The Potential of the Cyclotide Scaffold for Drug Development


Abstract
:
1. Introduction
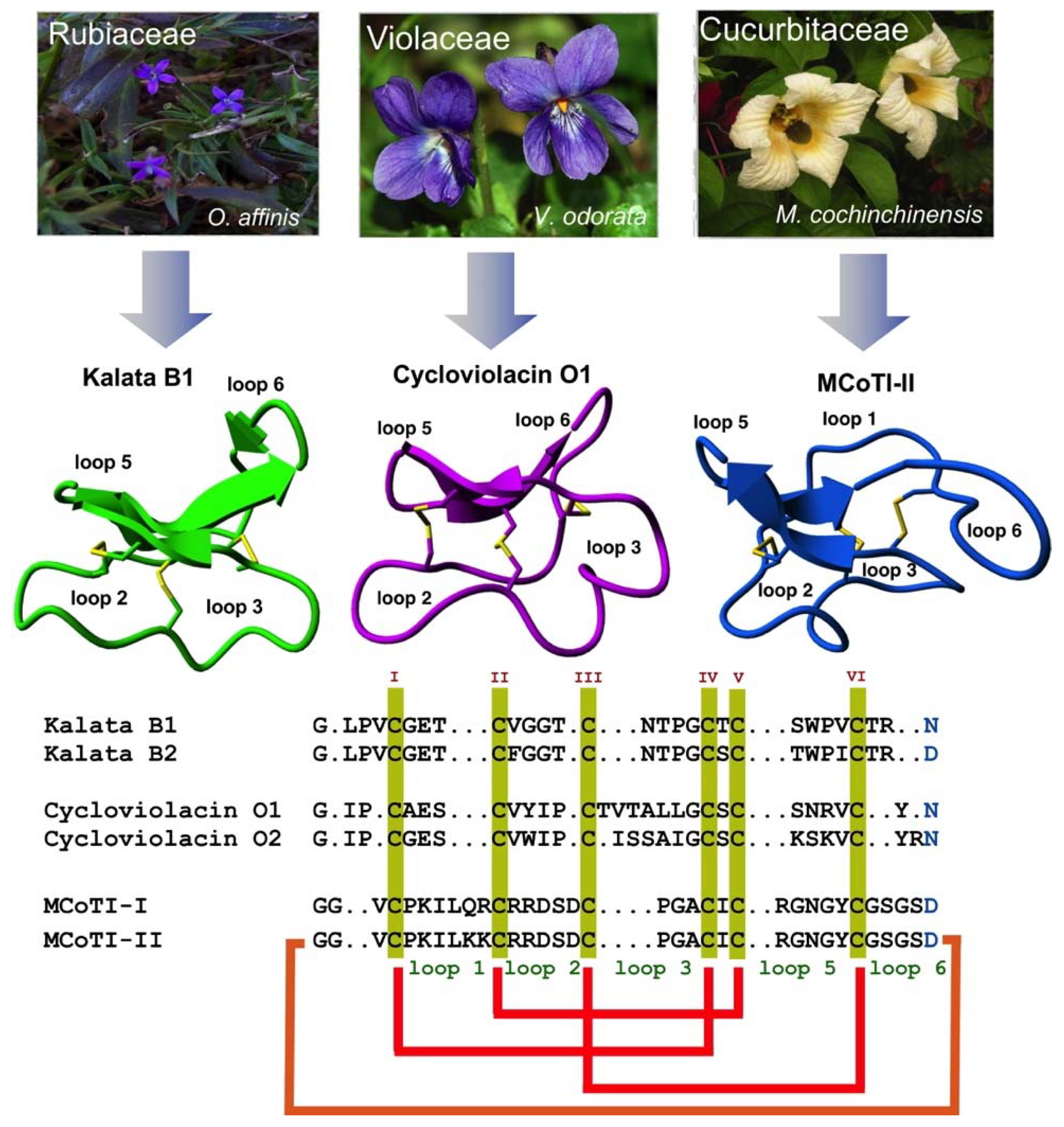
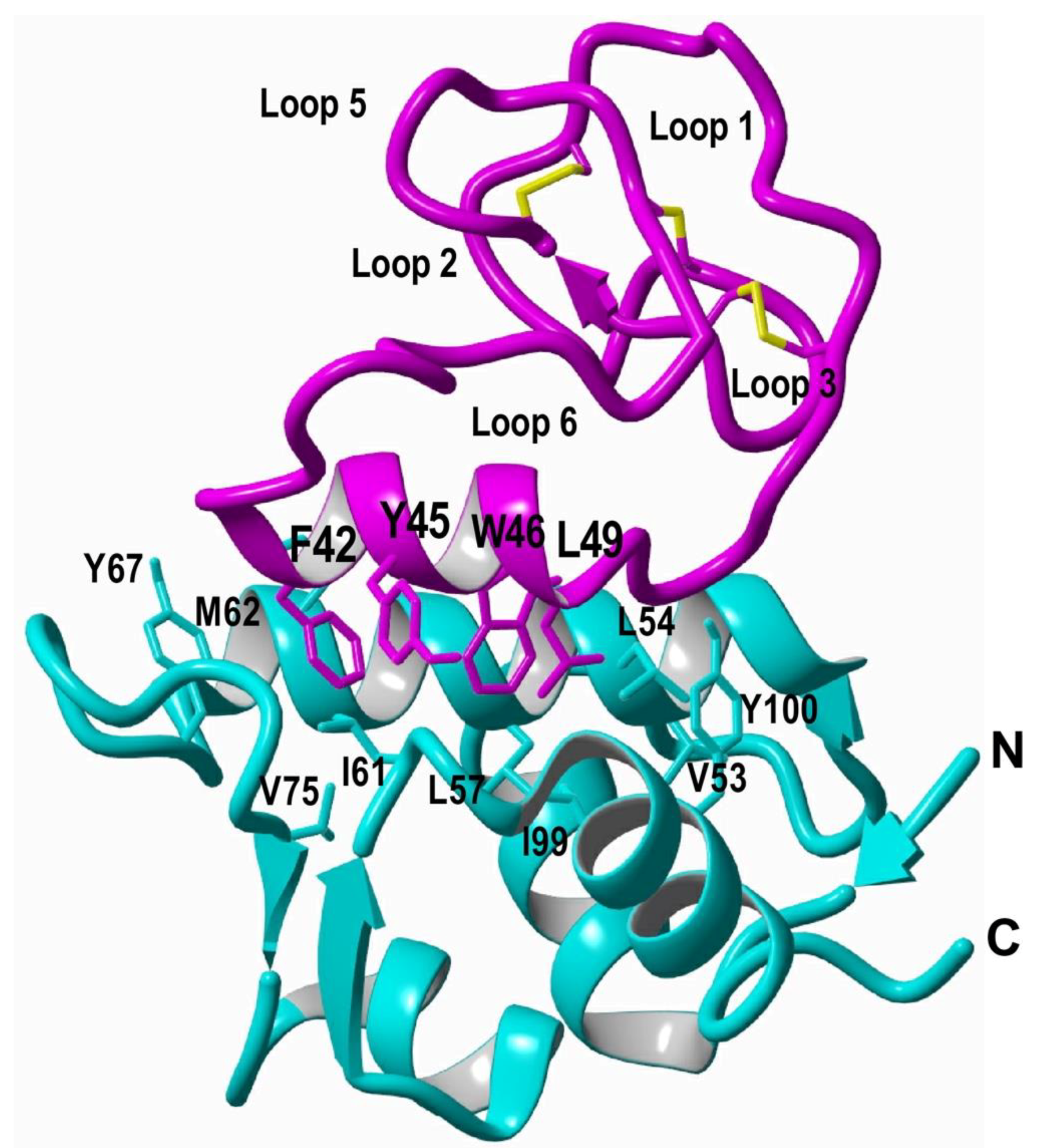
2. Structure
3. Biosynthesis
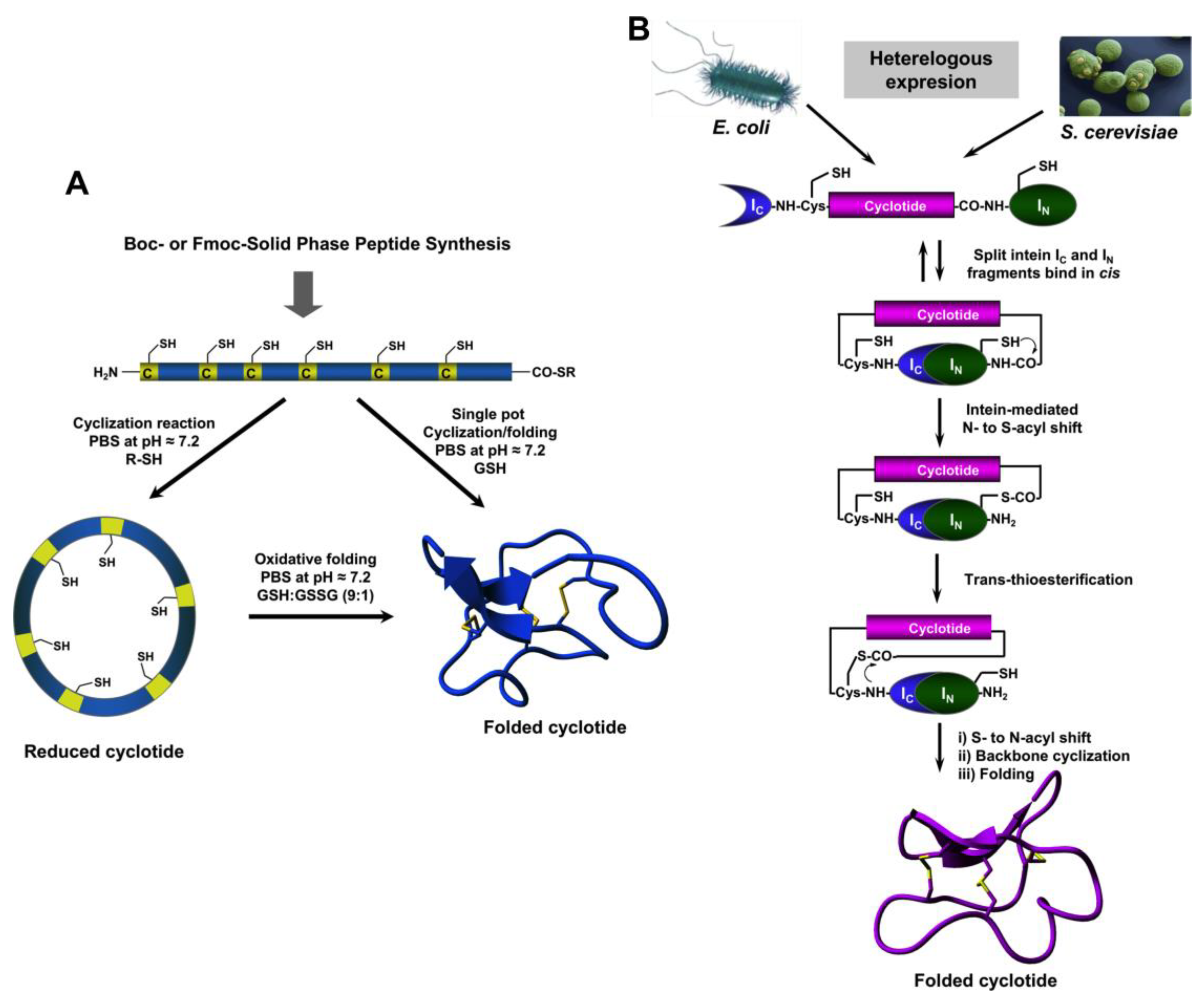
4. Chemical Synthesis
5. Recombinant Expression
6. Biological Activities of Naturally-occurring Cyclotides
7. Cyclotides with Novel Biological Activities
8. Biodistribution Studies on Cyclotides
9. Summary
10. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Jubb, H.; Higueruelo, A.P.; Winter, A.; Blundell, T.L. Structural biology and drug discovery for protein-protein interactions. Trends Pharmacol. Sci. 2012, 33, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Zinzalla, G.; Thurston, D.E. Targeting protein-protein interactions for therapeutic intervention: A challenge for the future. Future Med. Chem. 2009, 1, 65–93. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.C.; Vassilev, L.T. Targeting protein-protein interactions for cancer therapy. J. Mol. Med. 2005, 83, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Dougherty, P.G.; Pei, D. Targeting intracellular protein-protein interactions with cell-permeable cyclic peptides. Curr. Opin. Chem. Biol. 2017, 38, 80–86. [Google Scholar] [CrossRef]
- Ji, Y.; Majumder, S.; Millard, M.; Borra, R.; Bi, T.; Elnagar, A.Y.; Neamati, N.; Shekhtman, A.; Camarero, J.A. In vivo activation of the p53 tumor suppressor pathway by an engineered cyclotide. J. Am. Chem. Soc. 2013, 135, 11623–11633. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-art strategies for targeting protein–protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef] [PubMed]
- Laraia, L.; McKenzie, G.; Spring, D.R.; Venkitaraman, A.R.; Huggins, D.J. Overcoming chemical, biological, and computational challenges in the development of inhibitors targeting protein-protein interactions. Chem. Biol. 2015, 22, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Stumpp, M.T.; Binz, H.K.; Amstutz, P. Darpins: A new generation of protein therapeutics. Drug Discov. Today 2008, 13, 695–701. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-vegf antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Klint, J.K.; Senff, S.; Rupasinghe, D.B.; Er, S.Y.; Herzig, V.; Nicholson, G.M.; King, G.F. Spider-venom peptides that target voltage-gated sodium channels: Pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. [Google Scholar] [CrossRef]
- Wurch, T.; Pierre, A.; Depil, S. Novel protein scaffolds as emerging therapeutic proteins: From discovery to clinical proof-of-concept. Trends Biotechnol. 2012, 30, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J. Discovery and development of the chi-conopeptide class of analgesic peptides. Toxicon 2012, 59, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Sancheti, H.; Camarero, J.A. “Splicing up” drug discovery. Cell-based expression and screening of genetically-encoded libraries of backbone-cyclized polypeptides. Adv. Drug Deliv. Rev. 2009, 61, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Bloom, L.; Calabro, V. Fn3: A new protein scaffold reaches the clinic. Drug Discov. Today 2009, 14, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J. Conotoxin venom peptide therapeutics. Adv. Exp. Med. Biol. 2009, 655, 44–48. [Google Scholar] [PubMed]
- Chaudhuri, D.; Aboye, T.; Camarero, J.A. Using backbone-cyclized cys-rich polypeptides as molecular scaffolds to target protein-protein interactions. Biochem. J 2019, 476, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.; Craik, D.J. Designing macrocyclic disulfide-rich peptides for biotechnological applications. Nat. Chem. Biol. 2018, 14, 417–427. [Google Scholar] [CrossRef]
- Craik, D.J.; Lee, M.H.; Rehm, F.B.H.; Tombling, B.; Doffek, B.; Peacock, H. Ribosomally-synthesised cyclic peptides from plants as drug leads and pharmaceutical scaffolds. Biorg. Med. Chem. 2018, 26, 2727–2737. [Google Scholar] [CrossRef] [PubMed]
- Poth, A.G.; Colgrave, M.L.; Lyons, R.E.; Daly, N.L.; Craik, D.J. Discovery of an unusual biosynthetic origin for circular proteins in legumes. Proc. Natl. Acad. Sci. USA 2011, 108, 10127–10132. [Google Scholar] [CrossRef]
