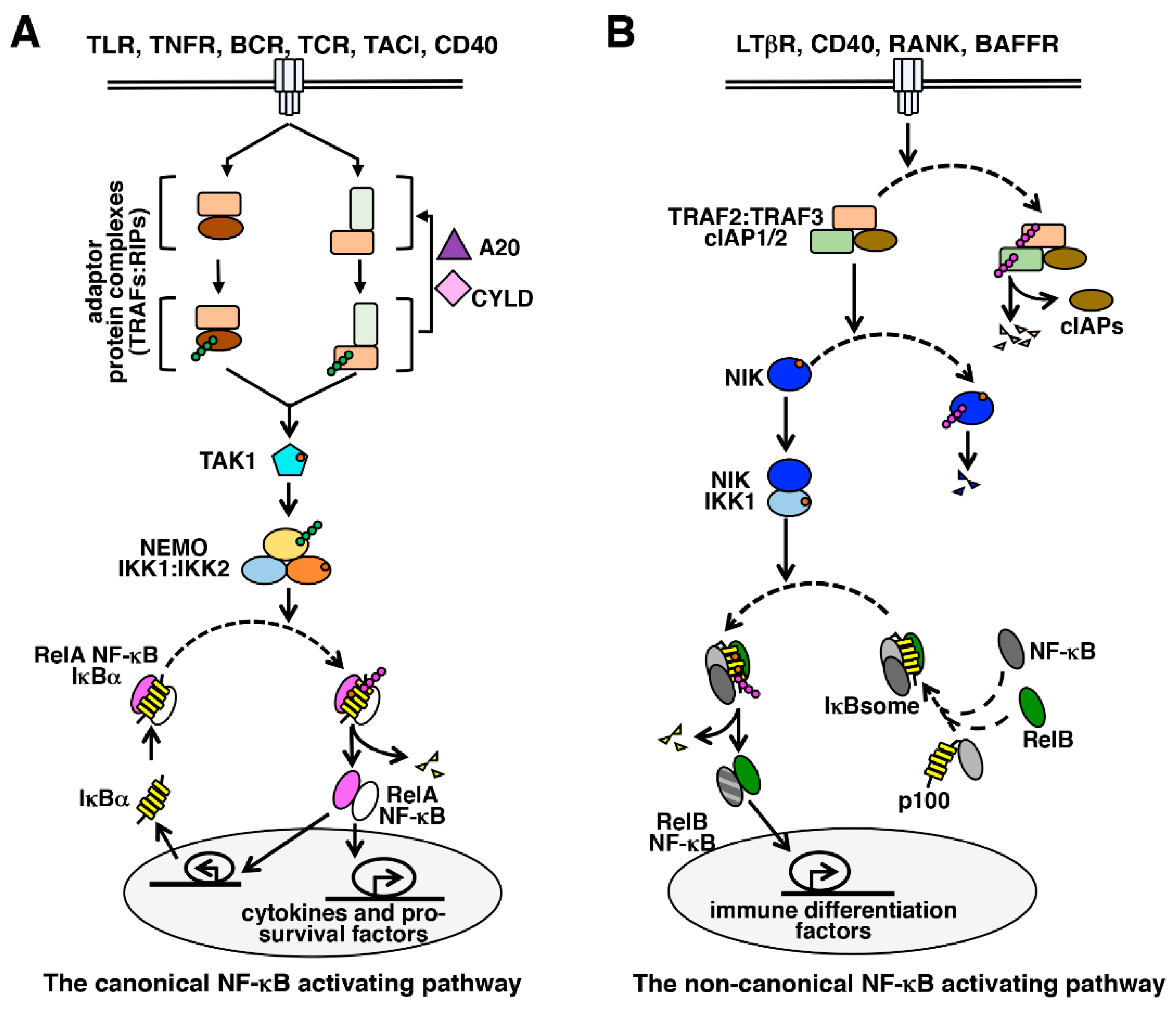
3. The NF-κB Signaling System
4. The Canonical NF-κB Activation Pathway
5. The Non-Canonical NF-κB Activation Pathway
6. NF-κB Deregulating Mutations in Multiple Myeloma
7. NF-κB-Related Microenvironmental Cues in Multiple Myeloma
8. Interactions between NF-κB Signaling Pathways and Multiple Myeloma
9. NF-κB Driven Gene Expressions in Myeloma Cells
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: Globocan 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Flores-Montero, J.; de Tute, R.; Paiva, B.; Perez, J.J.; Bottcher, S.; Wind, H.; Sanoja, L.; Puig, N.; Lecrevisse, Q.; Vidriales, M.B.; et al. Immunophenotype of normal vs. Myeloma plasma cells: Toward antibody panel specifications for mrd detection in multiple myeloma. Cytom. B Clin. Cytom. 2016, 90, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.; van der Burg, M.; Garcia-Sanz, R.; Fenton, J.A.; Langerak, A.W.; Gonzalez, M.; van Dongen, J.J.; San Miguel, J.F.; Morgan, G.J. Immunoglobulin gene rearrangements and the pathogenesis of multiple myeloma. Blood 2007, 110, 3112–3121. [Google Scholar] [CrossRef] [PubMed]
- Mahindra, A.; Hideshima, T.; Anderson, K.C. Multiple myeloma: Biology of the disease. Blood Rev. 2010, 24 (Suppl. 1), S5–S11. [Google Scholar] [CrossRef]
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. N. Engl. J. Med. 2004, 351, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.S.; Kumar, S.K.; Orlowski, R.Z.; Cook, G.; Richardson, P.G.; Gertz, M.A.; Giralt, S.; Mateos, M.V.; Leleu, X.; Anderson, K.C. Management of relapsed and refractory multiple myeloma: Novel agents, antibodies, immunotherapies and beyond. Leukemia 2018, 32, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; Van Ness, B.; Chesi, M.; Minvielle, S.; et al. International myeloma working group molecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Attal, M.; Moreau, P.; Charbonnel, C.; Garban, F.; Hulin, C.; Leyvraz, S.; Michallet, M.; Yakoub-Agha, I.; Garderet, L.; et al. Genetic abnormalities and survival in multiple myeloma: The experience of the intergroupe francophone du myelome. Blood 2007, 109, 3489–3495. [Google Scholar] [CrossRef] [PubMed]
- Corre, J.; Munshi, N.; Avet-Loiseau, H. Genetics of multiple myeloma: Another heterogeneity level? Blood 2015, 125, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Robiou du Pont, S.; Cleynen, A.; Fontan, C.; Attal, M.; Munshi, N.; Corre, J.; Avet-Loiseau, H. Genomics of multiple myeloma. J. Clin. Oncol. 2017, 35, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.R.; Waldron, J.A.; Jagannath, S.; Barlogie, B. Cytogenetic findings in 200 patients with multiple myeloma. Cancer Genet. Cytogenet. 1995, 82, 41–49. [Google Scholar] [CrossRef]
- Chesi, M.; Bergsagel, P.L.; Brents, L.A.; Smith, C.M.; Gerhard, D.S.; Kuehl, W.M. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines. Blood 1996, 88, 674–681. [Google Scholar] [PubMed]
- Chesi, M.; Nardini, E.; Brents, L.A.; Schrock, E.; Ried, T.; Kuehl, W.M.; Bergsagel, P.L. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat. Genet. 1997, 16, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Nardini, E.; Lim, R.S.; Smith, K.D.; Kuehl, W.M.; Bergsagel, P.L. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in igH/MMSET hybrid transcripts. Blood 1998, 92, 3025–3034. [Google Scholar] [PubMed]
