Identification of Toxicity Parameters Associated with Combustion Produced Soot Surface Chemistry and Particle Structure by in Vitro Assays

 ,
, 
Abstract
:1. Introduction
2. Experimental Section
2.1. Synthesis and Characterization of Soot
- (1)
- Nascent soot (S1): Unmodified, non-oxidized, R250. This commercially produced carbon black was used as the model nascent soot form given its chemical purity and absence of organic content;
- (2)
- Nitric acid-treated soot (S2): Wet chemical treatment of R250 was conducted by treating 1 g of carbon black with 100 ml of laboratory-grade concentrated nitric acid (HNO3, >90%) at 80 °C under reflux for 24 h, just below the acid’s boiling point of 83 °C. The carbon–acid mixture was continuously stirred for uniform oxidation and functionalization. The mixture was maintained at a consistent simmer, and thereafter, it was washed with distilled water, filtered, and dried to obtain functionalized carbon black as synthetic soot;
- (3)
- Ozone-treated soot (S3): Dry gaseous treatment of carbon black was performed via exposure to ozone (O3) generated by subjecting oxygen (O2) to ultraviolet (UV) light. Ozone, a reactive gas, interacts with the carbon at room temperature and mildly oxidizes it, thereby functionalizing the carbon in the process. This method is a comparatively mild oxidative treatment than the wet acid reflux;
- (4)
- Nitric acid and heat-treated soot (S4): The powdered form of HNO3-treated carbon black was subjected to isothermal heat treatment at 300 °C in a hot-wall furnace for 1 h under an inert (Ar) environment.
2.2. X-Ray Photoelectron Spectroscopy (XPS)
2.3. Thermogravimetric Analysis (TGA)
2.4. Cell Culture and Soot Exposure
2.4.1. Cell and Soot Exposure for Cell Viability Assessment
