Key Milestones Contributing to the Understanding of the Mechanisms Underlying Fibromyalgia


Abstract
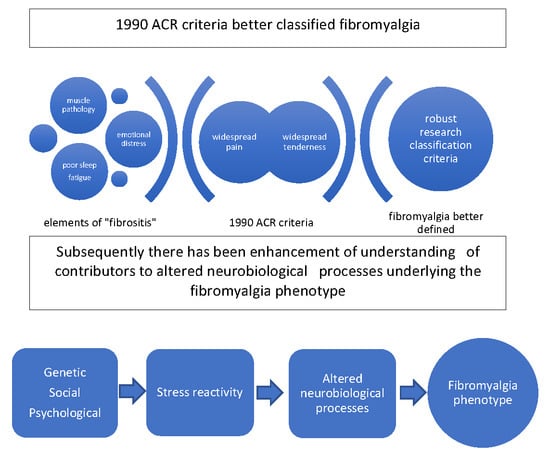
:
1. Describing Fibromyalgia
2. Evolution of Classification and Diagnostic Criteria
3. Exploration of Peripheral Muscle Mechanisms was Prominent Prior to the 1990 ACR Criteria
4. Neuroinflammation as a Peripheral Pain Mechanism
5. Referred Pain as a Peripheral Pain Mechanism
6. Characterization of Central Sensitization in Fibromyalgia after the 1990 ACR Criteria
7. Neurotransmitters
8. Descending Pathways in Fibromyalgia
9. The Brain in Fibromyalgia
10. Genetic Factors
11. Psychological Factors in Fibromyalgia
12. Sleep in Fibromyalgia
13. Stress Reactivity in Fibromyalgia
14. Social Factors
15. Summary
Funding
Conflicts of Interest
References
- Wolfe, F.; Smythe, H.A.; Yunus, M.B.; Bennett, R.M.; Bombardier, C.; Goldenberg, D.L.; Tugwell, P.; Campbell, S.M.; Abeles, M.; Clark, P.; et al. The American college of rheumatology 1990 criteria for the classification of fibromyalgia. Report of the multicenter criteria committee. Arthritis Rheum. 1990, 33, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Hauser, W.; Ablin, J.; Fitzcharles, M.A.; Littlejohn, G.; Luciano, J.V.; Usui, C.; Walitt, B. Fibromyalgia. Nat. Rev. Dis. Primers 2015, 1, 15022. [Google Scholar] [CrossRef] [PubMed]
- Inanici, F.; Yunus, M.B. History of fibromyalgia: Past to present. Curr. Pain Headache Rep. 2004, 8, 369–378. [Google Scholar] [CrossRef]
- Wallace, D. History of fibromyalgia. In Fibromyalgia. The Essential Clinicians Guide; Clauw, D., Wallace, D., Eds.; Oxford University Press: Oxford, UK, 2009; pp. 1–5. [Google Scholar]
- Smythe, H.A.; Moldofsky, H. Two contributions to understanding of the “fibrositis” syndrome. Bull. Rheum. Dis. 1977, 28, 928–931. [Google Scholar] [PubMed]
- Yunus, M.; Masi, A.T.; Calabro, J.J.; Miller, K.A.; Feigenbaum, S.L. Primary fibromyalgia (fibrositis): Clinical study of 50 patients with matched normal controls. Semin. Arthritis Rheum. 1981, 11, 151–171. [Google Scholar] [CrossRef]
- Wolfe, F. The clinical syndrome of fibrositis. Am. J. Med. 1986, 81, 7–14. [Google Scholar] [CrossRef]
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.A.; Goldenberg, D.L.; Katz, R.S.; Mease, P.; Russell, A.S.; Russell, I.J.; Winfield, J.B.; Yunus, M.B. The American college of rheumatology preliminary diagnostic criteria for fibromyalgia and measurement of symptom severity. Arthritis Care Res. 2010, 62, 600–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.A.; Goldenberg, D.L.; Hauser, W.; Katz, R.S.; Mease, P.; Russell, A.S.; Russell, I.J.; Winfield, J.B. Fibromyalgia criteria and severity scales for clinical and epidemiological studies: A modification of the acr preliminary diagnostic criteria for fibromyalgia. J. Rheumatol. 2011, 38, 1113–1122. [Google Scholar] [CrossRef]
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.A.; Goldenberg, D.L.; Hauser, W.; Katz, R.L.; Mease, P.J.; Russell, A.S.; Russell, I.J.; Walitt, B. 2016 revisions to the 2010/2011 fibromyalgia diagnostic criteria. Semin. Arthritis Rheum. 2016, 46, 319–329. [Google Scholar] [CrossRef]
- Wolfe, F.; Butler, S.H.; Fitzcharles, M.; Hauser, W.; Katz, R.L.; Mease, P.J.; Rasker, J.J.; Russell, A.S.; Russell, I.J.; Walitt, B. Revised chronic widespread pain criteria: Development from and integration with fibromyalgia criteria. Scand. J. Pain 2019. [Google Scholar] [CrossRef]
- Littlejohn, G. Fibromyalgia: Honing fibromyalgia diagnosis. Nat. Rev. Rheumatol. 2014, 10, 267–269. [Google Scholar] [CrossRef]