- Gould, A.; Camarero, J.A. Cyclotides: Overview and biotechnological applications. ChemBioChem 2017, 18, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Du, J. Cyclotides as drug design scaffolds. Curr. Opin. Chem. Biol. 2017, 38, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Camarero, J.A. Cyclotides, a versatile ultrastable micro-protein scaffold for biotechnological applications. Bioorg. Med. Chem. Lett. 2017, 27, 5089–5099. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, K.J.; Daly, N.L.; Plan, M.R.; Waine, C.; Craik, D.J. Twists, knots, and rings in proteins. Structural definition of the cyclotide framework. J. Biol. Chem. 2003, 278, 8606–8616. [Google Scholar] [CrossRef] [PubMed]
- Felizmenio-Quimio, M.E.; Daly, N.L.; Craik, D.J. Circular proteins in plants: Solution structure of a novel macrocyclic trypsin inhibitor from momordica cochinchinensis. J. Biol. Chem. 2001, 276, 22875–22882. [Google Scholar] [CrossRef] [PubMed]
- Saether, O.; Craik, D.J.; Campbell, I.D.; Sletten, K.; Juul, J.; Norman, D.G. Elucidation of the primary and three-dimensional structure of the uterotonic polypeptide kalata b1. Biochemistry 1995, 34, 4147–4158. [Google Scholar] [CrossRef]
- Li, Y.; Bi, T.; Camarero, J.A. Chemical and biological production of cyclotides. Adv. Bot. Res. 2015, 76, 271–303. [Google Scholar] [PubMed]
- Contreras, J.; Elnagar, A.Y.; Hamm-Alvarez, S.F.; Camarero, J.A. Cellular uptake of cyclotide mcoti-i follows multiple endocytic pathways. J. Control. Release 2011, 155, 134–143. [Google Scholar] [CrossRef]
- Cascales, L.; Henriques, S.T.; Kerr, M.C.; Huang, Y.H.; Sweet, M.J.; Daly, N.L.; Craik, D.J. Identification and characterization of a new family of cell-penetrating peptides: Cyclic cell-penetrating peptides. J. Biol. Chem. 2011, 286, 36932–36943. [Google Scholar] [CrossRef]
- Wong, C.T.; Rowlands, D.K.; Wong, C.H.; Lo, T.W.; Nguyen, G.K.; Li, H.Y.; Tam, J.P. Orally active peptidic bradykinin b1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angew. Chem. Int. Ed. Engl. 2012, 51, 5620–5624. [Google Scholar] [CrossRef]
- Thell, K.; Hellinger, R.; Sahin, E.; Michenthaler, P.; Gold-Binder, M.; Haider, T.; Kuttke, M.; Liutkeviciute, Z.; Goransson, U.; Grundemann, C.; et al. Oral activity of a nature-derived cyclic peptide for the treatment of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 3960–3965. [Google Scholar] [CrossRef] [PubMed]
- Puttamadappa, S.S.; Jagadish, K.; Shekhtman, A.; Camarero, J.A. Backbone dynamics of cyclotide mcoti-i free and complexed with trypsin. Angew. Chem. Int. Ed. Engl. 2010, 49, 7030–7034. [Google Scholar] [CrossRef] [PubMed]
- Colgrave, M.L.; Craik, D.J. Thermal, chemical, and enzymatic stability of the cyclotide kalata b1: The importance of the cyclic cystine knot. Biochemistry 2004, 43, 5965–5975. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.E.; Camarero, J.A. Biological activities of natural and engineered cyclotides, a novel molecular scaffold for peptide-based therapeutics. Curr. Mol. Pharmacol. 2010, 3, 153–163. [Google Scholar] [CrossRef]
- Gran, L. Oxytocic principles of oldenlandia affinis. Lloydia 1973, 36, 174–178. [Google Scholar]
- Gran, L. On the effect of a polypeptide isolated from “kalata-kalata” (oldenlandia affinis dc) on the oestrogen dominated uterus. Acta Pharmacol. Toxicol. 1973, 33, 400–408. [Google Scholar] [CrossRef]
- Weidmann, J.; Craik, D.J. Discovery, structure, function, and applications of cyclotides: Circular proteins from plants. J. Exp. Bot. 2016, 67, 4801–4812. [Google Scholar] [CrossRef]
- Wang, C.K.; Kaas, Q.; Chiche, L.; Craik, D.J. Cybase: A database of cyclic protein sequences and structures, with applications in protein discovery and engineering. Nucleic Acids Res. 2008, 36, D206–D210. [Google Scholar] [CrossRef]
- Aboye, T.L.; Clark, R.J.; Burman, R.; Roig, M.B.; Craik, D.J.; Goransson, U. Interlocking disulfides in circular proteins: Toward efficient oxidative folding of cyclotides. Antioxid. Redox Signal. 2011, 14, 77–86. [Google Scholar] [CrossRef]
- Heitz, A.; Hernandez, J.F.; Gagnon, J.; Hong, T.T.; Pham, T.T.; Nguyen, T.M.; Le-Nguyen, D.; Chiche, L. Solution structure of the squash trypsin inhibitor mcoti-ii. A new family for cyclic knottins. Biochemistry 2001, 40, 7973–7983. [Google Scholar] [CrossRef]
- Mylne, J.S.; Chan, L.Y.; Chanson, A.H.; Daly, N.L.; Schaefer, H.; Bailey, T.L.; Nguyencong, P.; Cascales, L.; Craik, D.J. Cyclic peptides arising by evolutionary parallelism via asparaginyl-endopeptidase-mediated biosynthesis. Plant Cell 2012, 24, 2765–2778. [Google Scholar] [CrossRef]