- Hurt, E.M.; Wiestner, A.; Rosenwald, A.; Shaffer, A.L.; Campo, E.; Grogan, T.; Bergsagel, P.L.; Kuehl, W.M.; Staudt, L.M. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 2004, 5, 191–199. [Google Scholar] [CrossRef]
- Hanamura, I.; Iida, S.; Akano, Y.; Hayami, Y.; Kato, M.; Miura, K.; Harada, S.; Banno, S.; Wakita, A.; Kiyoi, H.; et al. Ectopic expression of MAFB gene in human myeloma cells carrying (14;20)(q32;q11) chromosomal translocations. Jpn. J. Cancer Res. 2001, 92, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, P.L.; Kuehl, W.M.; Zhan, F.; Sawyer, J.; Barlogie, B.; Shaughnessy, J., Jr. Cyclin d dysregulation: An early and unifying pathogenic event in multiple myeloma. Blood 2005, 106, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Li, C.; Magrangeas, F.; Gouraud, W.; Charbonnel, C.; Harousseau, J.L.; Attal, M.; Marit, G.; Mathiot, C.; Facon, T.; et al. Prognostic significance of copy-number alterations in multiple myeloma. J. Clin. Oncol. 2009, 27, 4585–4590. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Leone, P.E.; Jenner, M.W.; Li, C.; Gonzalez, D.; Johnson, D.C.; Ross, F.M.; Davies, F.E.; Morgan, G.J. Integration of global snp-based mapping and expression arrays reveals key regions, mechanisms, and genes important in the pathogenesis of multiple myeloma. Blood 2006, 108, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Hanamura, I.; Stewart, J.P.; Huang, Y.; Zhan, F.; Santra, M.; Sawyer, J.R.; Hollmig, K.; Zangarri, M.; Pineda-Roman, M.; van Rhee, F.; et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: Incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood 2006, 108, 1724–1732. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Bergsagel, P.L. Molecular pathogenesis of multiple myeloma: Basic and clinical updates. Int. J. Hematol. 2013, 97, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed]
- Neri, A.; Baldini, L.; Trecca, D.; Cro, L.; Polli, E.; Maiolo, A.T. p53 gene mutations in multiple myeloma are associated with advanced forms of malignancy. Blood 1993, 81, 128–135. [Google Scholar] [PubMed]
- Affer, M.; Chesi, M.; Chen, W.G.; Keats, J.J.; Demchenko, Y.N.; Roschke, A.V.; Van Wier, S.; Fonseca, R.; Bergsagel, P.L.; Kuehl, W.M. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate myc expression in multiple myeloma. Leukemia 2014, 28, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Neri, A. Utilizing next-generation sequencing in the management of multiple myeloma. Expert Rev. Mol. Diagn. 2017, 17, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Bustoros, M.; Mouhieddine, T.H.; Detappe, A.; Ghobrial, I.M. Established and novel prognostic biomarkers in multiple myeloma. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Kim, S.; Gould, J.; Knoechel, B.; Drier, Y.; Cotton, M.J.; Gray, D.; Birrer, N.; Wong, B.; Ha, G.; et al. Genetic interrogation of circulating multiple myeloma cells at single-cell resolution. Sci. Transl. Med. 2016, 8, 363ra147. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.J.; Van Wier, S.; Tiedemann, R.; Shi, C.X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Troppan, K.; Hofer, S.; Wenzl, K.; Lassnig, M.; Pursche, B.; Steinbauer, E.; Wiltgen, M.; Zulus, B.; Renner, W.; Beham-Schmid, C.; et al. Frequent down regulation of the tumor suppressor gene a20 in multiple myeloma. PLoS ONE 2015, 10, e0123922. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-kappaB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Ubiquitination in signaling to and activation of ikk. Immunol. Rev. 2012, 246, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the nfkappab system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. Bcma is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Hatzoglou, A.; Roussel, J.; Bourgeade, M.F.; Rogier, E.; Madry, C.; Inoue, J.; Devergne, O.; Tsapis, A. TNF receptor family member BCMA (B cell maturation) associates with tnf receptor-associated factor (TRAF) 1, TRAF2, and TRAF3 and activates NF-kappa B, elk-1, c-jun N-terminal kinase, and p38 mitogen-activated protein kinase. J. Immunol. 2000, 165, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, M.; Cren, M.; Robert, N.; Bollore, K.; Fest, T.; Duperray, C.; Guilloton, F.; Hose, D.; Tarte, K.; Klein, B. IL-6 supports the generation of human long-lived plasma cells in combination with either april or stromal cell-soluble factors. Leukemia 2014, 28, 1647–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.C. The noncanonical NF-kappaB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Savinova, O.V.; Hoffmann, A.; Ghosh, G. The NFKB1 and NFKB2 proteins p105 and p100 function as the core of high-molecular-weight heterogeneous complexes. Mol. Cell 2009, 34, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Fusco, A.; Huang, D.B.; Gupta, K.; Young Kim, D.; Ware, C.F.; Van Duyne, G.D.; Ghosh, G. p100/ikappabdelta sequesters and inhibits NF-kappaB through kappaBsome formation. Proc. Natl. Acad. Sci. USA 2014, 111, 15946–15951. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Polley, S.; Passos, D.O.; Huang, D.B.; Mulero, M.C.; Mazumder, A.; Biswas, T.; Verma, I.M.; Lyumkis, D.; Ghosh, G. Structural basis for the activation of IKK1/alpha. Cell Rep. 2016, 17, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Fong, A.; Sun, S.C. Genetic evidence for the essential role of beta-transducin repeat-containing protein in the inducible processing of NF-kappa B2/p100. J. Biol. Chem. 2002, 277, 22111–22114. [Google Scholar] [CrossRef] [PubMed]
- Mills, D.M.; Bonizzi, G.; Karin, M.; Rickert, R.C. Regulation of late B cell differentiation by intrinsic IKKalpha-dependent signals. Proc. Natl. Acad. Sci. USA 2007, 104, 6359–6364. [Google Scholar] [CrossRef] [PubMed]
- De Silva, N.S.; Anderson, M.M.; Carette, A.; Silva, K.; Heise, N.; Bhagat, G.; Klein, U. Transcription factors of the alternative NF-kappaB pathway are required for germinal center B-cell development. Proc. Natl. Acad. Sci. USA 2016, 113, 9063–9068. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.; Tseng, P.H.; Keats, J.J.; Wang, H.; Vignali, D.A.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.C.; Mak, T.W.; et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors CIAP1, CIAP2, TRAF2 and TRAF3 and the kinase nik. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Zarnegar, B.; Ytterberg, A.J.; Shiba, T.; Dempsey, P.W.; Ware, C.F.; Loo, J.A.; Cheng, G. Negative feedback in noncanonical NF-kappaB signaling modulates NIK stability through IKKalpha-mediated phosphorylation. Sci. Signal. 2010, 3, ra41. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Bebien, M.; Otero, D.C.; Johnson-Vroom, K.E.; Cao, Y.; Vu, D.; Jegga, A.G.; Aronow, B.J.; Ghosh, G.; Rickert, R.C.; et al. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:P52 dimers. EMBO J. 2004, 23, 4202–4210. [Google Scholar] [CrossRef] [PubMed]