- Clauw, D. Time to stop the fm wars–and refocus on identifying and treating individuals with this type of pain earlier in their illnesso. Arthritis Care Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Galvez-Sanchez, C.M.; Reyes Del Paso, G.A. Diagnostic criteria for fibromyalgia: Critical review and future perspectives. J. Clin. Med. 2020, 9, 1219. [Google Scholar] [CrossRef] [PubMed]
- Hench, P.K.; Mitler, M.M. Fibromyalgia. 1. Review of a common rheumatologic syndrome. Postgrad. Med. 1986, 80, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Balfour, W. Observations on the pathology and cure of rheumatism. Edinb. Med. Surg. J. 1815, 11, 168–187. [Google Scholar]
- Scudamore, C. A Treatise on the Nature and Cure of Rheumatism; Longman, Rees, Orme, Brown & Gren, Paternoster Row: London, UK, 1827. [Google Scholar]
- Stockman, R. The causes, pathology, and treatment of chronic rheumatism. Edinb. Med. J. 1904, 15, 107–116. [Google Scholar]
- Collins, D.H. Infection and fibrositis. Ann. Rheum. Dis. 1940, 2, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Yunus, M.B.; Kalyan-Raman, U.P. Muscle biopsy findings in primary fibromyalgia and other forms of nonarticular rheumatism. Rheum. Dis. Clin. N. Am. 1989, 15, 115–134. [Google Scholar]
- Simms, R.W. Fibromyalgia is not a muscle disorder. Am. J. Med. Sci. 1998, 315, 346–350. [Google Scholar]
- Ruggiero, L.; Manganelli, F.; Santoro, L. Muscle pain syndromes and fibromyalgia: The role of muscle biopsy. Curr. Opin. Support. Palliat. Care 2018, 12, 382–387. [Google Scholar] [CrossRef]
- Klaver-Krol, E.G.; Rasker, J.J.; Klaver, M.M.; Ten Klooster, P.M.; Zwarts, M.J. Fibromyalgia: Increased reactivity of the muscle membrane and a role of central regulation. Clin. Neurophysiol. 2019, 130, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Affaitati, G.; Costantini, R.; Fabrizio, A.; Lapenna, D.; Tafuri, E.; Giamberardino, M.A. Effects of treatment of peripheral pain generators in fibromyalgia patients. Eur. J. Pain 2011, 15, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Littlejohn, G.O.; Weinstein, C.; Helme, R.D. Increased neurogenic inflammation in fibrositis syndrome. J. Rheumatol. 1987, 14, 1022–1025. [Google Scholar] [PubMed]
- Littlejohn, G.; Guymer, E. Neurogenic inflammation in fibromyalgia. Semin. Immunopathol. 2018, 40, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Grayston, R.; Czanner, G.; Elhadd, K.; Goebel, A.; Frank, B.; Uceyler, N.; Malik, R.A.; Alam, U. A systematic review and meta-analysis of the prevalence of small fiber pathology in fibromyalgia: Implications for a new paradigm in fibromyalgia etiopathogenesis. Semin. Arthritis Rheum. 2019, 48, 933–940. [Google Scholar] [CrossRef]
- Serra, J.; Collado, A.; Sola, R.; Antonelli, F.; Torres, X.; Salgueiro, M.; Quiles, C.; Bostock, H. Hyperexcitable c nociceptors in fibromyalgia. Ann. Neurol. 2014, 75, 196–208. [Google Scholar] [CrossRef]
- Yunus, M.B. Fibromyalgia and overlapping disorders: The unifying concept of central sensitivity syndromes. Semin. Arthritis Rheum. 2007, 36, 339–356. [Google Scholar] [CrossRef]
- Kellgren, J.H. On distribution of pain arising from deep somatic structures with charts of segmental pain areas. Clin. Sci. 1939, 4, 35–46. [Google Scholar]
- Graham, W. Fibrositis. In Arthritis and Allied Conditions; Hollander, J.L., Ed.; Lea and Febiger: Philadelphia, PA, USA, 1949; pp. 647–662. [Google Scholar]
- Smythe, H. Referred pain and tender points. Am. J. Med. 1986, 81, 90–92. [Google Scholar] [CrossRef]
- Arroyo, J.F.; Cohen, M.L. Abnormal responses to electrocutaneous stimulation in fibromyalgia. J. Rheumatol. 1993, 20, 1925–1931. [Google Scholar]