- Du, J.; Chan, L.Y.; Poth, A.G.; Craik, D.J. Discovery and characterization of cyclic and acyclic trypsin inhibitors from momordica dioica. J. Nat. Prod. 2019, 82, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Quimbar, P.; Malik, U.; Sommerhoff, C.P.; Kaas, Q.; Chan, L.Y.; Huang, Y.H.; Grundhuber, M.; Dunse, K.; Craik, D.J.; Anderson, M.A.; et al. High-affinity cyclic peptide matriptase inhibitors. J. Biol. Chem. 2013, 288, 13885–13896. [Google Scholar] [CrossRef]
- Chiche, L.; Heitz, A.; Gelly, J.C.; Gracy, J.; Chau, P.T.; Ha, P.T.; Hernandez, J.F.; Le-Nguyen, D. Squash inhibitors: From structural motifs to macrocyclic knottins. Curr. Protein Pept. Sci. 2004, 5, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Ravipati, A.S.; Henriques, S.T.; Poth, A.G.; Kaas, Q.; Wang, C.K.; Colgrave, M.L.; Craik, D.J. Lysine-rich cyclotides: A new subclass of circular knotted proteins from violaceae. ACS Chem. Biol. 2015, 10, 2491–2500. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Malik, U. Cyclotide biosynthesis. Curr. Opin. Chem. Biol. 2013, 17, 546–554. [Google Scholar] [CrossRef]
- Jennings, C.; West, J.; Waine, C.; Craik, D.; Anderson, M. Biosynthesis and insecticidal properties of plant cyclotides: The cyclic knotted proteins from oldenlandia affinis. Proc. Natl. Acad. Sci. USA 2001, 98, 10614–10619. [Google Scholar] [CrossRef]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef]
- Saska, I.; Gillon, A.D.; Hatsugai, N.; Dietzgen, R.G.; Hara-Nishimura, I.; Anderson, M.A.; Craik, D.J. An asparaginyl endopeptidase mediates in vivo protein backbone cyclization. J. Biol. Chem. 2007, 282, 29721–29728. [Google Scholar] [CrossRef]
- Poth, A.G.; Mylne, J.S.; Grassl, J.; Lyons, R.E.; Millar, A.H.; Colgrave, M.L.; Craik, D.J. Cyclotides associate with leaf vasculature and are the products of a novel precursor in petunia (solanaceae). J. Biol. Chem. 2012, 287, 27033–27046. [Google Scholar] [CrossRef]
- Nguyen, G.K.; Zhang, S.; Nguyen, N.T.; Nguyen, P.Q.; Chiu, M.S.; Hardjojo, A.; Tam, J.P. Discovery and characterization of novel cyclotides originated from chimeric precursors consisting of albumin-1 chain a and cyclotide domains in the fabaceae family. J. Biol. Chem. 2011, 286, 24275–24287. [Google Scholar] [CrossRef] [PubMed]
- Gillon, A.D.; Saska, I.; Jennings, C.V.; Guarino, R.F.; Craik, D.J.; Anderson, M.A. Biosynthesis of circular proteins in plants. Plant J. 2008, 53, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.K.; Wang, S.; Qiu, Y.; Hemu, X.; Lian, Y.; Tam, J.P. Butelase 1 is an asx-specific ligase enabling peptide macrocyclization and synthesis. Nat. Chem. Biol. 2014, 10, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.S.; Durek, T.; Kaas, Q.; Poth, A.G.; Gilding, E.K.; Conlan, B.F.; Saska, I.; Daly, N.L.; van der Weerden, N.L.; Craik, D.J.; et al. Efficient backbone cyclization of linear peptides by a recombinant asparaginyl endopeptidase. Nat. Commun. 2015, 6, 10199. [Google Scholar] [CrossRef] [PubMed]
- Poon, S.; Harris, K.S.; Jackson, M.A.; McCorkelle, O.C.; Gilding, E.K.; Durek, T.; van der Weerden, N.L.; Craik, D.J.; Anderson, M.A. Co-expression of a cyclizing asparaginyl endopeptidase enables efficient production of cyclic peptides in planta. J. Exp. Bot. 2018, 69, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Bernath-Levin, K.; Nelson, C.; Elliott, A.G.; Jayasena, A.S.; Millar, A.H.; Craik, D.J.; Mylne, J.S. Peptide macrocyclization by a bifunctional endoprotease. Chem. Biol. 2015, 22, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Hemu, X.; Qiu, Y.; Nguyen, G.K.; Tam, J.P. Total synthesis of circular bacteriocins by butelase 1. J. Am. Chem. Soc. 2016, 138, 6968–6971. [Google Scholar] [CrossRef]
- Nguyen, G.K.; Hemu, X.; Quek, J.P.; Tam, J.P. Butelase-mediated macrocyclization of d-amino-acid-containing peptides. Angew. Chem. Int. Ed. Engl. 2016, 55, 12802–12806. [Google Scholar] [CrossRef]
- Nguyen, G.K.; Qiu, Y.; Cao, Y.; Hemu, X.; Liu, C.F.; Tam, J.P. Butelase-mediated cyclization and ligation of peptides and proteins. Nat. Protoc. 2016, 11, 1977–1988. [Google Scholar] [CrossRef]
- Jackson, M.A.; Gilding, E.K.; Shafee, T.; Harris, K.S.; Kaas, Q.; Poon, S.; Yap, K.; Jia, H.; Guarino, R.; Chan, L.Y.; et al. Molecular basis for the production of cyclic peptides by plant asparaginyl endopeptidases. Nat. Commun. 2018, 9, 2411. [Google Scholar] [CrossRef]
- Zauner, F.B.; Elsasser, B.; Dall, E.; Cabrele, C.; Brandstetter, H. Structural analyses of arabidopsis thaliana legumain gamma reveal differential recognition and processing of proteolysis and ligation substrates. J. Biol. Chem. 2018, 293, 8934–8946. [Google Scholar] [CrossRef] [PubMed]