- Fusco, A.J.; Huang, D.B.; Miller, D.; Wang, V.Y.; Vu, D.; Ghosh, G. NF-kappaB p52:ReLB heterodimer recognizes two classes of kappaB sites with two distinct modes. EMBO Rep. 2009, 10, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.; Chatterjee, B.; Dhar, A.; Bais, S.S.; Chawla, M.; Roy, P.; George, A.; Bal, V.; Rath, S.; Basak, S. A TNF-p100 pathway subverts noncanonical NF-kappaB signaling in inflamed secondary lymphoid organs. EMBO J. 2017, 36, 3501–3516. [Google Scholar] [CrossRef] [PubMed]
- Siggers, T.; Chang, A.B.; Teixeira, A.; Wong, D.; Williams, K.J.; Ahmed, B.; Ragoussis, J.; Udalova, I.A.; Smale, S.T.; Bulyk, M.L. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-kappaB family DNA binding. Nat. Immunol. 2011, 13, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Barrera, L.A.; Ersing, I.; Willox, B.; Schmidt, S.C.; Greenfeld, H.; Zhou, H.; Mollo, S.B.; Shi, T.T.; Takasaki, K.; et al. The NF-kappaB genomic landscape in lymphoblastoid B cells. Cell Rep. 2014, 8, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, K.A.; Kaergel, E.; Heinig, M.; Fontaine, J.F.; Patone, G.; Muro, E.M.; Mathas, S.; Hummel, M.; Andrade-Navarro, M.A.; Hubner, N.; et al. A roadmap of constitutive NF-kappaB activity in hodgkin lymphoma: Dominant roles of p50 and p52 revealed by genome-wide analyses. Genome Med. 2016, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Mukherjee, T.; Chatterjee, B.; Vijayaragavan, B.; Banoth, B.; Basak, S. Non-canonical nfkappab mutations reinforce pro-survival tnf response in multiple myeloma through an autoregulatory RelB:P50 NFkappaB pathway. Oncogene 2017, 36, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.; Davis-Turak, J.; Macal, M.; Huang, J.Q.; Ponomarenko, J.; Kearns, J.D.; Yu, T.; Fagerlund, R.; Asagiri, M.; Zuniga, E.I.; et al. Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-kappaB pathways. Nat. Immunol. 2012, 13, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Cormier, F.; Monjanel, H.; Fabre, C.; Billot, K.; Sapharikas, E.; Chereau, F.; Bordereaux, D.; Molina, T.J.; Avet-Loiseau, H.; Baud, V. Frequent engagement of RelB activation is critical for cell survival in multiple myeloma. PLoS ONE 2013, 8, e59127. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Dimer-specific regulatory mechanisms within the NF-kappaB family of transcription factors. Immunol. Rev. 2012, 246, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational spectrum, copy number changes, and outcome: Results of a sequencing study of patients with newly diagnosed myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Kumar, S.; Vanwier, S.; Ahmann, G.; Price-Troska, T.; Henderson, K.; Chung, T.H.; Kim, S.; Mulligan, G.; Bryant, B.; et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007, 67, 2982–2989. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Fairfield, H.; Falank, C.; Avery, L.; Reagan, M.R. Multiple myeloma in the marrow: Pathogenesis and treatments. Ann. N. Y. Acad. Sci. 2016, 1364, 32–51. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Uchiyama, H.; Akbarali, Y.; Urashima, M.; Yamamoto, K.; Libermann, T.A.; Anderson, K.C. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood 1996, 87, 1104–1112. [Google Scholar] [PubMed]
- Costes, V.; Portier, M.; Lu, Z.Y.; Rossi, J.F.; Bataille, R.; Klein, B. Interleukin-1 in multiple myeloma: Producer cells and their role in the control of IL-6 production. Br. J. Haematol. 1998, 103, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Anderson, K.C. Emerging therapies targeting tumor vasculature in multiple myeloma and other hematologic and solid malignancies. Curr. Cancer Drug Targets 2011, 11, 1005–1024. [Google Scholar] [CrossRef] [PubMed]