- Granges, G.; Littlejohn, G. Pressure pain threshold in pain-free subjects, in patients with chronic regional pain syndromes, and in patients with fibromyalgia syndrome. Arthritis Rheum. 1993, 36, 642–646. [Google Scholar] [CrossRef]
- Gibson, S.J.; Littlejohn, G.O.; Gorman, M.M.; Helme, R.D.; Granges, G. Altered heat pain thresholds and cerebral event-related potentials following painful co2 laser stimulation in subjects with fibromyalgia syndrome. Pain 1994, 58, 185–193. [Google Scholar] [CrossRef]
- Staud, R.; Robinson, M.E.; Price, D.D. Temporal summation of second pain and its maintenance are useful for characterizing widespread central sensitization of fibromyalgia patients. J. Pain 2007, 8, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmeules, J.A.; Cedraschi, C.; Rapiti, E.; Baumgartner, E.; Finckh, A.; Cohen, P.; Dayer, P.; Vischer, T.L. Neurophysiologic evidence for a central sensitization in patients with fibromyalgia. Arthritis Rheum. 2003, 48, 1420–1429. [Google Scholar] [CrossRef]
- Banic, B.; Petersen-Felix, S.; Andersen, O.K.; Radanov, B.P.; Villiger, P.M.; Arendt-Nielsen, L.; Curatolo, M. Evidence for spinal cord hypersensitivity in chronic pain after whiplash injury and in fibromyalgia. Pain 2004, 107, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J. Central sensitization: Implications for the diagnosis and treatment of pain. Pain 2011, 152, 2–15. [Google Scholar] [CrossRef]
- Staud, R. Is it all central sensitization. Role of peripheral tissue nociception in chronic musculoskeletal pain. Curr. Rheumatol. Rep. 2010, 12, 448–454. [Google Scholar] [CrossRef]
- DeSantana, J.M.; Sluka, K.A. Central mechanisms in the maintenance of chronic widespread noninflammatory muscle pain. Curr. Pain Headache Rep. 2008, 12, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Russell, I.J.; Vaeroy, H.; Javors, M.; Nyberg, F. Cerebrospinal fluid biogenic amine metabolites in fibromyalgia/fibrositis syndrome and rheumatoid arthritis. Arthritis Rheum. 1992, 35, 550–556. [Google Scholar] [CrossRef]
- Becker, S.; Schweinhardt, P. Dysfunctional neurotransmitter systems in fibromyalgia, their role in central stress circuitry and pharmacological actions on these systems. Pain Res. Treat 2012. [Google Scholar] [CrossRef] [PubMed]
- Littlejohn, G.; Guymer, E. Modulation of nmda receptor activity in fibromyalgia. Biomedicines 2017, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, P.; Cohen, M.; Quintner, J. An evolutionary stress-response hypothesis for chronic widespread pain (fibromyalgia syndrome). Pain Med. 2011, 12, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Vaeroy, H.; Helle, R.; Forre, O.; Kass, E.; Terenius, L. Elevated csf levels of substance p and high incidence of raynaud phenomenon in patients with fibromyalgia: New features for diagnosis. Pain 1988, 32, 21–26. [Google Scholar] [CrossRef]
- Russell, I.J.; Orr, M.D.; Littman, B.; Vipraio, G.A.; Alboukrek, D.; Michalek, J.E.; Lopez, Y.; MacKillip, F. Elevated cerebrospinal fluid levels of substance p in patients with the fibromyalgia syndrome. Arthritis Rheum. 1994, 37, 1593–1601. [Google Scholar] [CrossRef]
- Sarchielli, P.; Di Filippo, M.; Nardi, K.; Calabresi, P. Sensitization, glutamate, and the link between migraine and fibromyalgia. Curr. Pain Headache Rep. 2007, 11, 343–351. [Google Scholar] [CrossRef]
- Graven-Nielsen, T.; Aspegren Kendall, S.; Henriksson, K.G.; Bengtsson, M.; Sorensen, J.; Johnson, A.; Gerdle, B.; Arendt-Nielsen, L. Ketamine reduces muscle pain, temporal summation, and referred pain in fibromyalgia patients. Pain 2000, 85, 483–491. [Google Scholar] [CrossRef]
- Giovengo, S.L.; Russell, I.J.; Larson, A.A. Increased concentrations of nerve growth factor in cerebrospinal fluid of patients with fibromyalgia. J. Rheumatol. 1999, 26, 1564–1569. [Google Scholar]