- Aboye, T.; Kuang, Y.; Neamati, N.; Camarero, J.A. Rapid parallel synthesis of bioactive folded cyclotides by using a tea-bag approach. ChemBioChem 2015, 16, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, W.G.; Aboye, T.; Chatterjee, S.; Camarero, J.A.; Nimmagadda, S. In vivo evaluation of an engineered cyclotide as specific cxcr4 imaging reagent. Chemistry 2017, 23, 14469–14475. [Google Scholar] [CrossRef] [PubMed]
- Aboye, T.; Meeks, C.J.; Majumder, S.; Shekhtman, A.; Rodgers, K.; Camarero, J.A. Design of a mcoti-based cyclotide with angiotensin (1-7)-like activity. Molecules 2016, 21, 152. [Google Scholar] [CrossRef] [PubMed]
- Aboye, T.L.; Li, Y.; Majumder, S.; Hao, J.; Shekhtman, A.; Camarero, J.A. Efficient one-pot cyclization/folding of rhesus theta-defensin-1 (rtd-1). Bioorg. Med. Chem. Lett. 2012, 22, 2823–2826. [Google Scholar] [CrossRef]
- Li, Y.; Gould, A.; Aboye, T.; Bi, T.; Breindel, L.; Shekhtman, A.; Camarero, J.A. Full sequence amino acid scanning of theta-defensin rtd-1 yields a potent anthrax lethal factor protease inhibitor. J. Med. Chem. 2017, 60, 1916–1927. [Google Scholar] [CrossRef]
- Yang, R.; Wong, Y.H.; Nguyen, G.K.T.; Tam, J.P.; Lescar, J.; Wu, B. Engineering a catalytically efficient recombinant protein ligase. J. Am. Chem. Soc. 2017, 139, 5351–5358. [Google Scholar] [CrossRef]
- Thongyoo, P.; Roque-Rosell, N.; Leatherbarrow, R.J.; Tate, E.W. Chemical and biomimetic total syntheses of natural and engineered mcoti cyclotides. Org. Biomol. Chem. 2008, 6, 1462–1470. [Google Scholar] [CrossRef]
- Jia, X.; Kwon, S.; Wang, C.I.; Huang, Y.H.; Chan, L.Y.; Tan, C.C.; Rosengren, K.J.; Mulvenna, J.P.; Schroeder, C.I.; Craik, D.J. Semienzymatic cyclization of disulfide-rich peptides using sortase a. J. Biol. Chem. 2014, 289, 6627–6638. [Google Scholar] [CrossRef]
- Aboye, T.L.; Camarero, J.A. Biological synthesis of circular polypeptides. J. Biol. Chem. 2012, 287, 27026–27032. [Google Scholar] [CrossRef]
- Kimura, R.H.; Tran, A.T.; Camarero, J.A. Biosynthesis of the cyclotide kalata b1 by using protein splicing. Angew. Chem. Int. Ed. Engl. 2006, 45, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.; Kimura, R.H.; Woo, Y.H.; Camarero, J.A. In vivo biosynthesis of an ala-scan library based on the cyclic peptide sfti-1. Amino Acids 2010, 38, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Jagadish, K.; Borra, R.; Lacey, V.; Majumder, S.; Shekhtman, A.; Wang, L.; Camarero, J.A. Expression of fluorescent cyclotides using protein trans-splicing for easy monitoring of cyclotide-protein interactions. Angew. Chem. Int. Ed. Engl. 2013, 52, 3126–3131. [Google Scholar] [CrossRef] [PubMed]
- Jagadish, K.; Gould, A.; Borra, R.; Majumder, S.; Mushtaq, Z.; Shekhtman, A.; Camarero, J.A. Recombinant expression and phenotypic screening of a bioactive cyclotide against alpha-synuclein-induced cytotoxicity in baker’s yeast. Angew. Chem. Int. Ed. Engl. 2015, 54, 8390–8394. [Google Scholar] [CrossRef]
- Jagadish, K.; Camarero, J.A. Recombinant expression of cyclotides using split inteins. Methods Mol. Biol. 2017, 1495, 41–55. [Google Scholar]
- Seydel, P.; Dornenburg, H. Establishment of in vitro plants, cell and tissue cultures from oldenlandia affinis for the production of cyclic peptides. Plant Cell Tissue Organ Cult. 2006, 85, 247–255. [Google Scholar] [CrossRef]
- Jennings, C.V.; Rosengren, K.J.; Daly, N.L.; Plan, M.; Stevens, J.; Scanlon, M.J.; Waine, C.; Norman, D.G.; Anderson, M.A.; Craik, D.J. Isolation, solution structure, and insecticidal activity of kalata b2, a circular protein with a twist: Do mobius strips exist in nature? Biochemistry 2005, 44, 851–860. [Google Scholar] [CrossRef]
- Pinto, M.F.; Fensterseifer, I.C.; Migliolo, L.; Sousa, D.A.; de Capdville, G.; Arboleda-Valencia, J.W.; Colgrave, M.L.; Craik, D.J.; Magalhaes, B.S.; Dias, S.C.; et al. Identification and structural characterization of novel cyclotide with activity against an insect pest of sugar cane. J. Biol. Chem. 2012, 287, 134–147. [Google Scholar] [CrossRef]
- Craik, D.J. Host-defense activities of cyclotides. Toxins 2012, 4, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Gilding, E.K.; Jackson, M.A.; Poth, A.G.; Henriques, S.T.; Prentis, P.J.; Mahatmanto, T.; Craik, D.J. Gene coevolution and regulation lock cyclic plant defence peptides to their targets. New Phytol. 2016, 210, 717–730. [Google Scholar] [CrossRef]