- Chilov, D.; Kukk, E.; Taira, S.; Jeltsch, M.; Kaukonen, J.; Palotie, A.; Joukov, V.; Alitalo, K. Genomic organization of human and mouse genes for vascular endothelial growth factor C. J. Biol. Chem. 1997, 272, 25176–25183. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Mitsiades, N.; Poulaki, V.; Schlossman, R.; Akiyama, M.; Chauhan, D.; Hideshima, T.; Treon, S.P.; Munshi, N.C.; Richardson, P.G.; et al. Activation of NF-kappaB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/AKT signaling in human multiple myeloma cells: Therapeutic implications. Oncogene 2002, 21, 5673–5683. [Google Scholar] [CrossRef] [PubMed]
- Sprynski, A.C.; Hose, D.; Caillot, L.; Reme, T.; Shaughnessy, J.D., Jr.; Barlogie, B.; Seckinger, A.; Moreaux, J.; Hundemer, M.; Jourdan, M.; et al. The role of IGF-1 as a major growth factor for myeloma cell lines and the prognostic relevance of the expression of its receptor. Blood 2009, 113, 4614–4626. [Google Scholar] [CrossRef] [PubMed]
- Musolino, C.; Allegra, A.; Innao, V.; Allegra, A.G.; Pioggia, G.; Gangemi, S. Inflammatory and anti-inflammatory equilibrium, proliferative and antiproliferative balance: The role of cytokines in multiple myeloma. Med. Inflamm. 2017, 2017, 1852517. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Komai, M.; Itoh, T.; Imano, M.; Sakamoto, K.; Shimaoka, H.; Ogawa, N.; Mashimo, K.; Fujiwara, D.; Takeda, T.; et al. Inhibition of the tumour necrosis factor-alpha autocrine loop enhances the sensitivity of multiple myeloma cells to anticancer drugs. Eur. J. Cancer 2013, 49, 3708–3717. [Google Scholar] [CrossRef] [PubMed]
- Corral, L.G.; Haslett, P.A.; Muller, G.W.; Chen, R.; Wong, L.M.; Ocampo, C.J.; Patterson, R.T.; Stirling, D.I.; Kaplan, G. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J. Immunol. 1999, 163, 380–386. [Google Scholar] [PubMed]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMIDS) in multiple myeloma. Leukemia 2010, 24, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Reme, T.; Lugagne, C.; Moine, P.; Rossi, J.F.; Klein, B.; Tarte, K. Baff and april protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004, 103, 3148–3157. [Google Scholar] [CrossRef] [PubMed]
- Hengeveld, P.J.; Kersten, M.J. B-cell activating factor in the pathophysiology of multiple myeloma: A target for therapy? Blood Cancer J. 2015, 5, e282. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, B.J.; Scheible, C.A.; Nuebling, T.; Kopp, H.G.; Wirths, S.; Azuma, M.; Schneider, P.; Jung, G.; Grosse-Hovest, L.; Salih, H.R. Rankl expression, function, and therapeutic targeting in multiple myeloma and chronic lymphocytic leukemia. Cancer Res. 2013, 73, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Landowski, T.H.; Olashaw, N.E.; Agrawal, D.; Dalton, W.S. Cell adhesion-mediated drug resistance (CAM-DR) is associated with activation of NF-kappa B (RelB/p50) in myeloma cells. Oncogene 2003, 22, 2417–2421. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Purschke, W.G.; Moschetta, M.; Buchner, K.; Maasch, C.; Zboralski, D.; Zollner, S.; Vonhoff, S.; Mishima, Y.; et al. SDF-1 inhibition targets the bone marrow niche for cancer therapy. Cell Rep. 2014, 9, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Thu, Y.M.; Richmond, A. NF-kappaB inducing kinase: A key regulator in the immune system and in cancer. Cytokine Growth Factor Rev. 2010, 21, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single nfkappab system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Kim, H.; Kearns, J.D.; Tergaonkar, V.; O’Dea, E.; Werner, S.L.; Benedict, C.A.; Ware, C.F.; Ghosh, G.; Verma, I.M.; et al. A fourth IKappaB protein within the NF-kappaB signaling module. Cell 2007, 128, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.; Kearns, J.D.; Basak, S.; Savinova, O.V.; Ghosh, G.; Hoffmann, A. Kinetic control of negative feedback regulators of NF-kappaB/RelA determines their pathogen- and cytokine-receptor signaling specificity. Proc. Natl. Acad. Sci. USA 2009, 106, 9619–9624. [Google Scholar] [CrossRef] [PubMed]
- Almaden, J.V.; Tsui, R.; Liu, Y.C.; Birnbaum, H.; Shokhirev, M.N.; Ngo, K.A.; Davis-Turak, J.C.; Otero, D.; Basak, S.; Rickert, R.C.; et al. A pathway switch directs baff signaling to distinct NFkappaB transcription factors in maturing and proliferating B cells. Cell Rep. 2014, 9, 2098–2111. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.; Yamazaki, S.; He, J.Q.; Cheng, G. Control of canonical NF-kappaB activation through the nik-ikk complex pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 3503–3508. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, Y.N.; Glebov, O.K.; Zingone, A.; Keats, J.J.; Bergsagel, P.L.; Kuehl, W.M. Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood 2010, 115, 3541–3552. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Chauhan, D.; Kiziltepe, T.; Ikeda, H.; Okawa, Y.; Podar, K.; Raje, N.; Protopopov, A.; Munshi, N.C.; Richardson, P.G.; et al. Biologic sequelae of I{kappa}B kinase (IKK) inhibition in multiple myeloma: Therapeutic implications. Blood 2009, 113, 5228–5236. [Google Scholar] [CrossRef] [PubMed]
- Fabre, C.; Mimura, N.; Bobb, K.; Kong, S.Y.; Gorgun, G.; Cirstea, D.; Hu, Y.; Minami, J.; Ohguchi, H.; Zhang, J.; et al. Dual inhibition of canonical and noncanonical NF-kappaB pathways demonstrates significant antitumor activities in multiple myeloma. Clin. Cancer Res. 2012, 18, 4669–4681. [Google Scholar] [CrossRef] [PubMed]
- Derudder, E.; Dejardin, E.; Pritchard, L.L.; Green, D.R.; Korner, M.; Baud, V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-alpha and lymphotoxin-beta receptor activation: Critical roles for p100. J. Biol. Chem. 2003, 278, 23278–23284. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Shih, V.F.; Hoffmann, A. Generation and activation of multiple dimeric transcription factors within the NF-kappaB signaling system. Mol. Cell Biol. 2008, 28, 3139–3150. [Google Scholar] [CrossRef] [PubMed]
- Busino, L.; Millman, S.E.; Scotto, L.; Kyratsous, C.A.; Basrur, V.; O’Connor, O.; Hoffmann, A.; Elenitoba-Johnson, K.S.; Pagano, M. Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat. Cell Biol. 2012, 14, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Mineva, N.D.; Rothstein, T.L.; Meyers, J.A.; Lerner, A.; Sonenshein, G.E. CD40 ligand-mediated activation of the de novo RelB NF-kappaB synthesis pathway in transformed B cells promotes rescue from apoptosis. J. Biol. Chem. 2007, 282, 17475–17485. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Belguise, K.; Kersual, N.; Kirsch, K.H.; Mineva, N.D.; Galtier, F.; Chalbos, D.; Sonenshein, G.E. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell Biol. 2007, 9, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Dejardin, E.; Droin, N.M.; Delhase, M.; Haas, E.; Cao, Y.; Makris, C.; Li, Z.W.; Karin, M.; Ware, C.F.; Green, D.R. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity 2002, 17, 525–535. [Google Scholar] [CrossRef]