- Seidel, M.F.; Herguijuela, M.; Forkert, R.; Otten, U. Nerve growth factor in rheumatic diseases. Semin. Arthritis Rheum. 2010, 40, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Montoya, P.; Sitges, C.; Garcia-Herrera, M.; Rodriguez-Cotes, A.; Izquierdo, R.; Truyols, M.; Collado, D. Reduced brain habituation to somatosensory stimulation in patients with fibromyalgia. Arthritis Rheum. 2006, 54, 1995–2003. [Google Scholar] [CrossRef]
- Julien, N.; Goffaux, P.; Arsenault, P.; Marchand, S. Widespread pain in fibromyalgia is related to a deficit of endogenous pain inhibition. Pain 2005, 114, 295–302. [Google Scholar] [CrossRef]
- Yarnitsky, D. Conditioned pain modulation (the diffuse noxious inhibitory control-like effect): Its relevance for acute and chronic pain states. Curr. Opin. Anaesthesiol. 2010, 23, 611–615. [Google Scholar] [CrossRef]
- Schweinhardt, P.; Sauro, K.M.; Bushnell, M.C. Fibromyalgia: A disorder of the brain. Neuroscientist 2008, 14, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Normand, E.; Potvin, S.; Gaumond, I.; Cloutier, G.; Corbin, J.F.; Marchand, S. Pain inhibition is deficient in chronic widespread pain but normal in major depressive disorder. J. Clin. Psychiatry 2011, 72, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajah, A.S.; Guymer, E.K.; Leech, M.T.; Littlejohn, G.O. The relationship between fibromyalgia, stress and depression. Int. J. Clin. Rheumatol. 2014, 9, 371–384. [Google Scholar] [CrossRef]
- Jensen, K.B.; Kosek, E.; Petzke, F.; Carville, S.; Fransson, P.; Marcus, H.; Williams, S.C.; Choy, E.; Giesecke, T.; Mainguy, Y.; et al. Evidence of dysfunctional pain inhibition in fibromyalgia reflected in racc during provoked pain. Pain 2009, 144, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Ernberg, M.; Voog, U.; Alstergren, P.; Lundeberg, T.; Kopp, S. Plasma and serum serotonin levels and their relationship to orofacial pain and anxiety in fibromyalgia. J. Orofac. Pain 2000, 14, 37–46. [Google Scholar]
- Wolfe, F.; Russell, I.J.; Vipraio, G.; Ross, K.; Anderson, J. Serotonin levels, pain threshold, and fibromyalgia symptoms in the general population. J. Rheumatol. 1997, 24, 555–559. [Google Scholar]
- Northcott, M.J.; Guymer, E.; Littlejohn, G.O. Pharmacologic treatment options for fibromyalgia. Clin. Pharm. 2017. [Google Scholar] [CrossRef]
- Bosma, R.L.; Hemington, K.S.; Davis, K.D. Using magnetic resonance imaging to visualize the brain in chronic pain. Pain 2017, 158, 1192–1193. [Google Scholar] [CrossRef] [PubMed]
- Mountz, J.M.; Bradley, L.A.; Alarcon, G.S. Abnormal functional activity of the central nervous system in fibromyalgia syndrome. Am. J. Med. Sci. 1998, 315, 385–396. [Google Scholar] [PubMed]
- Guedj, E.; Cammilleri, S.; Niboyet, J.; Dupont, P.; Vidal, E.; Dropinski, J.P.; Mundler, O. Clinical correlate of brain spect perfusion abnormalities in fibromyalgia. J. Nucl. Med. 2008, 49, 1798–1803. [Google Scholar] [CrossRef] [Green Version]
- Nebel, M.B.; Gracely, R.H. Neuroimaging of fibromyalgia. Rheum. Dis. Clin. N. Am. 2009, 35, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Napadow, V.; LaCount, L.; Park, K.; As-Sanie, S.; Clauw, D.J.; Harris, R.E. Intrinsic brain connectivity in fibromyalgia is associated with chronic pain intensity. Arthritis Rheum. 2010, 62, 2545–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napadow, V.; Kim, J.; Clauw, D.J.; Harris, R.E. Decreased intrinsic brain connectivity is associated with reduced clinical pain in fibromyalgia. Arthritis Rheum. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, K.B.; Loitoile, R.; Kosek, E.; Petzke, F.; Carville, S.; Fransson, P.; Marcus, H.; Williams, S.C.; Choy, E.; Mainguy, Y.; et al. Patients with fibromyalgia display less functional connectivity in the brain’s pain inhibitory network. Mol. Pain 2012, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.E.; Sundgren, P.C.; Craig, A.D.; Kirshenbaum, E.; Sen, A.; Napadow, V.; Clauw, D.J. Elevated insular glutamate in fibromyalgia is associated with experimental pain. Arthritis Rheum. 2009, 60, 3146–3152. [Google Scholar] [CrossRef] [Green Version]
