17β-Estradiol Promotes Proinflammatory and Procoagulatory Phenotype of Innate Immune Cells in the Presence of Antiphospholipid Antibodies


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Isolation of Peripheral Blood Mononuclear Cells (PBMCs) and Culturing
2.3. Flow Cytometry Analysis
2.4. Cytokine Production
2.5. Statistical Analysis
3. Results
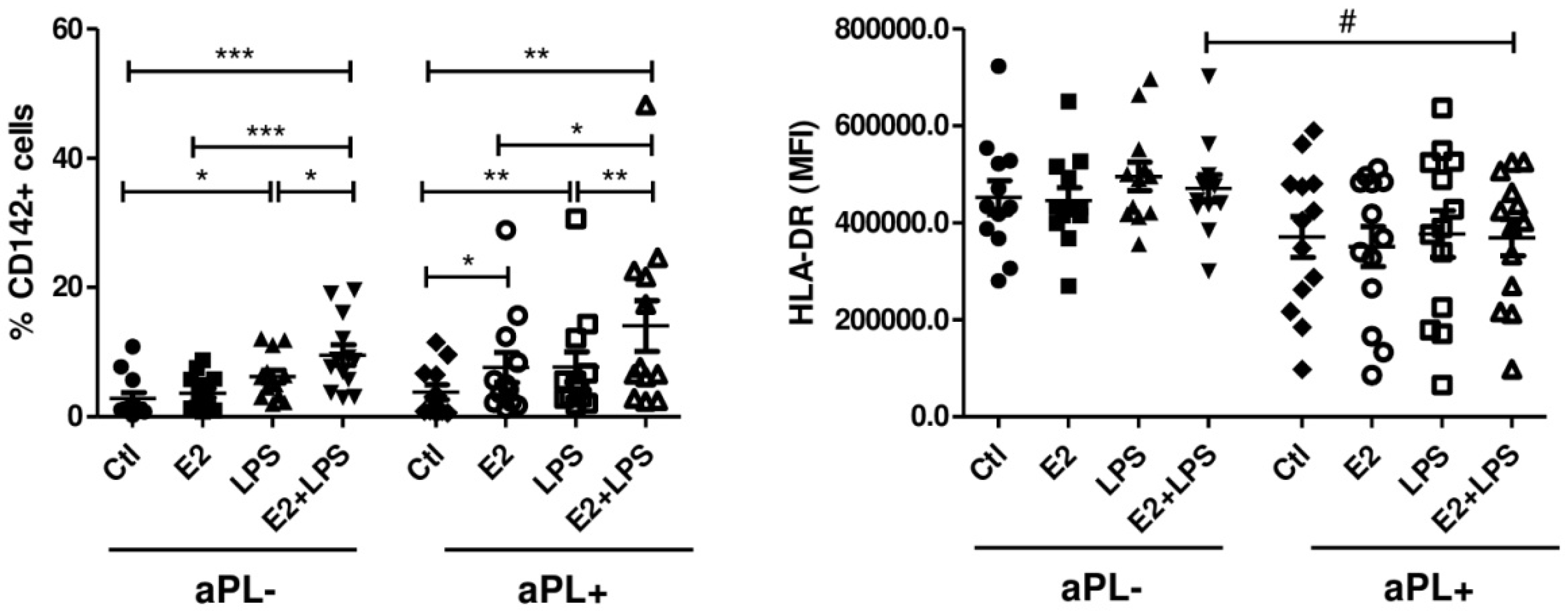
3.1. E2 Increases the Procoagulant Activity of Monocytes Isolated from aPL+ Patients
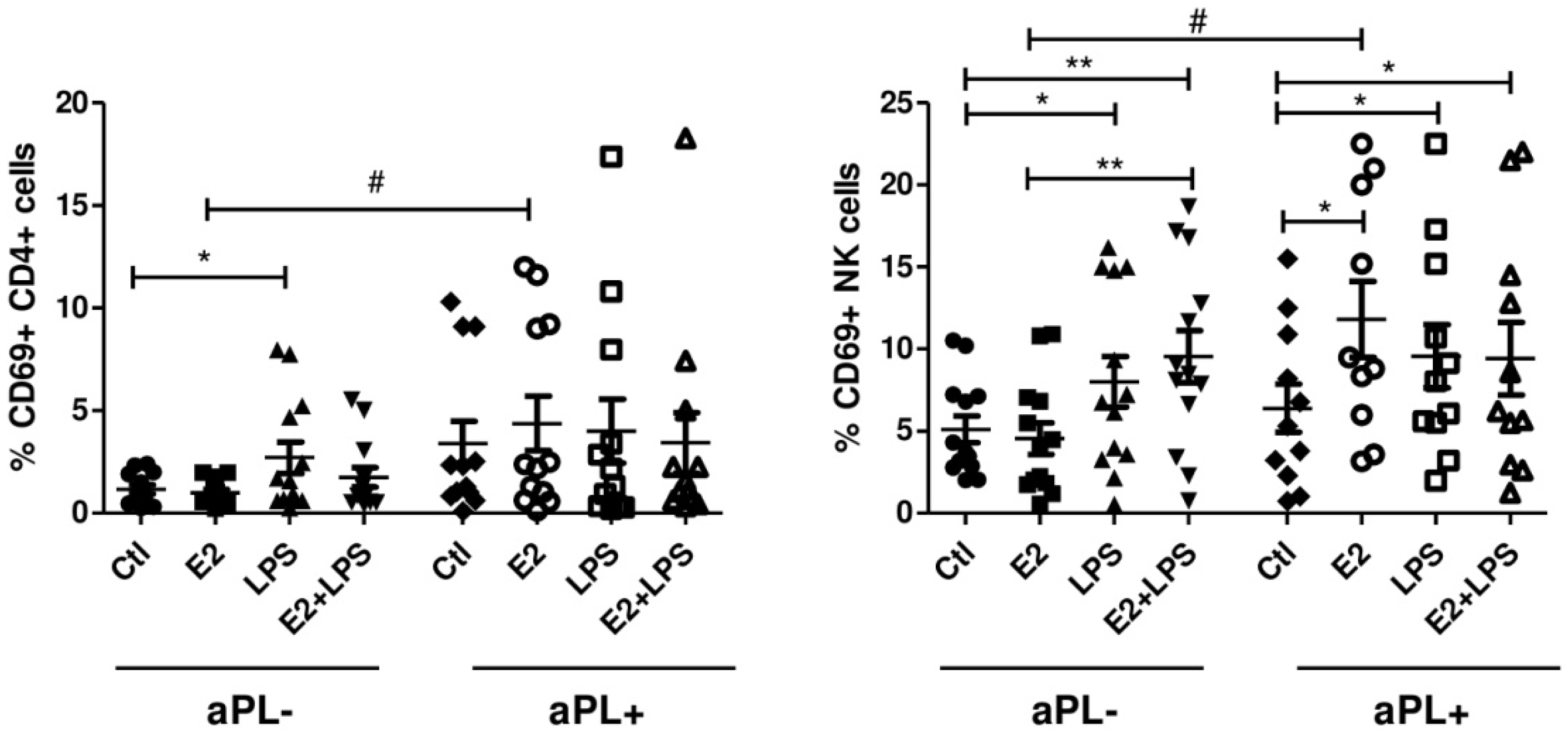
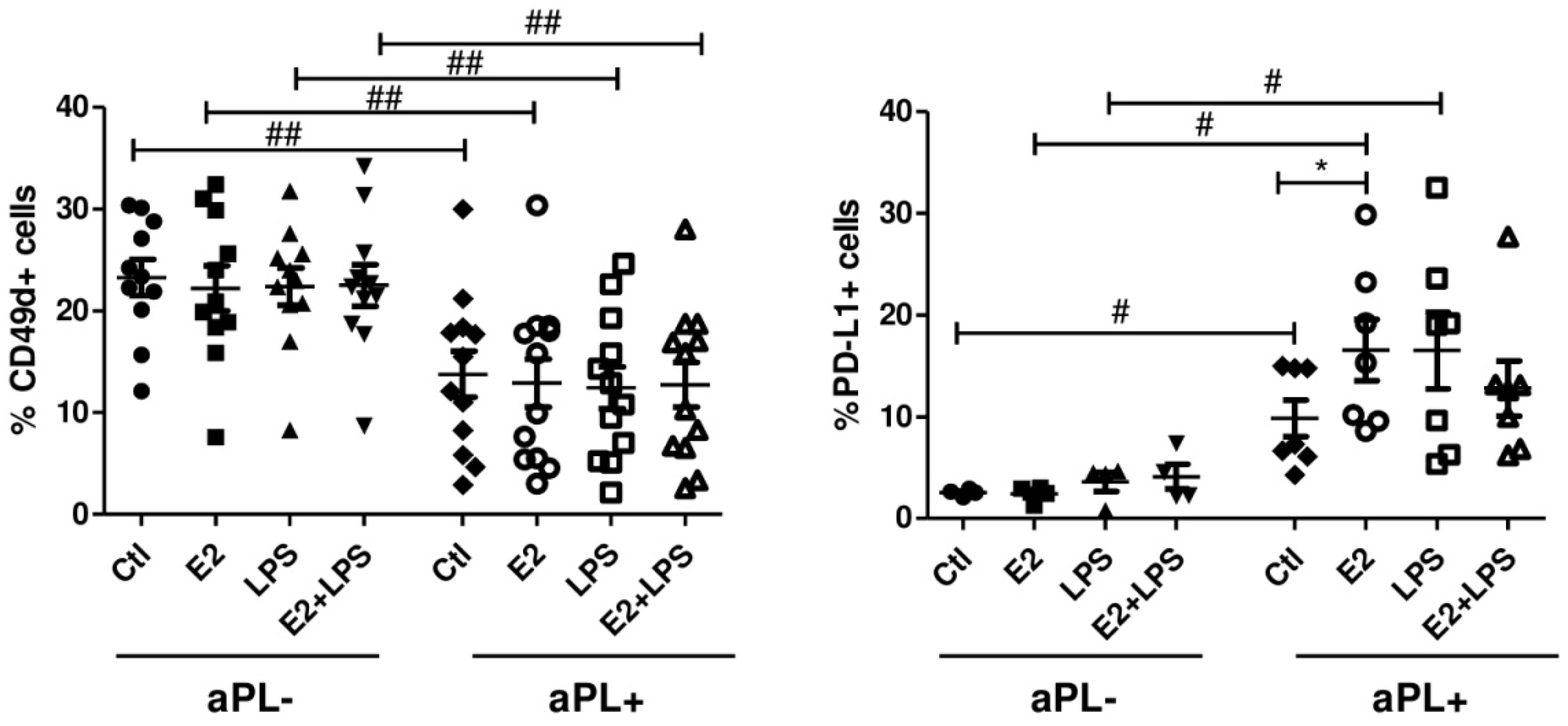
3.2. E2 Activates CD4+ Cells and NK Cells in aPL+ Group
3.3. Increased Percentage of PD-L1+ B Cells in Response to E2
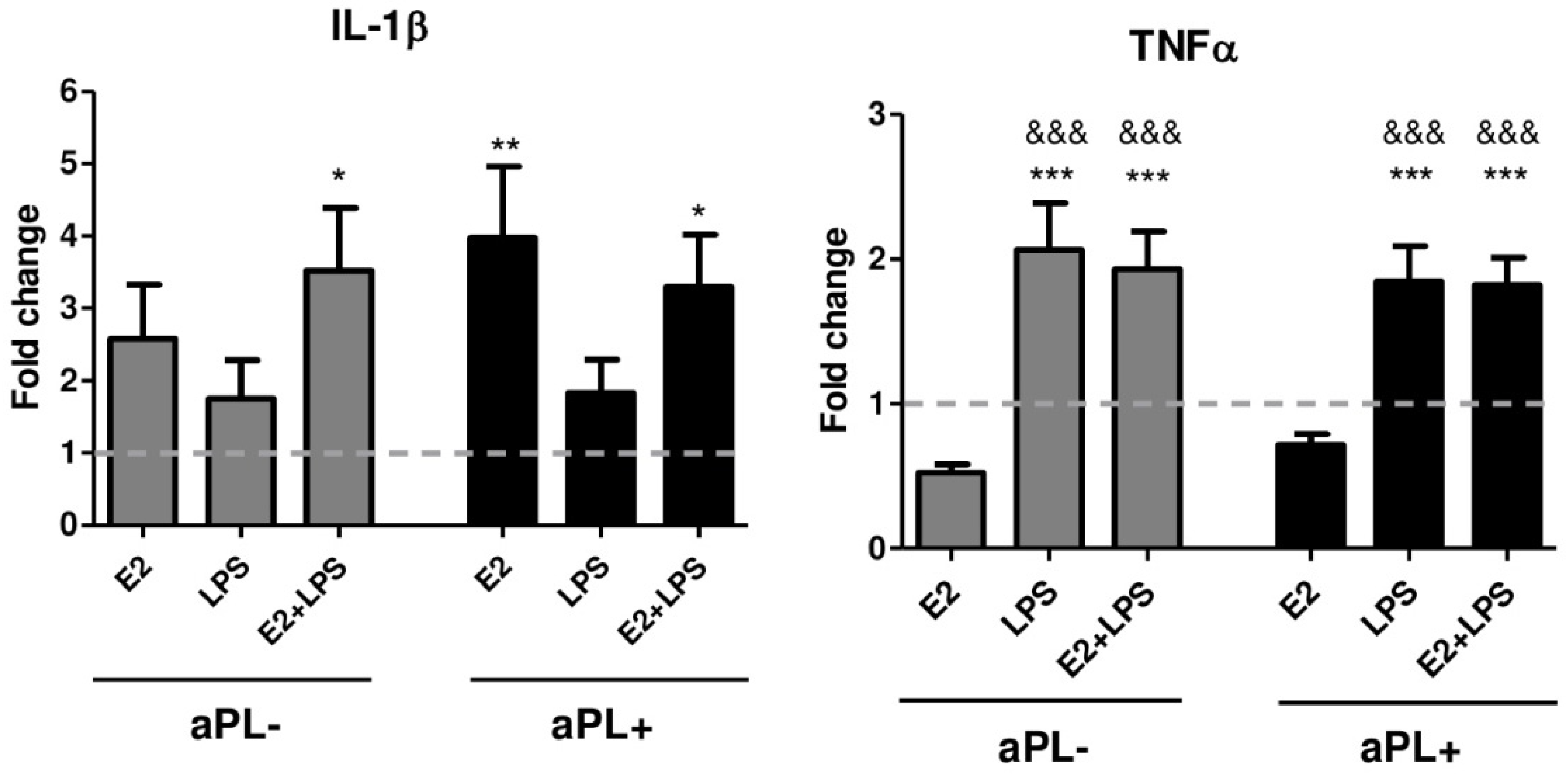
3.4. E2 Induces Increased Production of IL-1β by PBMCs from Patients with aPL Positivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schreiber, K.; Sciascia, S.; de Groot, P.G.; Devreese, K.; Jacobsen, S.; Ruiz-Irastorza, G.; Salmon, J.E.; Shoenfeld, Y.; Shovman, O.; Hunt, B.J. Antiphospholipid syndrome. Nat. Rev. Dis. Primers 2018, 4, 17103. [Google Scholar] [CrossRef]
- Martirosyan, A.; Aminov, R.; Manukyan, G. Environmental Triggers of Autoreactive Responses: Induction of Antiphospholipid Antibody Formation. Front. Immunol. 2019, 10, 1609. [Google Scholar] [CrossRef] [PubMed]
- Cervera, R. Antiphospholipid syndrome. Thromb. Res. 2017, 151, S43–S47. [Google Scholar] [CrossRef]
- Meroni, P.L.; Borghi, M.O.; Raschi, E.; Tedesco, F. Pathogenesis of antiphospholipid syndrome: Understanding the antibodies. Nat. Rev. Rheumatol. 2011, 7, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Manukyan, G.; Martirosyan, A.; Slavik, L.; Margaryan, S.; Ulehlova, J.; Mikulkova, Z.; Hlusi, A.; Papajik, T.; Kriegova, E. Anti-domain 1 β2 glycoprotein antibodies increase expression of tissue factor on monocytes and activate NK Cells and CD8 + cells in vitro. Auto. Highlights 2020, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Cervera, R.; Serrano, R.; Pons-Estel, G.J.; Ceberio-Hualde, L.; Shoenfeld, Y.; de Ramón, E.; Buonaiuto, V.; Jacobsen, S.; Zeher, M.M.; Tarr, T.; et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: A multicentre prospective study of 1000 patients. Ann. Rheum. Dis. 2015, 74, 1011–1018. [Google Scholar] [CrossRef]