- Colgrave, M.L.; Kotze, A.C.; Huang, Y.H.; O’Grady, J.; Simonsen, S.M.; Craik, D.J. Cyclotides: Natural, circular plant peptides that possess significant activity against gastrointestinal nematode parasites of sheep. Biochemistry 2008, 47, 5581–5589. [Google Scholar] [CrossRef]
- Colgrave, M.L.; Kotze, A.C.; Ireland, D.C.; Wang, C.K.; Craik, D.J. The anthelmintic activity of the cyclotides: Natural variants with enhanced activity. ChemBioChem 2008, 9, 1939–1945. [Google Scholar] [CrossRef]
- Malagon, D.; Botterill, B.; Gray, D.J.; Lovas, E.; Duke, M.; Gray, C.; Kopp, S.R.; Knott, L.M.; McManus, D.P.; Daly, N.L.; et al. Anthelminthic activity of the cyclotides (kalata b1 and b2) against schistosome parasites. Biopolymers 2013, 100, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Plan, M.R.; Saska, I.; Cagauan, A.G.; Craik, D.J. Backbone cyclised peptides from plants show molluscicidal activity against the rice pest pomacea canaliculata (golden apple snail). J. Agric. Food Chem. 2008, 56, 5237–5241. [Google Scholar] [CrossRef]
- Parsley, N.C.; Kirkpatrick, C.L.; Crittenden, C.M.; Rad, J.G.; Hoskin, D.W. Pepsavi-ms reveals anticancer and antifungal cycloviolacins in viola odorata. Phytochemistry 2018, 152, 61–70. [Google Scholar] [CrossRef]
- Barbeta, B.L.; Marshall, A.T.; Gillon, A.D.; Craik, D.J.; Anderson, M.A. Plant cyclotides disrupt epithelial cells in the midgut of lepidopteran larvae. Proc. Natl. Acad. Sci. USA 2008, 105, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Troeira Henriques, S.; Huang, Y.H.; Chaousis, S.; Wang, C.K.; Craik, D.J. Anticancer and toxic properties of cyclotides are dependent on phosphatidylethanolamine phospholipid targeting. ChemBioChem 2014, 15, 1956–1965. [Google Scholar] [CrossRef]
- Henriques, S.T.; Craik, D.J. Importance of the cell membrane on the mechanism of action of cyclotides. ACS Chem. Biol. 2012, 7, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.T.; Huang, Y.H.; Castanho, M.A.; Bagatolli, L.A.; Sonza, S.; Tachedjian, G.; Daly, N.L.; Craik, D.J. Phosphatidylethanolamine binding is a conserved feature of cyclotide-membrane interactions. J. Biol. Chem. 2012, 287, 33629–33643. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Colgrave, M.L.; Daly, N.L.; Keleshian, A.; Martinac, B.; Craik, D.J. The biological activity of the prototypic cyclotide kalata b1 is modulated by the formation of multimeric pores. J. Biol. Chem. 2009, 284, 20699–20707. [Google Scholar] [CrossRef]
- Henriques, S.T.; Huang, Y.H.; Rosengren, K.J.; Franquelim, H.G.; Carvalho, F.A.; Johnson, A.; Sonza, S.; Tachedjian, G.; Castanho, M.A.; Daly, N.L.; et al. Decoding the membrane activity of the cyclotide kalata b1: The importance of phosphatidylethanolamine phospholipids and lipid organization on hemolytic and anti-hiv activities. J. Biol. Chem. 2011, 286, 24231–24241. [Google Scholar] [CrossRef]
- Troeira Henriques, S.; Craik, D.J. Cyclotide structure and function: The role of membrane binding and permeation. Biochemistry 2017, 56, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Cranfield, C.G.; Henriques, S.T.; Martinac, B.; Duckworth, P.; Craik, D.J.; Cornell, B. Kalata b1 and kalata b2 have a surfactant-like activity in phosphatidylethanolomine-containing lipid membranes. Langmuir 2017, 33, 6630–6637. [Google Scholar] [CrossRef] [PubMed]
- Stromstedt, A.A.; Park, S.; Burman, R.; Goransson, U. Bactericidal activity of cyclotides where phosphatidylethanolamine-lipid selectivity determines antimicrobial spectra. Biochimica Et Biophysica Acta-Biomembranes 2017, 1859, 1986–2000. [Google Scholar] [CrossRef]
- Nguyen, G.K.; Zhang, S.; Wang, W.; Wong, C.T.; Nguyen, N.T.; Tam, J.P. Discovery of a linear cyclotide from the bracelet subfamily and its disulfide mapping by top-down mass spectrometry. J. Biol. Chem. 2011, 286, 44833–44844. [Google Scholar] [CrossRef]
- Wong, C.T.; Taichi, M.; Nishio, H.; Nishiuchi, Y.; Tam, J.P. Optimal oxidative folding of the novel antimicrobial cyclotide from hedyotis biflora requires high alcohol concentrations. Biochemistry 2011, 50, 7275–7283. [Google Scholar] [CrossRef] [PubMed]
- Pranting, M.; Loov, C.; Burman, R.; Goransson, U.; Andersson, D.I. The cyclotide cycloviolacin o2 from viola odorata has potent bactericidal activity against gram-negative bacteria. J. Antimicrob. Chemother. 2010, 65, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Fensterseifer, I.C.; Silva, O.N.; Malik, U.; Ravipati, A.S.; Novaes, N.R.; Miranda, P.R.; Rodrigues, E.A.; Moreno, S.E.; Craik, D.J.; Franco, O.L. Effects of cyclotides against cutaneous infections caused by staphylococcus aureus. Peptides 2015, 63, 38–42. [Google Scholar] [CrossRef]