- Stadanlick, J.E.; Kaileh, M.; Karnell, F.G.; Scholz, J.L.; Miller, J.P.; Quinn, W.J., 3rd; Brezski, R.J.; Treml, L.S.; Jordan, K.A.; Monroe, J.G.; et al. Tonic b cell antigen receptor signals supply an NF-kappaB substrate for prosurvival blys signaling. Nat. Immunol. 2008, 9, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Z.B.; Kofahl, B.; Beaudette, P.; Baum, K.; Ipenberg, I.; Weih, F.; Wolf, J.; Dittmar, G.; Scheidereit, C. Quantitative dissection and modeling of the nf-kappab p100-p105 module reveals interdependent precursor proteolysis. Cell Rep. 2014, 9, 1756–1769. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Li, X.; Tong, X.; Santos, D.; Otsuki, T.; Catley, L.; Tournilhac, O.; Podar, K.; Hideshima, T.; Schlossman, R.; et al. Human anti-CD40 antagonist antibody triggers significant antitumor activity against human multiple myeloma. Cancer Res. 2005, 65, 5898–5906. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.; He, J.Q.; Oganesyan, G.; Hoffmann, A.; Baltimore, D.; Cheng, G. Unique CD40-mediated biological program in B cell activation requires both type 1 and type 2 NF-kappaB activation pathways. Proc. Natl. Acad. Sci. USA 2004, 101, 8108–8113. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, W.; Maziarz, R.T.; Jagannath, S.; Spencer, A.; Durrant, S.; Becker, P.S.; Ewald, B.; Bilic, S.; Rediske, J.; Baeck, J.; et al. A phase 1 study of lucatumumab, a fully human anti-CD40 antagonist monoclonal antibody administered intravenously to patients with relapsed or refractory multiple myeloma. Br. J. Haematol. 2012, 159, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Sati, H.I.; Apperley, J.F.; Greaves, M.; Lawry, J.; Gooding, R.; Russell, R.G.; Croucher, P.I. Interleukin-6 is expressed by plasma cells from patients with multiple myeloma and monoclonal gammopathy of undetermined significance. Br. J. Haematol. 1998, 101, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, F.; Torcia, M.; Aldinucci, D.; Rubartelli, A.; Miliani, A.; Shaw, A.R.; Lansdorp, P.M.; Di Guglielmo, R. Production of interleukin-1 by bone marrow myeloma cells. Blood 1989, 74, 380–387. [Google Scholar] [PubMed]
- Frassanito, M.A.; Cusmai, A.; Iodice, G.; Dammacco, F. Autocrine interleukin-6 production and highly malignant multiple myeloma: Relation with resistance to drug-induced apoptosis. Blood 2001, 97, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Li, X.F.; Breitkreutz, I.; Song, W.; Neri, P.; Catley, L.; Podar, K.; Hideshima, T.; Chauhan, D.; Raje, N.; et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2006, 66, 6675–6682. [Google Scholar] [CrossRef] [PubMed]
- Tornatore, L.; Sandomenico, A.; Raimondo, D.; Low, C.; Rocci, A.; Tralau-Stewart, C.; Capece, D.; D’Andrea, D.; Bua, M.; Boyle, E.; et al. Cancer-selective targeting of the NF-kappaB survival pathway with GADD45beta/MKK7 inhibitors. Cancer Cell 2014, 26, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of akt in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Toth, C.R.; Hostutler, R.F.; Baldwin, A.S., Jr.; Bender, T.P. Members of the nuclear factor kappa B family transactivate the murine c-myb gene. J. Biol. Chem. 1995, 270, 7661–7671. [Google Scholar] [CrossRef] [PubMed]