2.4.2. Cell Culture and Soot Exposures for Gene Expression Assessment
2.4.3. Cell Culture and Soot Exposures for Protein Carbonylation Assessment
2.5. Cell Viability Assay
2.6. RNA Purification and cDNA synthesis
2.7. Real-Time PCR
2.8. Total Protein Determination
2.9. Protein Carbonylation Assay
2.10. Statistical Analysis
3. Results
3.1. Characterization of Soot
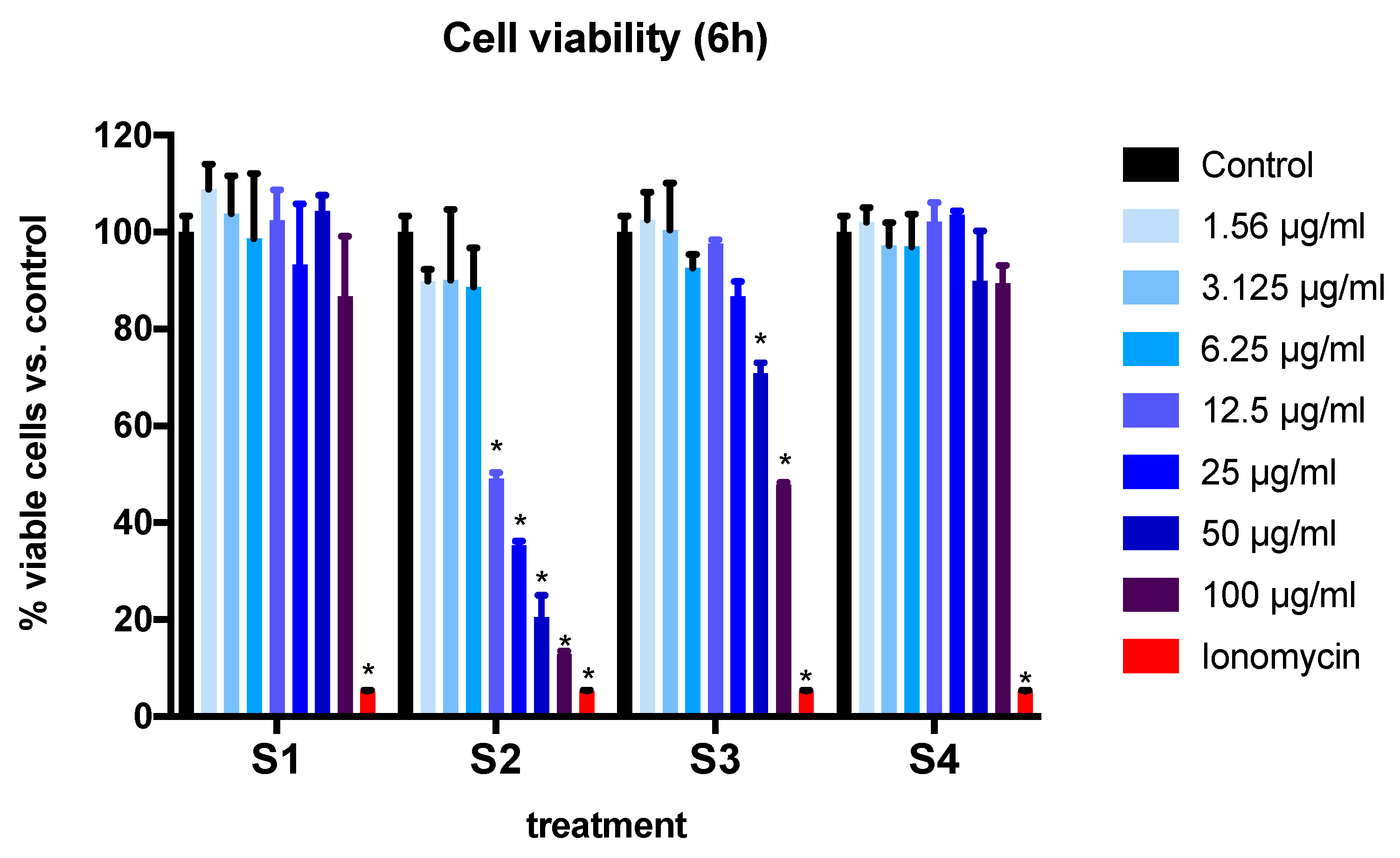
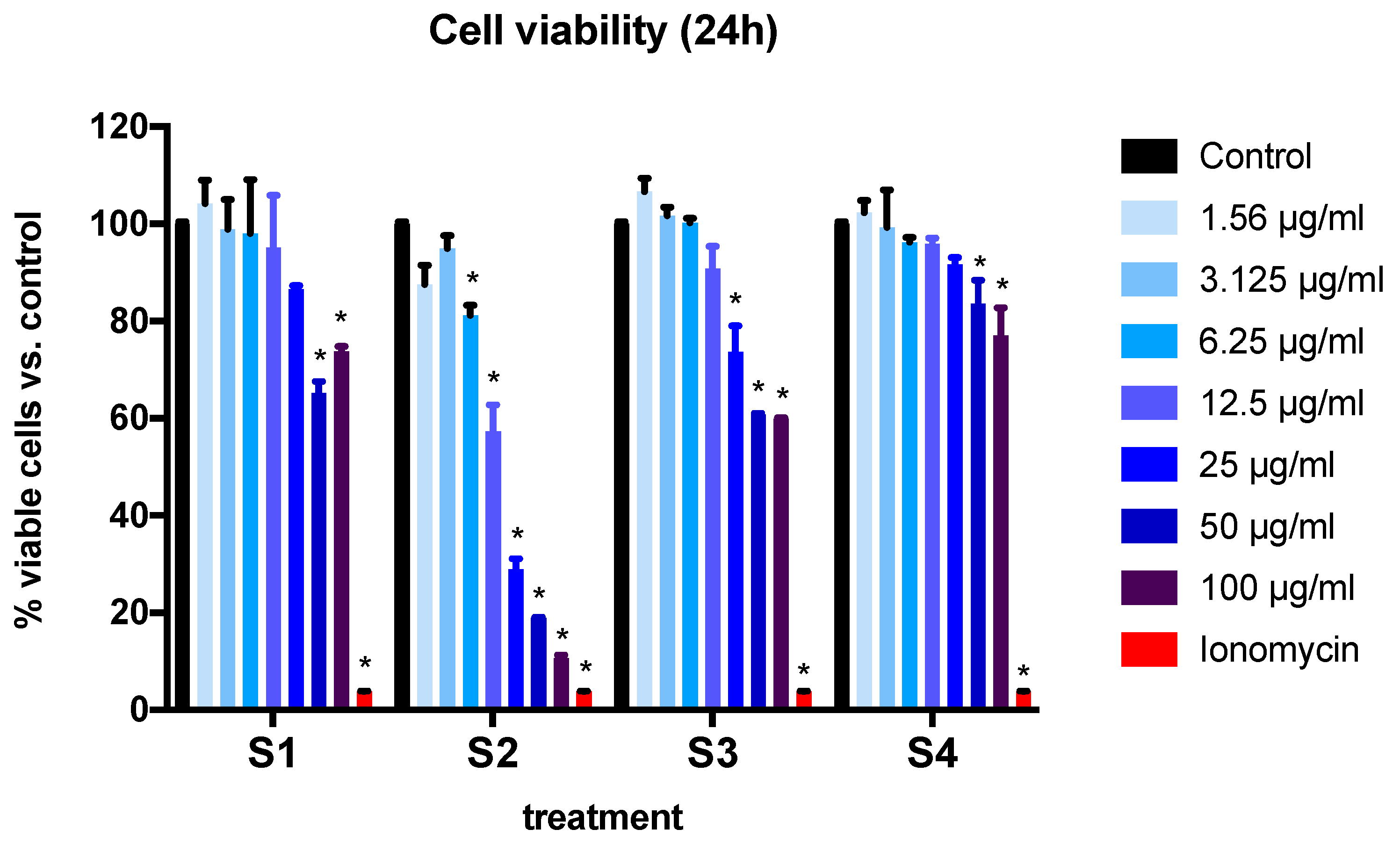
3.2. Cell Viability and Cytotoxicity in BEAS-2B Cells Exposed to Synthetic Soot
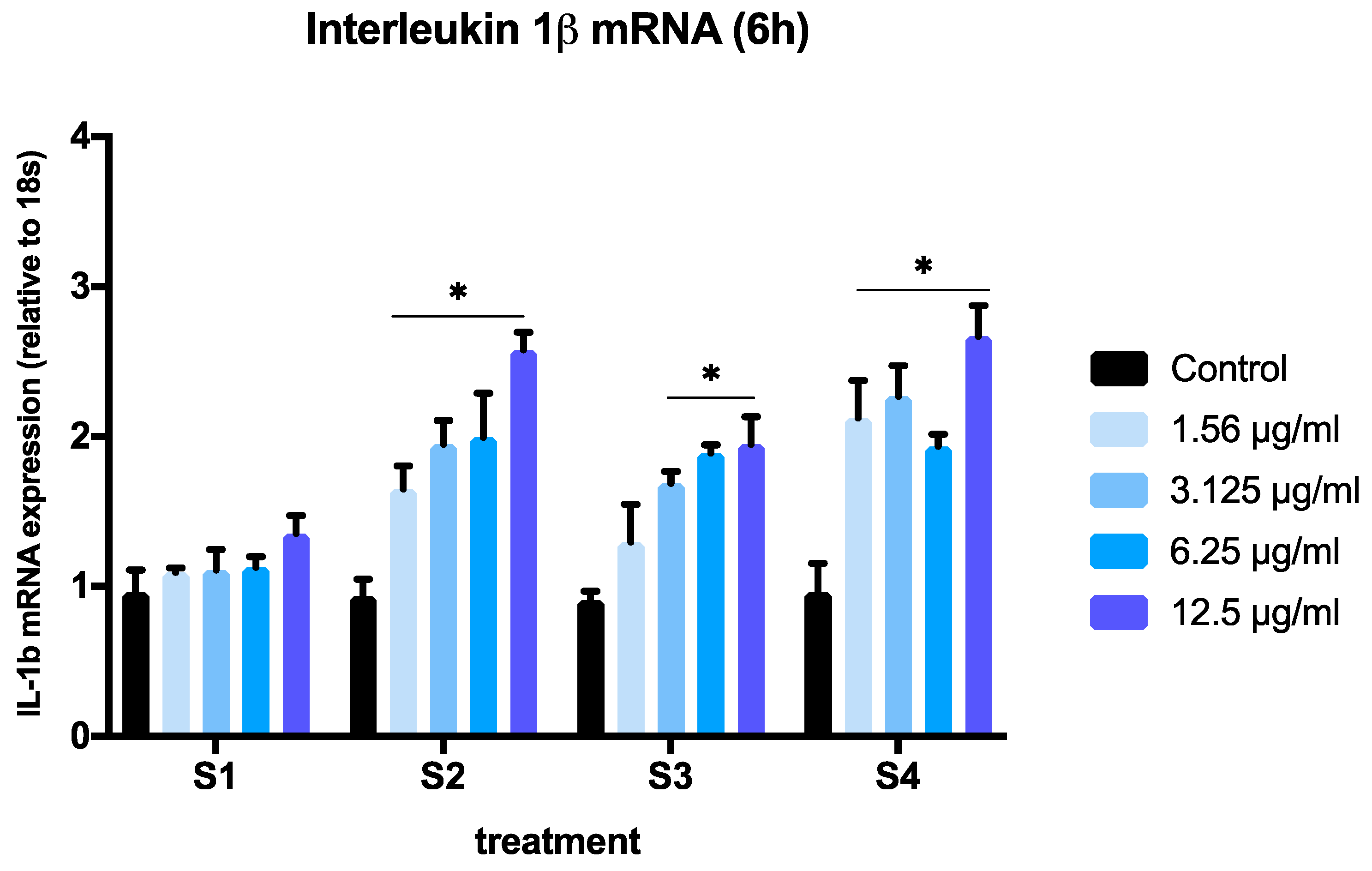
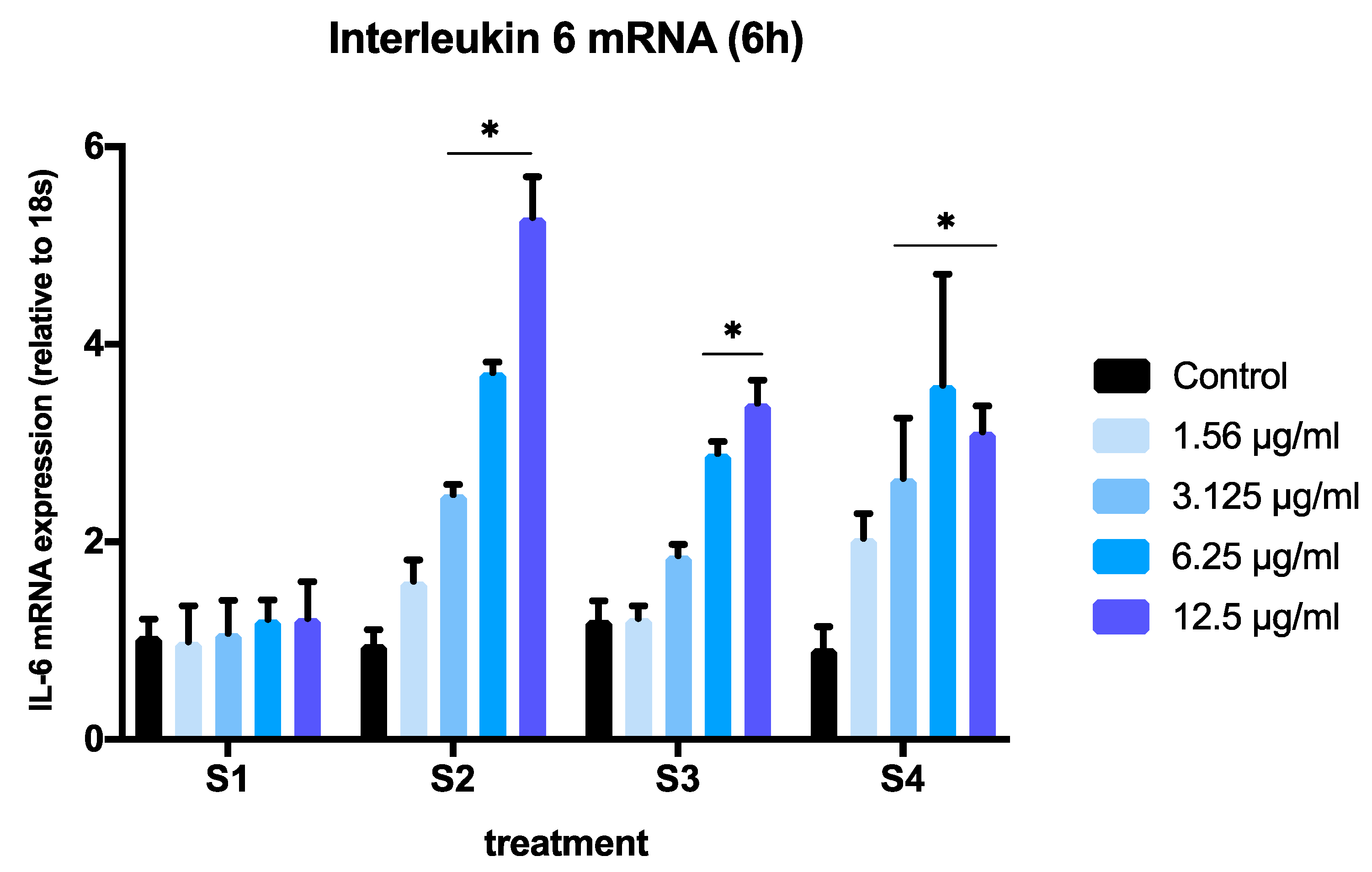
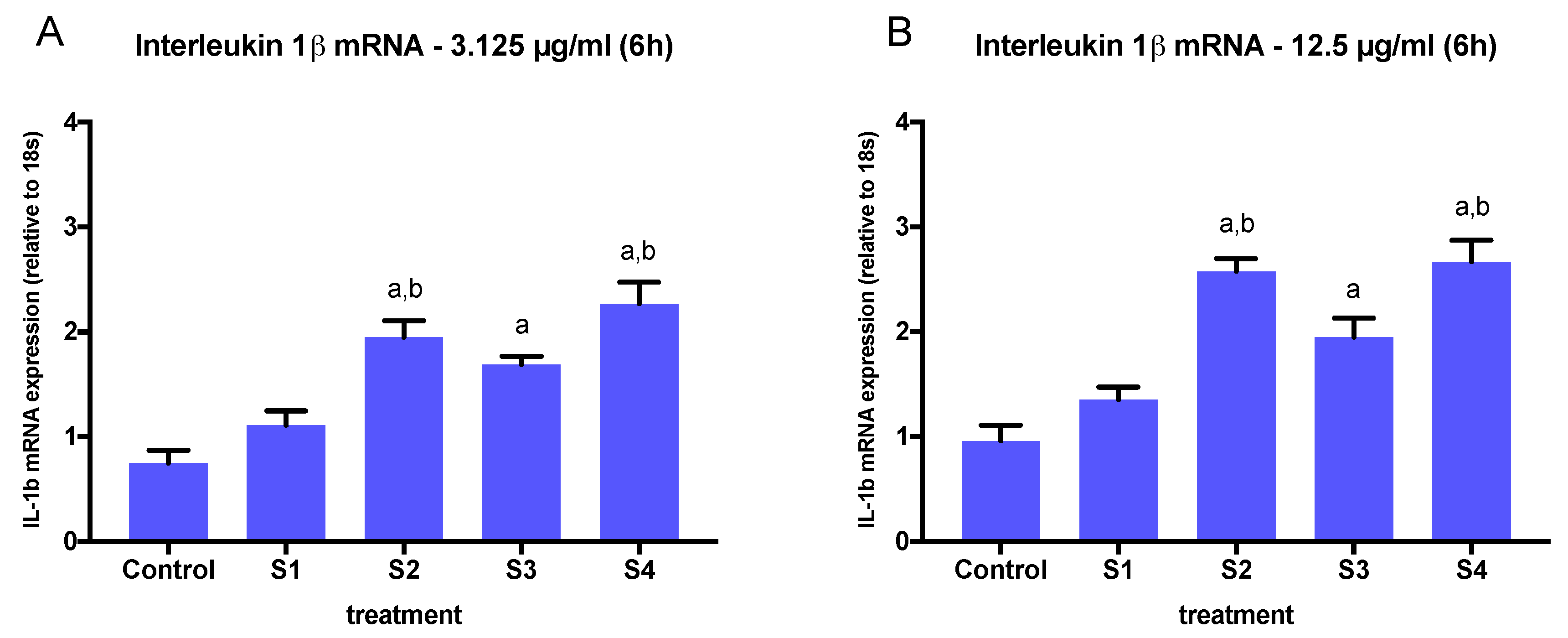
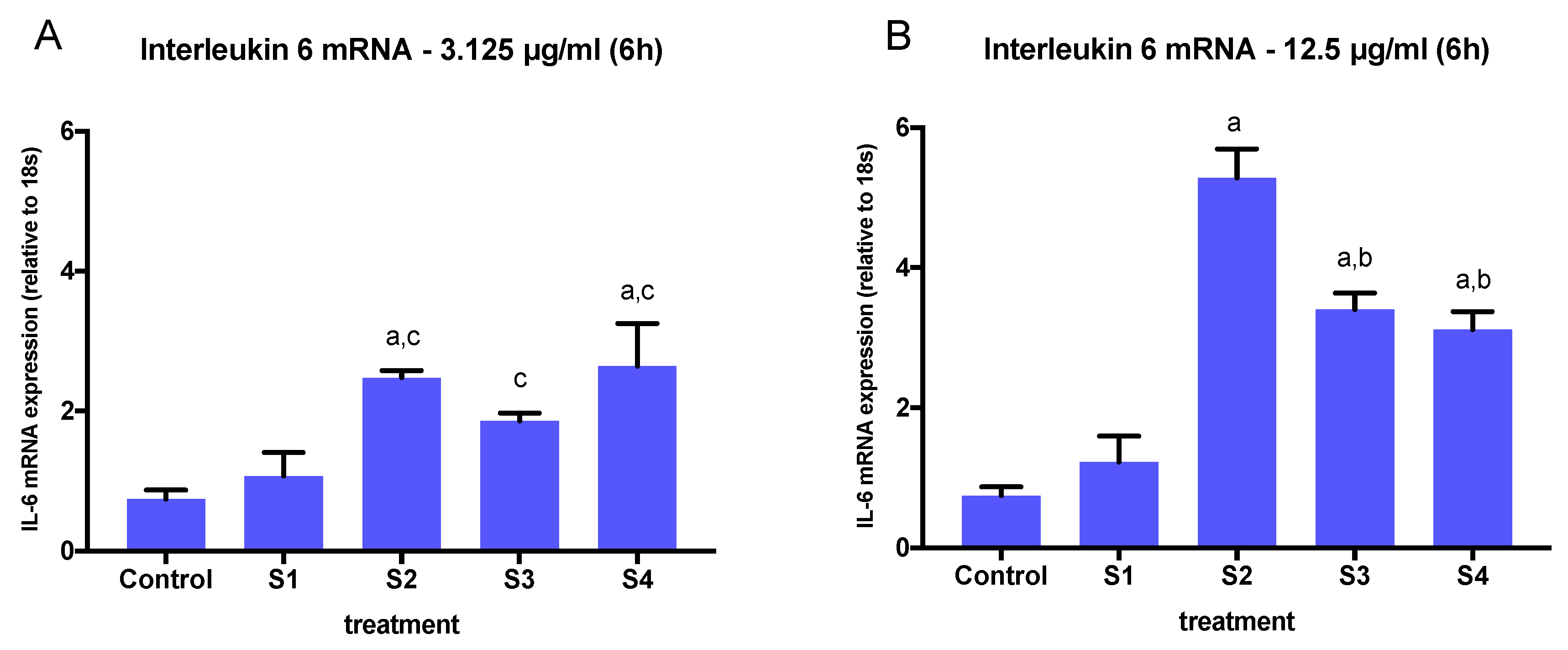
3.3. Expression of Pro-Inflammatory Genes in Cells Exposed to Synthetic Soot
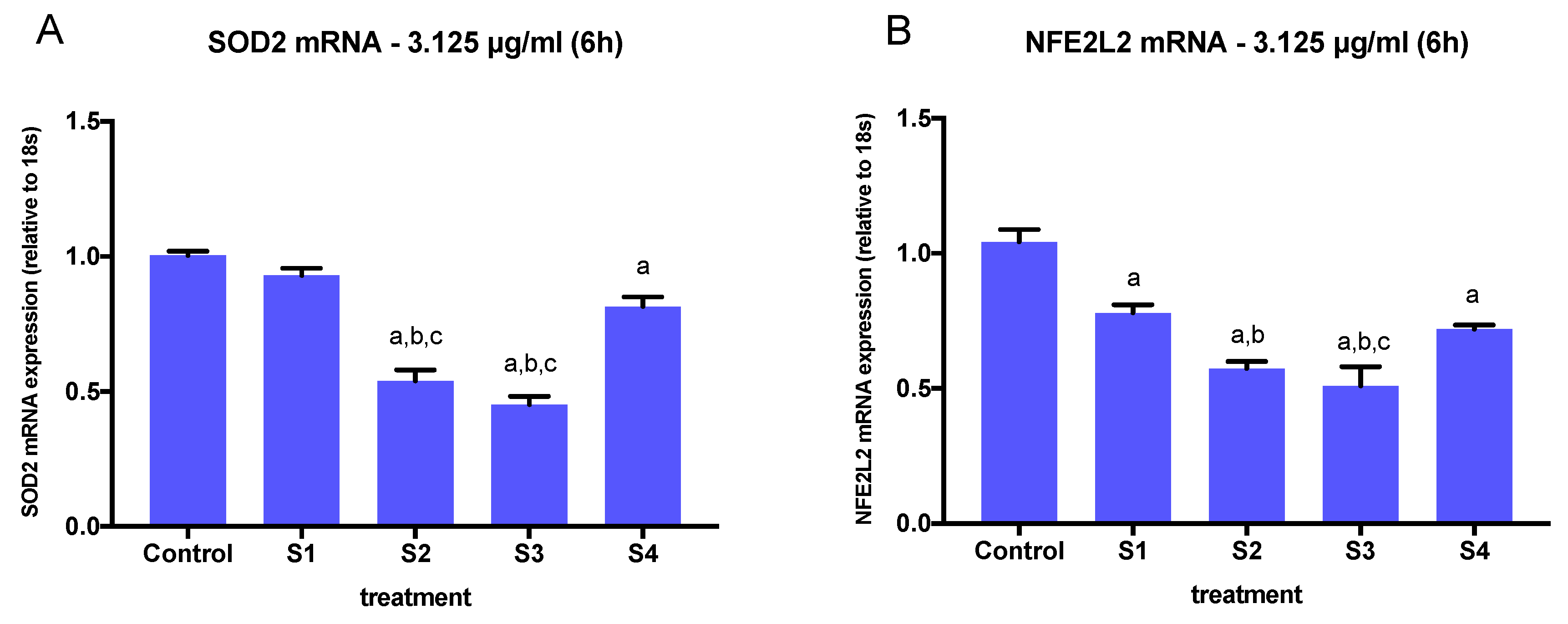
3.4. Expression of Oxidative Stress-Related Genes in Cells Exposed to Synthetic Soot