- Levine, J.S.; Branch, D.W.; Rauch, J. The antiphospholipid syndrome. N. Engl. J. Med. 2002, 346, 752–763. [Google Scholar] [CrossRef] [Green Version]
- Gerardi, M.C.; Fernandes, M.A.; Tincani, A.; Andreoli, L. Obstetric Anti-phospholipid Syndrome: State of the Art. Curr. Rheumatol. Rep. 2018, 20, 59. [Google Scholar] [CrossRef]
- Dragin, N.; Bismuth, J.; Cizeron-Clairac, G.; Biferi, M.G.; Berthault, C.; Serraf, A.; Nottin, R.; Klatzmann, D.; Cumano, A.; Barkats, M.; et al. Estrogen-mediated downregulation of AIRE influences sexual dimorphism in autoimmune diseases. J. Clin. Invest. 2016, 126, 1525–1537. [Google Scholar] [CrossRef] [Green Version]
- Moulton, V.R. Sex Hormones in Acquired Immunity and Autoimmune Disease. Front. Immunol. 2018, 9, 2279. [Google Scholar] [CrossRef]
- Tchaikovski, S.N.; Rosing, J. Mechanisms of estrogen-induced venous thromboembolism. Thromb. Res. 2010, 126, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, Y.; Coupland, C.; Hippisley-Cox, J. Use of hormone replacement therapy and risk of venous thromboembolism: Nested case-control studies using the QResearch and CPRD databases. BMJ 2019, 364, k4810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napso, T.; Yong, H.E.J.; Lopez-Tello, J.; Sferruzzi-Perri, A.N. The Role of Placental Hormones in Mediating Maternal Adaptations to Support Pregnancy and Lactation. Front. Physiol. 2018, 9, 1091. [Google Scholar] [CrossRef] [PubMed]
- James, A.H. Venous thromboembolism in pregnancy. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 326–331. [Google Scholar] [CrossRef]
- Webber, L.; Anderson, R.A.; Davies, M.; Janse, F.; Vermeulen, N. HRT for women with premature ovarian insufficiency: A comprehensive review. Hum. Reprod. Open 2017, 2017, hox007. [Google Scholar] [CrossRef] [Green Version]
