Temozolomide in Glioblastoma Therapy: Role of Apoptosis, Senescence and Autophagy. Comment on Strobel et al., Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69


{kind=link}
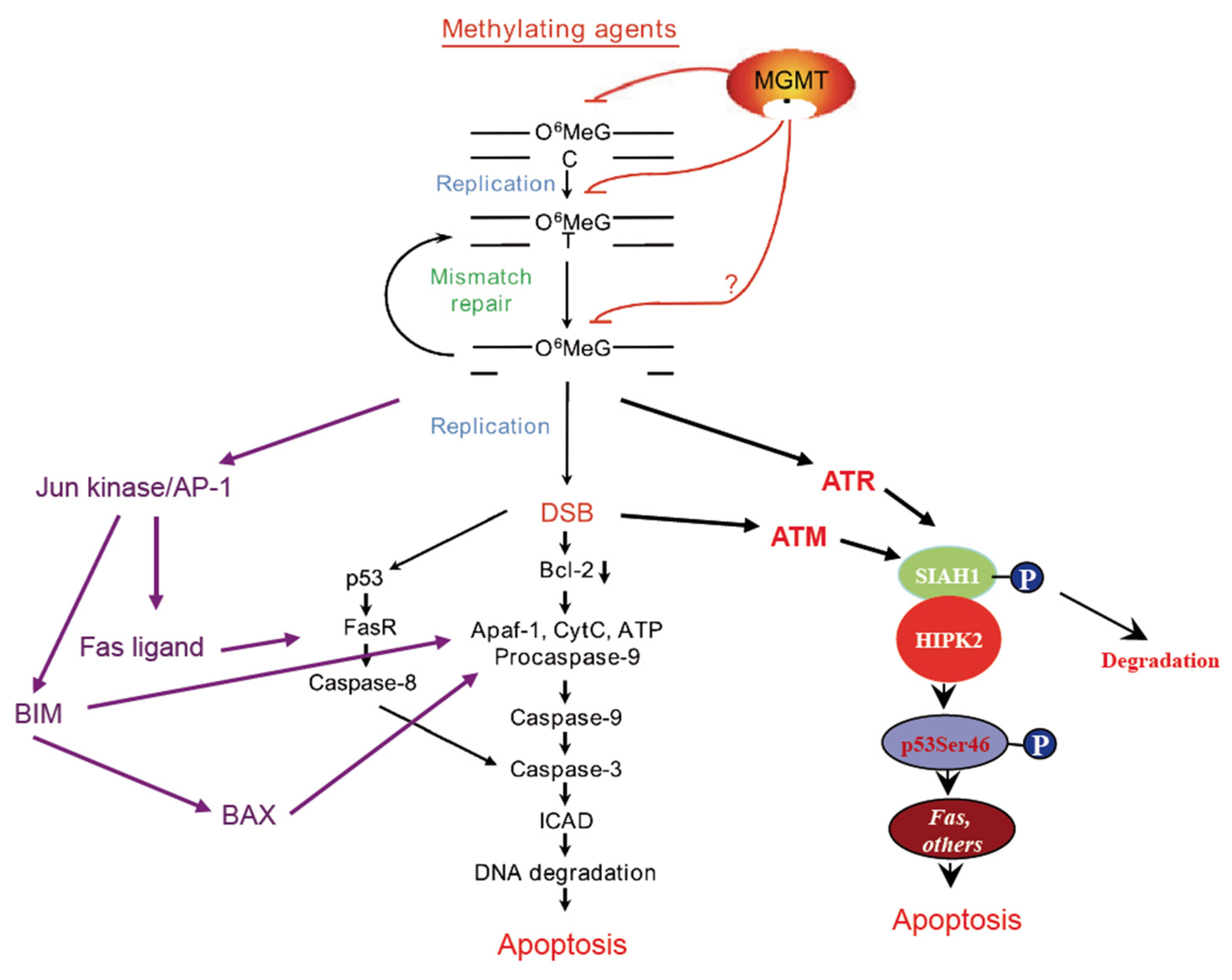
Abstract
:Funding
Conflicts of Interest
References
- Strobel, H.; Baisch, T.; Fitzel, R.; Schilberg, K.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.-M.; Westhoff, M.-A. Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Hogan, S.A.; Levesque, M.P.; Cheng, P.F. Melanoma Immunotherapy: Next-Generation Biomarkers. Front. Oncol. 2018, 8, 178. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair 2019, 78, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Ziouta, A.; Ochs, K.; Coquerelle, T. Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex−, Mex+ and methylation-tolerant mismatch repair compromised cells: Facts and models. Mutat. Res. 1997, 381, 227–241. [Google Scholar] [CrossRef]
- Meikrantz, W. O6-alkylguanine DNA lesions trigger apoptosis. Carcinogenesis 1998, 19, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Ochs, K.; Kaina, B. Apoptosis induced by DNA damage O6-methylguanine is Bcl-2 and caspase-9/3 regulated and Fas/caspase-8 independent. Cancer Res. 2000, 60, 5815–5824. [Google Scholar] [PubMed]
- Quiros, S.; Roos, W.P.; Kaina, B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle 2010, 9, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Eich, M.; Roos, W.; Nikolova, T.; Kaina, B. Contribution of ATM and ATR to the Resistance of Glioblastoma and Malignant Melanoma Cells to the Methylating Anticancer Drug Temozolomide. Mol. Cancer Ther. 2013, 12, 2529–2540. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Batista, L.F.Z.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.M.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quirós, S.; Tomaszowski, K.-H.; Christmann, M.; Kaina, B. Survival and Death Strategies in Glioma Cells: Autophagy, Senescence and Apoptosis Triggered by a Single Type of Temozolomide-Induced DNA Damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef] [PubMed]
- Aasland, D.; Götzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Marzolini, C.; Shen, F.; Bauer, J.; Buclin, T.; Decosterd, L.A.; Lejeune, F.; Gander, M.; Leyvraz, S. Pharmacokinetics of temozolomide in association with fotemustine in malignant melanoma and malignant glioma patients: Comparison of oral, intravenous, and hepatic intra-arterial administration. Cancer Chemother. Pharmacol. 1998, 42, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Portnow, J.; Badie, B.; Chen, M.; Liu, A.; Blanchard, S.; Synold, T.W. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: Potential implications for the current approach to chemoradiation. Clin. Cancer Res. 2009, 15, 7092–7098. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and Cerebrospinal Fluid Population Pharmacokinetics of Temozolomide in Malignant Glioma Patients. Clin. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Kaina, B. Are There Thresholds in Glioblastoma Cell Death Responses Triggered by Temozolomide? Int. J. Mol. Sci. 2019, 20, 1562. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Roos, W.P.; Wu, Q.; Hofmann, T.G.; Kaina, B. The SIAH1–HIPK2–p53ser46 Damage Response Pathway is Involved in Temozolomide-Induced Glioblastoma Cell Death. Mol. Cancer Res. 2019, 17, 1129–1141. [Google Scholar] [CrossRef] [PubMed]