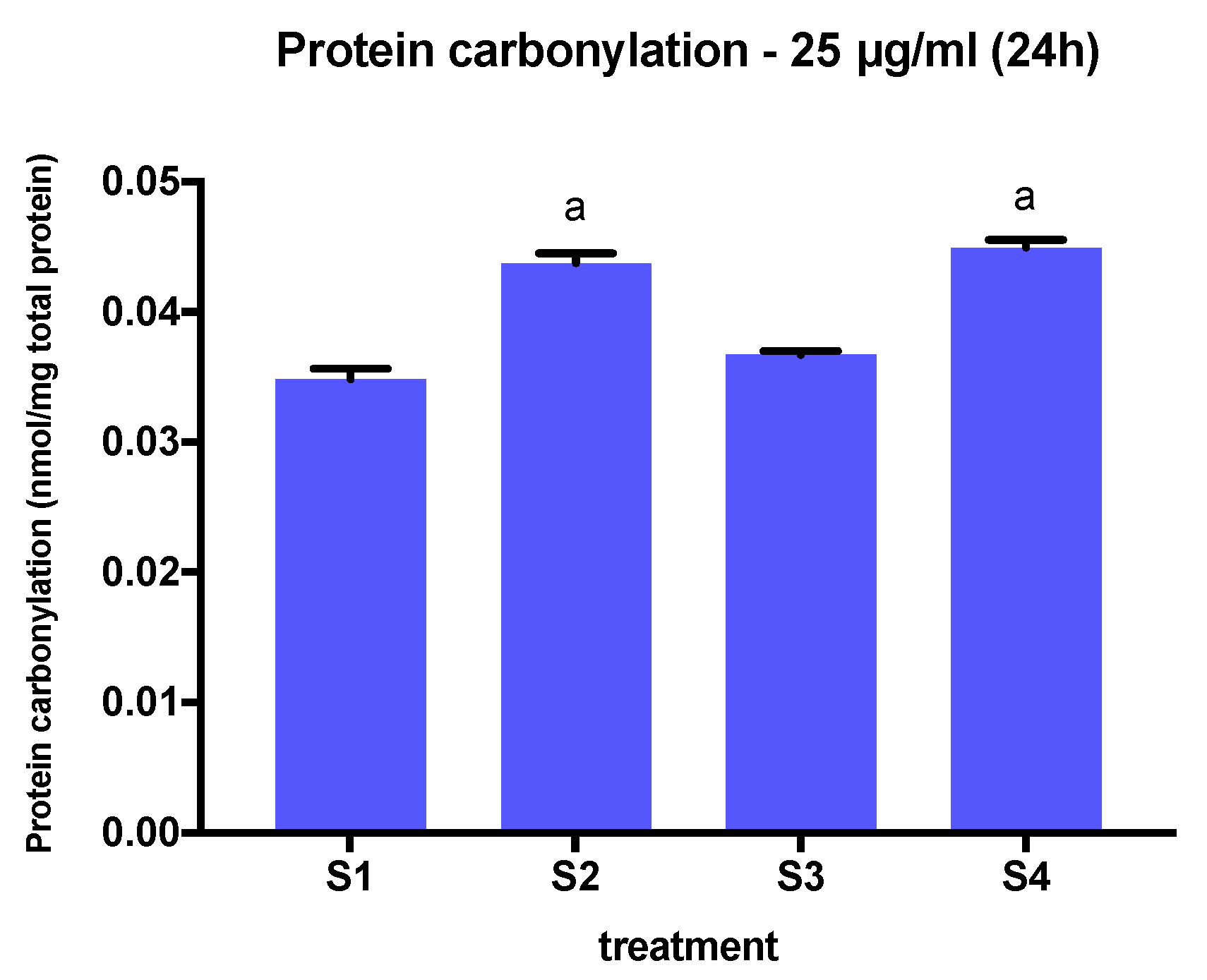
3.5. Protein Carbonylation in Cells Exposed to Synthetic Soot
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, R.; Wang, G.; Guo, S.; Zamora, M.; Ying, Q.; Lin, Y.; Wang, W.; Hu, M.; Wang, Y. Formation of Urban Fine Particulate Matter. Chem. Rev. 2015, 115, 3803–3855. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Zanobetti, A.; Koutrakis, P.; Schwartz, J.D. Associations of fine particulate matter species with mortality in the united states: A multicity time-series analysis. Environ. Health Perspect. 2014, 122, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, J.; Pozzer, A.; Pöschl, U.; Fnais, M.; Haines, A.; Münzel, T. Loss of life expectancy from air pollution compared to other risk factors: A worldwide perspective. Cardiovas. Res. 2020. [Google Scholar] [CrossRef]