- Delgado, B.J.; Lopez-Ojeda, W. Estrogen. StatPearls. Treasure Island (FL): StatPearls Publishing. Available online: https://www.ncbi.nlm.nih.gov/books/NBK538260/ (accessed on 12 March 2019).
- Khan, D.; Ansar Ahmed, S. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front. Immunol. 2016, 6, 635. [Google Scholar] [CrossRef] [Green Version]
- Karpuzoglu, E.; Phillips, R.A.; Dai, R.; Graniello, C.; Gogal, R.M., Jr.; Ahmed, S.A. Signal transducer and activation of transcription (STAT) 4beta, a shorter isoform of interleukin-12-induced STAT4, is preferentially activated by estrogen. Endocrinology 2009, 150, 1310–1320. [Google Scholar] [CrossRef]
- Lelu, K.; Laffont, S.; Delpy, L.; Paulet, P.E.; Périnat, T.; Tschanz, S.A.; Pelletier, L.; Engelhardt, B.; Guéry, J.C. Estrogen receptor alpha signaling in T lymphocytes is required for estradiol-mediated inhibition of Th1 and Th17 cell differentiation and protection against experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 2386–2393. [Google Scholar] [CrossRef] [Green Version]
- Grimaldi, C.M.; Jeganathan, V.; Diamond, B. Hormonal regulation of B cell development: 17 beta-estradiol impairs negative selection of high-affinity DNA-reactive B cells at more than one developmental checkpoint. J. Immunol. 2006, 176, 2703–2710. [Google Scholar] [CrossRef]
- Liu, H.B.; Loo, K.K.; Palaszynski, K.; Ashouri, J.; Lubahn, D.B.; Voskuhl, R.R. Estrogen receptor alpha mediates estrogen’s immune protection in autoimmune disease. J. Immunol. 2003, 171, 6936–6940. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.M.; McMullen, P.D.; Andersen, M.E.; Clewell, R.A. Multiple receptors shape the estrogen response pathway and are critical considerations for the future of in vitro-based risk assessment efforts. Crit. Rev. Toxicol. 2017, 47, 564–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, S.E. Estrogen and autoimmune disease. Clin. Rev. Allergy Immunol. 2011, 40, 60–65. [Google Scholar] [CrossRef] [PubMed]