- Gardam, S.; Beyaert, R. The kinase NIK as a therapeutic target in multiple myeloma. Expert Opin. Ther. Targets 2011, 15, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Mailankody, S.; Kazandjian, D.; Korde, N.; Roschewski, M.; Manasanch, E.; Bhutani, M.; Tageja, N.; Kwok, M.; Zhang, Y.; Zingone, A.; et al. Baseline mutational patterns and sustained MRD negativity in patients with high-risk smoldering myeloma. Blood Adv. 2017, 1, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Mikulasova, A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Jackson, G.H.; Smetana, J.; Kufova, Z.; Pour, L.; Sandecka, V.; Almasi, M.; et al. The spectrum of somatic mutations in monoclonal gammopathy of undetermined significance indicates a less complex genomic landscape than that in multiple myeloma. Haematologica 2017, 102, 1617–1625. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
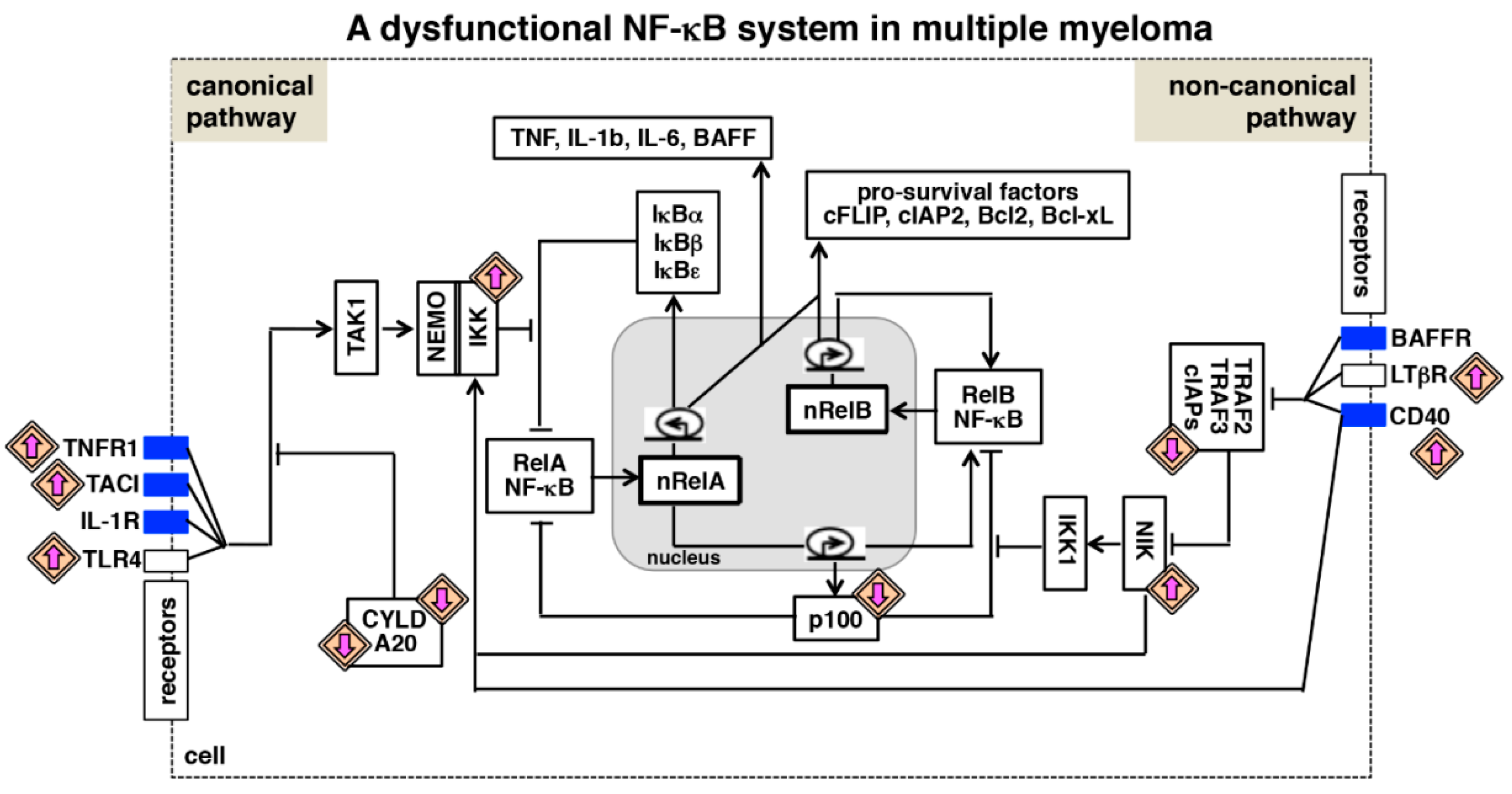{kind=link}
| Genetic Abnormalities | Genes or Chromosomes Affected | Comments | References |
|---|---|---|---|
| Hyperdiploidy | chromosomes 3, 5, 7, 9, 11, 15, 19, 21 | Functional role in the pathogenesis of MM remains elusive. | [12,14] |
| Monosomy | chromosome 13 | Functional role of remains unclear. | [11] |
| Frequent IGH translocations | t(11;14)(q13;q32) CCND1 t(4;14)(p16;q32) FGFR3 | Upregulate the expression of oncogenes encoding cyclin D1 and fibroblast growth factor receptor 3. | [15,16] |
| t(4;14)(p16;q32) MMSET/WHSC1 | Upregulate the expression of MMSET, which methylates chromatin-associated proteins and modulates their functions. | [17] | |
| Relatively rare translocations | t(14;16)(q32;q23) MAF t(14;20)(q32;q11) MAFB t(6;14)(p21;q32) CCND3 | These chromosomal translocations cause deregulated expressions of cell cycle regulators, including cyclin D2 and cyclin D3. | [18,19,20] |
| Duplication | chromosome 1 (1q) | Increased incidences in advanced MM, functional roles are unclear. | [23] |
| Deletions | 1p, 6q, 8p, 12p, 14q, 16q, 17p, 20p | Functional role remains unclear. | [21,22] |
| BIRC2 BIRC3 TRAF3 CYLD | Frequent homozygous deletions, which disrupt the function of various inhibitors of the NF-κB system. | [33,34] | |
| TNFAIP3 | Heterozygous deletions, which inactivate the inhibitor of IKK, A20. | [35] | |
| CDKN2C | Abrogate the function of the tumor suppressor protein cyclin-dependent kinase inhibitor 2C. | [26] | |
| Gene mutations | NRAS KRAS BRAF | Gain-of-function mutations in NRAS and KRAS trigger aberrant MAPK activity and are associated with the progression of MM, mutations in the BRAF oncogene promote myeloma growth. | [25] |
| TP53 | Abrogate the expression of p53 tumor suppressor in advanced MM. | [27] | |
| 8q24 locus rearrangements | MYC | Upregulate the expression of the MYC oncogene. | [28] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, P.; Sarkar, U.A.; Basak, S. The NF-κB Activating Pathways in Multiple Myeloma. Biomedicines 2018, 6, 59. https://doi.org/10.3390/biomedicines6020059
Roy P, Sarkar UA, Basak S. The NF-κB Activating Pathways in Multiple Myeloma. Biomedicines. 2018; 6(2):59. https://doi.org/10.3390/biomedicines6020059
Chicago/Turabian StyleRoy, Payel, Uday Aditya Sarkar, and Soumen Basak. 2018. "The NF-κB Activating Pathways in Multiple Myeloma" Biomedicines 6, no. 2: 59. https://doi.org/10.3390/biomedicines6020059
APA StyleRoy, P., Sarkar, U. A., & Basak, S. (2018). The NF-κB Activating Pathways in Multiple Myeloma. Biomedicines, 6(2), 59. https://doi.org/10.3390/biomedicines6020059