- Babatola, S.S. Global burden of diseases attributable to air pollution. J. Public Health Afr. 2018, 9, 813. [Google Scholar] [CrossRef] [Green Version]
- Schwarze, P.E.; Ovrevik, J.; Låg, M.; Refsnes, M.; Nafstad, P.; Hetland, R.B.; Dybing, E. Particulate matter properties and health effects: Consistency of epidemiological and toxicological studies. Hum. Exp. Toxicol. 2006, 25, 559–579. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.L.; Dominici, F.; Ebisu, K.; Zeger, S.L.; Samet, J.M. Spatial and temporal variation in PM2.5 chemical composition in the United States for health effects studies. Environ. Health Perspect. 2007, 115, 989–995. [Google Scholar] [CrossRef] [Green Version]
- Cassee, F.R.; Héroux, M.E.; Gerlofs-Nijland, M.E.; Kelly, F.J. Particulate matter beyond mass: Recent health evidence on the role of fractions, chemical constituents and sources of emission. Inhal. Toxicol. 2013, 25, 802–812. [Google Scholar] [CrossRef]
- Xing, Y.F.; Xu, Y.H.; Shi, M.H.; Lian, Y.X. The impact of PM2.5 on the human respiratory system. J. Thorac. Dis. 2016, 8, E69–E74. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, C.; Ji, G.; Shao, W.; Zhang, C.; Gu, A.; Zhao, P. Effect of exposure to ambient PM2.5 pollution on the risk of respiratory tract diseases: A meta-analysis of cohort studies. J. Biomed. Res. 2017, 31, 130–142. [Google Scholar] [CrossRef]
- Du, Y.; Xu, X.; Chu, M.; Guo, Y.; Wang, J. Air particulate matter and cardiovascular disease: The epidemiological, biomedical and clinical evidence. J. Thorac. Dis. 2016, 8, E8–E19. [Google Scholar] [CrossRef]
- Li, D.; Li, Y.; Li, G.; Zhang, Y.; Li, J.; Chen, H. Fluorescent reconstitution on deposition of PM 2.5 in lung and extrapulmonary organs. Proc. Natl. Acad. Sci. USA 2019, 116, 