- López-Pedrera, C.; Buendía, P.; Cuadrado, M.J.; Siendones, E.; Aguirre, M.A.; Barbarroja, N.; Montiel-Duarte, C.; Torres, A.; Khamashta, M.; Velasco, F. Antiphospholipid antibodies from patients with the antiphospholipid syndrome induce monocyte tissue factor expression through the simultaneous activation of NF-kappaB/Rel proteins via the p38 mitogen-activated protein kinase pathway, and of the MEK-1/ERK pathway. Arthritis Rheum. 2006, 54, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Lv, P.P.; Meng, Y.; Lv, M.; Feng, C.4; Liu, Y.; Li, J.Y.; Yu, D.Q.; Shen, Y.; Hu, X.L.; Gao, Q.; et al. Altered thyroid hormone profile in offspring after exposure to high estradiol environment during the first trimester of pregnancy: A cross-sectional study. BMC Med. 2014, 12, 240. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Lv, P.P.; Ding, G.L.; Yu, T.T.; Liu, Y.; Shen, Y.; Hu, X.L.; Lin, X.H.; Tian, S.; Lv, M.; et al. High Maternal Serum Estradiol Levels Induce Dyslipidemia in Human Newborns via a Hepatic HMGCR Estrogen Response Element. Sci. Rep. 2015, 5, 10086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Høibraaten, E.; Abdelnoor, M.; Sandset, P.M. Hormone replacement therapy with estradiol and risk of venous thromboembolism--a population-based case-control study. Thromb. Haemost. 1999, 82, 1218–1221. [Google Scholar]
- Gomes, M.P.V.; Deitcher, S.R. Risk of venous thromboembolic disease associated with hormonal contraceptives and hormone replacement therapy: A clinical review. Arch. Intern. Med. 2004, 164, 1965. [Google Scholar] [CrossRef]
- Sandset, P.M.; Høibraaten, E.; Eilertsen, A.L.; Dahm, A. Mechanisms of thrombosis related to hormone therapy. Thromb. Res. 2009, 123, S70–S73. [Google Scholar] [CrossRef]
- Straub, R.H. The complex role of estrogens in inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef] [Green Version]
- Loy, R.A.; Loukides, J.A.; Polan, M.L. Ovarian steroids modulate human monocyte tumor necrosis factor alpha messenger ribonucleic acid levels in cultured human peripheral monocytes. Fertil. Steril. 1992, 58, 733–739. [Google Scholar] [CrossRef]