- He, W.; Chan, L.Y.; Zeng, G.; Daly, N.L.; Craik, D.J.; Tana, N. Isolation and characterization of cytotoxic cyclotides from viola philippica. Peptides 2011, 32, 1719–1723. [Google Scholar] [CrossRef]
- Lindholm, P.; Goransson, U.; Johansson, S.; Claeson, P.; Gullbo, J.; Larsson, R.; Bohlin, L.; Backlund, A. Cyclotides: A novel type of cytotoxic agents. Mol. Cancer Ther. 2002, 1, 365–369. [Google Scholar]
- Svangard, E.; Goransson, U.; Hocaoglu, Z.; Gullbo, J.; Larsson, R.; Claeson, P.; Bohlin, L. Cytotoxic cyclotides from viola tricolor. J. Nat. Prod. 2004, 67, 144–147. [Google Scholar] [CrossRef]
- Herrmann, A.; Burman, R.; Mylne, J.S.; Karlsson, G.; Gullbo, J.; Craik, D.J.; Clark, R.J.; Goransson, U. The alpine violet, viola biflora, is a rich source of cyclotides with potent cytotoxicity. Phytochemistry 2008, 69, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, M.A.; Abagheri-Mahabadi, N.; Hashempour, H.; Farhadpour, M.; Gruber, C.W.; Ghassempour, A. Viola plant cyclotide vigno 5 induces mitochondria-mediated apoptosis via cytochrome c release and caspases activation in cervical cancer cells. Fitoterapia 2016, 109, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Wang, D.; Chen, J.; Tao, X. Novel cyclotides from hedyotis diffusa induce apoptosis and inhibit proliferation and migration of prostate cancer cells. Int. J. Clin. Exp. Med. 2015, 8, 4059–4065. [Google Scholar] [PubMed]
- Zhang, H.; Song, T.; Yang, Y.; Fu, C.; Li, J. Exploring the interaction mechanism between cyclopeptide dc3 and androgen receptor using molecular dynamics simulations and free energy calculations. Front. Chem. 2018, 6, 119. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.E.; Najas, J.Z.; Magalhães, L.G.; Bobey, A.F.; Mendonça, J.N.; Lopes, N.P.; Leme, F.M.; Teixeira, S.P.; Trovó, M.; Andricopulo, A.D.; et al. Inhibition of breast cancer cell migration by cyclotides isolated from pombalia calceolaria. J. Nat. Prod. 2018, 81, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Koehbach, J.; O’Brien, M.; Muttenthaler, M.; Miazzo, M.; Akcan, M.; Elliott, A.G.; Daly, N.L.; Harvey, P.J.; Arrowsmith, S.; Gunasekera, S.; et al. Oxytocic plant cyclotides as templates for peptide g protein-coupled receptor ligand design. Proc. Natl. Acad. Sci. USA 2013, 110, 21183–21188. [Google Scholar] [CrossRef]
- Keov, P.; Liutkeviciute, Z.; Hellinger, R.; Clark, R.J.; Gruber, C.W. Discovery of peptide probes to modulate oxytocin-type receptors of insects. Sci. Rep. 2018, 8, 10020. [Google Scholar] [CrossRef]
- Stromstedt, A.A.; Kristiansen, P.E.; Gunasekera, S.; Grob, N.; Skjeldal, L.; Goransson, U. Selective membrane disruption by the cyclotide kalata b7: Complex ions and essential functional groups in the phosphatidylethanolamine binding pocket. Biochim. Biophys. Acta 2016, 1858, 1317–1327. [Google Scholar] [CrossRef]
- Fahradpour, M.; Keov, P.; Tognola, C.; Perez-Santamarina, E.; McCormick, P.J.; Ghassempour, A.; Gruber, C.W. Cyclotides isolated from an ipecac root extract antagonize the corticotropin releasing factor type 1 receptor. Front. Pharmacol. 2017, 8, 616. [Google Scholar] [CrossRef]
- Hellinger, R.; Koehbach, J.; Puigpinos, A.; Clark, R.J.; Tarrago, T.; Giralt, E.; Gruber, C.W. Inhibition of human prolyl oligopeptidase activity by the cyclotide psysol 2 isolated from psychotria solitudinum. J. Nat. Prod. 2015, 78, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Nworu, C.S.; Ejikeme, T.I.; Ezike, A.C.; Ndu, O.; Akunne, T.C.; Onyeto, C.A.; Okpalanduka, P.; Akah, P.A. Anti-plasmodial and anti-inflammatory activities of cyclotide-rich extract and fraction of oldenlandia affinis (R. & S.) D.C. (rubiaceae). Afr. Health Sci. 2017, 17, 827–843. [Google Scholar]
- Gunasekera, S.; Foley, F.M.; Clark, R.J.; Sando, L.; Fabri, L.J.; Craik, D.J.; Daly, N.L. Engineering stabilized vascular endothelial growth factor-a antagonists: Synthesis, structural characterization, and bioactivity of grafted analogues of cyclotides. J. Med. Chem. 2008, 51, 7697–7704. [Google Scholar] [CrossRef] [PubMed]
- Thongyoo, P.; Bonomelli, C.; Leatherbarrow, R.J.; Tate, E.W. Potent inhibitors of beta-tryptase and human leukocyte elastase based on the mcoti-ii scaffold. J. Med. Chem. 2009, 52, 6197–6200. [Google Scholar] [CrossRef]