- Kadetoff, D.; Lampa, J.; Westman, M.; Andersson, M.; Kosek, E. Evidence of central inflammation in fibromyalgia-increased cerebrospinal fluid interleukin-8 levels. J. Neuroimmunol. 2012, 242, 33–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosek, E.; Martinsen, S.; Gerdle, B.; Mannerkorpi, K.; Lofgren, M.; Bileviciute-Ljungar, I.; Fransson, P.; Schalling, M.; Ingvar, M.; Ernberg, M.; et al. The translocator protein gene is associated with symptom severity and cerebral pain processing in fibromyalgia. Brain Behav. Immun. 2016, 58, 218–227. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, D.S.; Forsberg, A.; Sandstrom, A.; Bergan, C.; Kadetoff, D.; Protsenko, E.; Lampa, J.; Lee, Y.C.; Hoglund, C.O.; Catana, C.; et al. Brain glial activation in fibromyalgia—A multi-site positron emission tomography investigation. Brain Behav. Immun. 2019, 75, 72–83. [Google Scholar] [CrossRef]
- Jung, C.; Ichesco, E.; Ratai, E.-M.; Gonzalez, R.G.; Ramon, G.; Burdo, T.; Loggia, M.L.; Harris, R.E.; Napadow, V. Magnetic resonance imaging of neuroinflammation in chronic pain: A role for astrogliosis. Pain 2020, 161, 1555–1564. [Google Scholar] [CrossRef]
- Bennett, R. Fibromyalgia: Present to future. Curr. Pain Headache Rep. 2004, 8, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Stormorken, H.; Brosstad, F. Fibromyalgia: Family clustering and sensory urgency with early onset indicate genetic predisposition and thus a “true” disease. Scand. J. Rheumatol. 1992, 21, 207. [Google Scholar] [CrossRef]
- Arnold, L.M.; Hudson, J.I.; Hess, E.V.; Ware, A.E.; Fritz, D.A.; Auchenbach, M.B.; Starck, L.O.; Keck, P.E., Jr. Family study of fibromyalgia. Arthritis Rheum. 2004, 50, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Ablin, J.N.; Buskila, D. Update on the genetics of the fibromyalgia syndrome. Best Pract. Res. Clin. Rheumatol. 2015, 29, 20–28. [Google Scholar] [CrossRef]
- Dutta, D.; Brumett, C.; Moser, S.; Fritsche, L.; Tsodikov, A.; Clauw, D.; Scott, L. Heritability of the fibromyalgia phenotype varies by age. Arthritis Rheumatol. 2020, 72, 815–823. [Google Scholar] [CrossRef]
- Fitzcharles, M.A.; Yunus, M.B. The clinical concept of fibromyalgia as a changing paradigm in the past 20 years. Pain Res. Treat. 2012, 2012, 184835. [Google Scholar] [CrossRef] [Green Version]
- Ablin, K.; Clauw, D.J. From fibrositis to functional somatic syndromes to a bell-shaped curve of pain and sensory sensitivity: Evolution of a clinical construct. Rheum. Dis. Clin. N. Am. 2009, 35, 233–251. [Google Scholar] [CrossRef]
- Malin, K.; Littlejohn, G.O. Neuroticism in young women with fibromyalgia links to key clinical features. Pain Res. Treat. 2012, 2012, 730–741. [Google Scholar] [CrossRef] [Green Version]
- McBeth, J.; Macfarlane, G.J.; Benjamin, S.; Morris, S.; Silman, A.J. The association between tender points, psychological distress, and adverse childhood experiences: A community-based study. Arthritis Rheum. 1999, 42, 1397–1404. [Google Scholar] [CrossRef]
- Littlejohn, G.; Guymer, E. Central processes underlying fibromyalgia. Eur. Med. J. 2018, 3, 79–86. [Google Scholar]