- Konecna, L.; Yan, M.S.; Miller, L.E.; Schölmerich, J.; Falk, W.; Straub, R.H. Modulation of IL-6 production during the menstrual cycle in vivo and in vitro. Brain Behav. Immun. 2000, 14, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Polan, M.L.; Loukides, J.; Nelson, P.; Carding, S.; Diamond, M.; Walsh, A.; Bottomly, K. Progesterone and estradiol modulate interleukin-1 beta messenger ribonucleic acid levels in cultured human peripheral monocytes. J. Clin. Endocrinol. Metab. 1989, 69, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K.; Marshall, G.D., Jr. Perimenstrual alterations in type-1/type-2 cytokine balance of normal women. Ann. Allergy Asthma Immunol. 1999, 83, 222–228. [Google Scholar] [CrossRef]
- Kanda, N.; Tamaki, K. Estrogen enhances immunoglobulin production by human PBMCs. J. Allergy Clin. Immunol. 1999, 103, 282–288. [Google Scholar] [CrossRef]
- Rogers, A.; Eastell, R. The effect of 17beta-estradiol on production of cytokines in cultures of peripheral blood. Bone 2001, 29, 30–34. [Google Scholar] [CrossRef]
- Pierangeli, S.S.; Vega-Ostertag, M.; Liu, X.; Girardi, G. Complement activation: A novel pathogenic mechanism in the antiphospholipid syndrome. Ann. N. Y. Acad. Sci. 2005, 1051, 413–420. [Google Scholar] [CrossRef]
- Girardi, G.; Berman, J.; Redecha, P.; Spruce, L.; Thurman, J.M.; Kraus, D.; Hollmann, T.J.; Casali, P.; Caroll, M.C.; Wetsel, R.A.; et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J. Clin. Invest. 2003, 112, 1644–1654. [Google Scholar] [CrossRef] [Green Version]
- Confavreux, C.; Hutchinson, M.; Hours, M.M.; Cortinovis-Tourniaire, P.; Moreau, T. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in multiple sclerosis group. N. Engl. J. Med. 1998, 339, 285–291. [Google Scholar] [CrossRef]
- McCombe, P.A. The Short and Long-Term Effects of Pregnancy on Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. J. Clin. Med. 2018, 7, E494. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.L.; Ostensen, M. Pregnancy and rheumatoid arthritis. Rheum. Dis. Clin. New Am. 1997, 23, 195–212. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef] [PubMed]