- Eliasen, R.; Daly, N.L.; Wulff, B.S.; Andresen, T.L.; Conde-Frieboes, K.W.; Craik, D.J. Design, synthesis, structural and functional characterization of novel melanocortin agonists based on the cyclotide kalata b1. J. Biol. Chem. 2012, 287, 40493–40501. [Google Scholar] [CrossRef] [PubMed]
- Aboye, T.L.; Ha, H.; Majumder, S.; Christ, F.; Debyser, Z.; Shekhtman, A.; Neamati, N.; Camarero, J.A. Design of a novel cyclotide-based cxcr4 antagonist with anti-human immunodeficiency virus (hiv)-1 activity. J. Med. Chem. 2012, 55, 10729–10734. [Google Scholar] [CrossRef]
- Balkwill, F. The significance of cancer cell expression of the chemokine receptor cxcr4. Semin. Cancer Biol. 2004, 14, 171–179. [Google Scholar] [CrossRef]
- Culp, E.; Wright, G.D. Bacterial proteases, untapped antimicrobial drug targets. J. Antibiot. 2017, 70, 366–377. [Google Scholar] [CrossRef]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef]
- Sommerhoff, C.P.; Avrutina, O.; Schmoldt, H.U.; Gabrijelcic-Geiger, D.; Diederichsen, U.; Kolmar, H. Engineered cystine knot miniproteins as potent inhibitors of human mast cell tryptase beta. J. Mol. Biol. 2010, 395, 167–175. [Google Scholar] [CrossRef]
- Swedberg, J.E.; Ghani, H.A.; Harris, J.M.; de Veer, S.J.; Craik, D.J. Potent, selective, and cell-penetrating inhibitors of kallikrein-related peptidase 4 based on the cyclic peptide mcoti-ii. ACS Med. Chem. Lett. 2018, 9, 1258–1262. [Google Scholar] [CrossRef]
- Huang, Y.H.; Henriques, S.T.; Wang, C.K.; Thorstholm, L.; Daly, N.L.; Kaas, Q.; Craik, D.J. Design of substrate-based bcr-abl kinase inhibitors using the cyclotide scaffold. Sci. Rep. 2015, 5, 12974. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Zeuner, K.E.; Schaffer, E.; Hopfner, F.; Bruggemann, N.; Berg, D. Progress of pharmacological approaches in parkinson’s disease. Clin. Pharmacol. Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, C.; Henriques, S.T.; Wang, C.K.; Cheneval, O.; Chan, L.Y.; Bokil, N.J.; Sweet, M.J.; Craik, D.J. Using the mcoti-ii cyclotide scaffold to design a stable cyclic peptide antagonist of set, a protein overexpressed in human cancer. Biochemistry 2016, 55, 396–405. [Google Scholar] [CrossRef]
- Thell, K.; Hellinger, R.; Schabbauer, G.; Gruber, C.W. Immunosuppressive peptides and their therapeutic applications. Drug Discov. Today 2014, 19, 645–653. [Google Scholar] [CrossRef]
- Hellinger, R.; Thell, K.; Vasileva, M.; Muhammad, T.; Gunasekera, S.; Kummel, D.; Goransson, U.; Becker, C.W.; Gruber, C.W. Chemical proteomics for target discovery of head-to-tail cyclized mini-proteins. Front. Chem. 2017, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.; Wang, W.; Puttamadappa, S.; Shekhtman, A.; Camarero, J.A. Biosynthesis and biological screening of a genetically encoded library based on the cyclotide mcoti-i. ChemBioChem 2009, 10, 2663–2670. [Google Scholar] [CrossRef]
- Getz, J.A.; Cheneval, O.; Craik, D.J.; Daugherty, P.S. Design of a cyclotide antagonist of neuropilin-1 and -2 that potently inhibits endothelial cell migration. ACS Chem. Biol. 2013, 8, 1147–1154. [Google Scholar] [CrossRef]
- Maass, F.; Wustehube-Lausch, J.; Dickgiesser, S.; Valldorf, B.; Reinwarth, M.; Schmoldt, H.U.; Daneschdar, M.; Avrutina, O.; Sahin, U.; Kolmar, H. Cystine-knot peptides targeting cancer-relevant human cytotoxic t lymphocyte-associated antigen 4 (ctla-4). J. Pept. Sci. 2015, 21, 651–660. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Wang, C.K.; Stalmans, S.; De Spiegeleer, B.; Craik, D.J. Biodistribution of the cyclotide mcoti-ii, a cyclic disulfide-rich peptide drug scaffold. J. Pept. Sci. 2016, 22, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Melander, E.; Eriksson, C.; Jansson, B.; Goransson, U.; Hammarlund-Udenaes, M. Improved method for quantitative analysis of the cyclotide kalata b1 in plasma and brain homogenate. Biopolymers 2016, 106, 910–916. [Google Scholar] [CrossRef]