- Staud, R. Biology and therapy of fibromyalgia: Pain in fibromyalgia syndrome. Arthritis Res. Ther. 2006, 8, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldofsky, H.; Scarisbrick, P.; England, R.; Smythe, H. Musculosketal symptoms and non-rem sleep disturbance in patients with “fibrositis syndrome” and healthy subjects. Psychosom. Med. 1975, 37, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H. The role of sleep in pain and fibromyalgia. Nat. Rev. Rheumatol. 2015, 11, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; Karaca, Z.; Unluhizarci, K.; Kelestimur, F. The hypothalamo-pituitary-adrenal axis in chronic fatigue syndrome and fibromyalgia syndrome. Stress 2007, 10, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Riedel, W.; Layka, H.; Neeck, G. Secretory pattern of gh, tsh, thyroid hormones, acth, cortisol, fsh, and lh in patients with fibromyalgia syndrome following systemic injection of the relevant hypothalamic-releasing hormones. Z Rheumatol. 1998, 57, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Adler, G.K.; Geenen, R. Hypothalamic-pituitary-adrenal and autonomic nervous system functioning in fibromyalgia. Rheum. Dis. Clin. N. Am. 2005, 31, 187–202. [Google Scholar] [CrossRef]
- Bennett, R.M.; Clark, S.C.; Walczyk, J. A randomized, double-blind, placebo-controlled study of growth hormone in the treatment of fibromyalgia. Am. J. Med. 1998, 104, 227–231. [Google Scholar] [CrossRef]
- Anderberg, U.M.; Uvnas-Moberg, K. Plasma oxytocin levels in female fibromyalgia syndrome patients. Z Rheumatol. 2000, 59, 373–379. [Google Scholar] [CrossRef]
- Lerma, C.; Martinez, A.; Ruiz, N.; Vargas, A.; Infante, O.; Martinez-Lavin, M. Nocturnal heart rate variability parameters as potential fibromyalgia biomarker: Correlation with symptoms severity. Arthritis Res. Ther. 2011, 13, 185. [Google Scholar] [CrossRef] [Green Version]
- Van Houdenhove, B.; Luyten, P. Stress, depression and fibromyalgia. Acta Neurol. Belg. 2006, 106, 149–156. [Google Scholar]
- Wolfe, F.; Ablin, J.; Guymer, E.K.; Littlejohn, G.O.; Rasker, J.J. The relation of physical comorbidity and multimorbidity to fibromyalgia, widespread pain, and fibromyalgia-related variables. J. Rheumatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hauser, W.; Henningsen, P. Fibromyalgia syndrome: A somatoform disorder. Eur. J. Pain 2014, 18, 1052–1059. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Neural Mechanisms | Type of Mechanism |
|---|---|
| Peripheral | Mechanoreceptor input |
| Referred pain | |
| Nociception | |
| Sympathetic nervous system | |
| Neuroinflammation | |
| Spinal cord | Central sensitization |
| Descending spinal cord control | |
| Brain | Neurotransmitter changes |
| Connectivity changes | |
| Neuroinflammation | |
| Other mechanisms | Genetic |
| Psychological | |
| Stress reactivity | |
| Social factors |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Littlejohn, G.; Guymer, E. Key Milestones Contributing to the Understanding of the Mechanisms Underlying Fibromyalgia. Biomedicines 2020, 8, 223. https://doi.org/10.3390/biomedicines8070223
Littlejohn G, Guymer E. Key Milestones Contributing to the Understanding of the Mechanisms Underlying Fibromyalgia. Biomedicines. 2020; 8(7):223. https://doi.org/10.3390/biomedicines8070223
Chicago/Turabian StyleLittlejohn, Geoffrey, and Emma Guymer. 2020. "Key Milestones Contributing to the Understanding of the Mechanisms Underlying Fibromyalgia" Biomedicines 8, no. 7: 223. https://doi.org/10.3390/biomedicines8070223
APA StyleLittlejohn, G., & Guymer, E. (2020). Key Milestones Contributing to the Understanding of the Mechanisms Underlying Fibromyalgia. Biomedicines, 8(7), 223. https://doi.org/10.3390/biomedicines8070223