- Whitacre, C.C. Sex differences in autoimmune disease. Nat. Immunol. 2001, 2, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Eudy, A.M.; Siega-Riz, A.M.; Engel, S.M.; Franceschini, N.; Howard, A.G.; Clowse, M.E.B.; Petri, M. Effect of pregnancy on disease flares in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Bodhankar, S.; Galipeau, D.; Vandenbark, A.A.; Murphy, S.J.; Offner, H. PD-1 Interaction with PD-L1 but not PD-L2 on B-cells Mediates Protective Effects of Estrogen against EAE. J. Clin. Cell Immunol. 2013, 4, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimaldi, C.M.; Cleary, J.; Dagtas, A.S.; Moussai, D.; Diamond, B. Estrogen alters thresholds for B cell apoptosis and activation. J. Clin. Invest. 2002, 109, 1625–1633. [Google Scholar] [CrossRef]
- Caruso, A.; De Carolis, S.; Di Simone, N. Antiphospholipid antibodies in obstetrics: New complexities and sites of action. Hum. Reprod. Update 1999, 5, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Perricone, C.; De Carolis, C.; Giacomelli, R.; Zaccari, G.; Cipriani, P.; Bizzi, E.; Perricone, R. High levels of NK cells in the peripheral blood of patients affected with anti-phospholipid syndrome and recurrent spontaneous abortion: A potential new hypothesis. Rheumatology 2007, 46, 1574–1578. [Google Scholar] [CrossRef] [Green Version]
- Gomaa, M.F.; Elkhouly, A.G.; Farghly, M.M.; Farid, L.A.; Awad, N.M. Uterine CD56dim and CD16+ Cells in Refractory Antiphospholipid Antibody-Related Pregnancy Loss and Chromosomally Intact Abortuses: A Case-Control Study. J. Hum. Reprod. Sci. 2017, 10, 18–23. [Google Scholar] [CrossRef]
- Carlino, C.; Stabile, H.; Morrone, S.; Bulla, R.; Soriani, A.; Agostinis, C.; Bossi, F.; Mocci, C.; Sarazani, F.; Tedesco, F. Recruitment of circulating NK cells through decidual tissues: A possible mechanism controlling NK cell accumulation in the uterus during early pregnancy. Blood 2008, 111, 3108–3115. [Google Scholar] [CrossRef] [Green Version]