- Gao, Y.; Cui, T.; Lam, Y. Synthesis and disulfide bond connectivity-activity studies of a kalata b1-inspired cyclopeptide against dengue ns2b-ns3 protease. Bioorg. Med. Chem. 2010, 18, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.; Gruber, C.W.; Cemazar, M.; Siatskas, C.; Tagore, P.; Payne, N.; Sun, G.; Wang, S.; Bernard, C.C.; Craik, D.J. Molecular grafting onto a stable framework yields novel cyclic peptides for the treatment of multiple sclerosis. ACS Chem. Biol. 2014, 9, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.Y.; Gunasekera, S.; Henriques, S.T.; Worth, N.F.; Le, S.J.; Clark, R.J.; Campbell, J.H.; Craik, D.J.; Daly, N.L. Engineering pro-angiogenic peptides using stable, disulfide-rich cyclic scaffolds. Blood 2011, 118, 6709–6717. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, J.E.; Mahatmanto, T.; Abdul Ghani, H.; de Veer, S.J.; Schroeder, C.I.; Harris, J.M.; Craik, D.J. Substrate-guided design of selective fxiia inhibitors based on the plant-derived momordica cochinchinensis trypsin inhibitor-ii (mcoti-ii) scaffold. J. Med. Chem. 2016, 59, 7287–7292. [Google Scholar] [CrossRef]
- Chan, L.Y.; Craik, D.J.; Daly, N.L. Cyclic thrombospondin-1 mimetics: Grafting of a thrombospondin sequence into circular disulfide-rich frameworks to inhibit endothelial cell migration. Biosci. Rep. 2015, 35, e00270. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.Y.; Craik, D.J.; Daly, N.L. Dual-targeting anti-angiogenic cyclic peptides as potential drug leads for cancer therapy. Sci. Rep. 2016, 6, 35347. [Google Scholar] [CrossRef]
- Slazak, B.; Kapusta, M.; Malik, S.; Bohdanowicz, J.; Kuta, E.; Malec, P.; Goransson, U. Immunolocalization of cyclotides in plant cells, tissues and organ supports their role in host defense. Planta 2016, 244, 1029–1040. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cyclotide | Biological Activity | Loop Modified | Application | Ref. |
|---|---|---|---|---|
| Möbius Subfamily | ||||
| Kalata B1 | VEGF-A antagonist | 2, 3, 5, and 6 | Anti-angiogenic, potential anti-cancer activity | [113] |
| Kalata B1 | Dengue NS2B-NS3 Protease inhibitor | 2 and 5 | Anti-viral for Dengue virus infections | [134] |
| Kalata B1 | Bradikynin B1 receptor antagonist | 6 | Chronic and inflammatory pain | [30] |
| Kalata B1 | Melanocortin 4 receptor Agonist | 6 | Obesity | [115] |
| Kalata B1 | Neuropilin-1/2 antagonist | 5 and 6 | Inhibition of endothelial cell migration and angiogenesis | [129] |
| Kalata B1 | Immunomodulator | 5 and 6 | Protecting against multiple sclerosis | [135] |
| Kalata B1 | Immunomodulator | 4 | Protecting against multiple sclerosis | [31] |
| Trypsin Inhibitor Subfamily | ||||
| MCoTI-I | CXCR4 antagonist | 6 | Anti-metastatic and anti-HIV PET-CT imaging | [62,63,116] |
| MCoTI-I | p53-Hdm2/HdmX | 6 | Anti-tumor by activation of p53 pathway | [5] |
| MCoTI-II | FMDV 3C protease Inhibitor | 1 | Anti-viral for foot-and-mouth disease | [68] |
| MCoTI-II | β-Tryptase inhibitor | 3, 5, and 6 | Inflammation diseases | [120] |
| MCoTI-II | β-Tryptase inhibitor Human elastase inhibitor | 1 | Inflammation diseases | [114] |
| MCoTI-II | CTLA-4 antagonist | 1,3, and 6 | Immunotherapy for cancer | [130] |
| MCoTI-II | Tryptase inhibitor | 1 | Anti-cancer | [43] |
| MCoTI-II | VEGF receptor agonist | 6 | Wound healing and cardiovascular damage | [136] |
| MCoTI-I | α-Synuclein-induced cytotoxicity inhibitor | 6 | Parkinson’s disease Validate phenotypic screening of genetically-encoded cyclotide libraries | [74] |
| MCoTI-II | BCR-Abl kinase Inhibitor | 1 and 6 | Chronic myeloid leukemia Attempt to graft both a cell penetrating peptide and kinase inhibitor | [122] |
| MCoTI-I | MAS1 receptor agonist | 6 | Lung cancer and myocardial infarction | [64] |
| MCoTI-II | SET antagonist | 6 | Potential anticancer | [125] |
| MCoTI-II | FXIIa and FXa inhibitors | 1 and 6 | Antithrombotic and cardiovascular disease | [137] |
| MCoTI-II | Thrombospondin-1 (TSP-1) agonist | 6 | Microvascular endothelial cell migration inhibition Anti-angiogenesis | [138] |
| MCoTI-II | Antiangiogenic | 5 and 6 | Anti-cancer | [139] |
| MCoTI-II | Kallikrein 4 (KLK4) inhibitor | 1 and 8 | Anti-cancer | [121] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camarero, J.A.; Campbell, M.J. The Potential of the Cyclotide Scaffold for Drug Development. Biomedicines 2019, 7, 31. https://doi.org/10.3390/biomedicines7020031
Camarero JA, Campbell MJ. The Potential of the Cyclotide Scaffold for Drug Development. Biomedicines. 2019; 7(2):31. https://doi.org/10.3390/biomedicines7020031
Chicago/Turabian StyleCamarero, Julio A., and Maria Jose Campbell. 2019. "The Potential of the Cyclotide Scaffold for Drug Development" Biomedicines 7, no. 2: 31. https://doi.org/10.3390/biomedicines7020031
APA StyleCamarero, J. A., & Campbell, M. J. (2019). The Potential of the Cyclotide Scaffold for Drug Development. Biomedicines, 7(2), 31. https://doi.org/10.3390/biomedicines7020031