- Matt, S.; Hofmann, T.G. The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cell. Mol. Life Sci. 2016, 73, 2829–2850. [Google Scholar] [CrossRef] [PubMed]
- Tomicic, M.T.; Meise, R.; Aasland, D.; Berte, N.; Kitzinger, R.; Krämer, O.H.; Kaina, B.; Christmann, M. Apoptosis induced by temozolomide and nimustine in glioblastoma cells is supported by JNK/c-Jun-mediated induction of the BH3-only protein BIM. Oncotarget 2015, 6, 33755–33768. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef] [PubMed]
- Lips, J.; Kaina, B. DNA double-strand breaks trigger apoptosis in p53-deficient fibroblasts. Carcinogenesis 2001, 22, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Bent, M.J.V.D. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Buhk, J.-H.; Wrede, A.; Hoffmann, A.L.; Bock, H.C.; Kaina, B.; Strik, H.M.; Christmann, M. Rechallenge with temozolomide with different scheduling is effective in recurrent malignant gliomas. Mol. Med. Rep. 2008, 1, 863–867. [Google Scholar]
- Samson, L.; Cairns, J. A new pathway for DNA repair in Escherichia coli. Nat. 1977, 267, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Bernd, K.; Fritz, G.; Mitra, S.; Coquerelle, T. Transfection and expression of human O6-methylguanine-DNA methyltransferase (MGMT) cDNA in Chinese hamster cells: The role of MGMT in protection against the genotoxic effects of alkylating agents. Carcinogenesis 1991, 12, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christmann, M. DNA repair in resistance to alkylating anticancer drugs. Int. J. Clin. Pharmacol. Ther. 2002, 40, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaina, B. Temozolomide in Glioblastoma Therapy: Role of Apoptosis, Senescence and Autophagy. Comment on Strobel et al., Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. Biomedicines 2019, 7, 90. https://doi.org/10.3390/biomedicines7040090
Kaina B. Temozolomide in Glioblastoma Therapy: Role of Apoptosis, Senescence and Autophagy. Comment on Strobel et al., Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. Biomedicines. 2019; 7(4):90. https://doi.org/10.3390/biomedicines7040090
Chicago/Turabian StyleKaina, Bernd. 2019. "Temozolomide in Glioblastoma Therapy: Role of Apoptosis, Senescence and Autophagy. Comment on Strobel et al., Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69" Biomedicines 7, no. 4: 90. https://doi.org/10.3390/biomedicines7040090
APA StyleKaina, B. (2019). Temozolomide in Glioblastoma Therapy: Role of Apoptosis, Senescence and Autophagy. Comment on Strobel et al., Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. Biomedicines, 7(4), 90. https://doi.org/10.3390/biomedicines7040090