- Chauleur, C.; Galanaud, J.P.; Alonso, S.; Cochery-Nouvellon, E.; Balducchi, J.P.; Marès, P.; Fabbro-Peray, P.; Gris, J.C. Observational study of pregnant women with a previous spontaneous abortion before the 10th gestation week with and without antiphospholipid antibodies. J. Thromb. Haemost. 2010, 8, 699–706. [Google Scholar] [CrossRef]
- Del Ross, T.; Ruffatti, A.; Visentin, M.S.; Tonello, M.; Calligaro, A.; Favaro, M.; Hoxha, A.; Punzi, L. Treatment of 139 pregnancies in antiphospholipid-positive women not fulfilling criteria for antiphospholipid syndrome: A retrospective study. J. Rheumatol. 2013, 40, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Riancho-Zarrabeitia, L.; Daroca, G.; Muñoz, P.; López-Hoyos, M.; Haya, A.; Martínez-Taboada, V.M. Serological evolution in women with positive antiphospholipid antibodies. Semin. Arthritis Rheum. 2017, 47, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Bala, M.M.; Paszek, E.; Lesniak, W.; Wloch-Kopec, D.; Jasinska, K.; Undas, A. Antiplatelet and anticoagulant agents for primary prevention of thrombosis in individuals with antiphospholipid antibodies. Cochrane Database Syst. Rev. 2018, 7, CD012534. [Google Scholar] [CrossRef] [PubMed]
- Gibbins, K.J.; Tebo, A.E.; Nielsen, S.K.; Branch, D.W. Antiphospholipid antibodies in women with severe preeclampsia and placental insufficiency: A case-control study. Lupus 2018, 27, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Alijotas-Reig, J.; Esteve-Valverde, E.; Ferrer-Oliveras, R.; LLurba, E.; Ruffatti, A.; Tincani, A.; Lefkou, E.; Bertero, M.T.; Espinosa, G.; de Carolis, S.; et al. Comparative study between obstetric antiphospholipid syndrome and obstetric morbidity related with antiphospholipid antibodies. Med. Clin. 2018, 151, 215–222. [Google Scholar] [CrossRef]
- WHO. Medical Eligibility Criteria for Contraceptive Use, 5th ed.; WHO: Geneva, Switzerland, 2015; Available online: http://www.who.int/reproductivehealth/publications/family_planning/Ex-Summ-MEC-5/en/ (accessed on 15 March 2015).
- Tektonidou, M.G.; Andreoli, L.; Limper, M.; Amoura, Z.; Cervera, R.; Costedoat-Chalumeau, N.; Cuadrado, M.J.; Dörner, T.; Ferrer-Oliveras, R.; Hambly, K.; et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann. Rheum. Dis. 2019, 78, 1296–1304. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| aPL- Group (Control Group) | aPL+ Group (Patients’ Group) | |
|---|---|---|
| Number of subjects | 13 | 14 |
| Age (years) | 34.6 (21–42) | 37.5 (23–46) |
| Clinical records | No history of thrombotic events, pregnancy loss, familial history of APS | 5/14- SLE 2/14- altered coagulation parameters 3/14- rheumatic diseases 4/14- other 2/14- pregnancy loses |
| LA (n = positive patients) | 0/14 | 10/14 |
| aCL (IgG) (U/ML) | <20 | 253.7 ± 621.8 |
| aCL (IgM) (U/ML) | <20 | 69.28 ± 70.17 |
| anti-β2GPI (IgG) (U/ML) | <20 | 651.0 ± 1808 |
| anti-β2GPI (IgM) (U/ML) | <20 | 142.3 ± 227.3 |
| anti-D1 β2GPI (CU/ML) | <20 | 175.6 ± 408 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manukyan, G.; Martirosyan, A.; Slavik, L.; Ulehlova, J.; Dihel, M.; Papajik, T.; Kriegova, E. 17β-Estradiol Promotes Proinflammatory and Procoagulatory Phenotype of Innate Immune Cells in the Presence of Antiphospholipid Antibodies. Biomedicines 2020, 8, 162. https://doi.org/10.3390/biomedicines8060162
Manukyan G, Martirosyan A, Slavik L, Ulehlova J, Dihel M, Papajik T, Kriegova E. 17β-Estradiol Promotes Proinflammatory and Procoagulatory Phenotype of Innate Immune Cells in the Presence of Antiphospholipid Antibodies. Biomedicines. 2020; 8(6):162. https://doi.org/10.3390/biomedicines8060162
Chicago/Turabian StyleManukyan, Gayane, Anush Martirosyan, Ludek Slavik, Jana Ulehlova, Martin Dihel, Tomas Papajik, and Eva Kriegova. 2020. "17β-Estradiol Promotes Proinflammatory and Procoagulatory Phenotype of Innate Immune Cells in the Presence of Antiphospholipid Antibodies" Biomedicines 8, no. 6: 162. https://doi.org/10.3390/biomedicines8060162
APA StyleManukyan, G., Martirosyan, A., Slavik, L., Ulehlova, J., Dihel, M., Papajik, T., & Kriegova, E. (2020). 17β-Estradiol Promotes Proinflammatory and Procoagulatory Phenotype of Innate Immune Cells in the Presence of Antiphospholipid Antibodies. Biomedicines, 8(6), 162. https://doi.org/10.3390/biomedicines8060162