2488–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourdrel, T.; Bind, M.A.; Béjot, Y.; Morel, O.; Argacha, J.F. Cardiovascular effects of air pollution. Arch. Cardiovasc. Dis. 2017, 110, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.; Woodruff, T.J.; Parker, J.D.; Schoendorf, K.C. Relationships between air pollution and preterm birth in California. Paediatr. Perinat. Epidemiol. 2006, 20, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Holgate, S.T. Mechanisms of particulate matter toxicity. Clin. Exp. Allergy 1999, 29, 1187–1194. [Google Scholar] [CrossRef]
- Martinelli, N.; Girelli, D.; Cigolini, D.; Sandri, M.; Ricci, G.; Rocca, G.; Olivieri, O. Access rate to the emergency department for venous thromboembolism in relationship with coarse and fine particulate matter air pollution. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, B.F.A.; Ignotti, E.; Artaxo, P.; Do Nascimento Saldiva, P.H.; Junger, W.L.; Hacon, S. Risk assessment of PM2.5 to child residents in Brazilian Amazon region with biofuel production. Environ. Health A Glob. Access Sci. Source 2012, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.F. Cytokines in chronic obstructive pulmonary disease. Eur. Respir. J. 2001, 18, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Delfino, R.J.; Staimer, N.; Tjoa, T.; Arhami, M.; Polidori, A.; Gillen, D.L.; George, S.C.; Shafer, M.M.; Schauer, J.J.; Sioutas, C. Associations of primary and secondary organic aerosols with airway and systemic inflammation in an elderly panel cohort. Epidemiology 2010, 21, 892–902. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Qiu, X.; Hu, X.; Shang, Y.; Pardo, M.; Fang, Y.; Wang, J.; Rudich, Y.; Zhu, T. Effects on IL-1Β signaling activation induced by water and organic extracts of fine particulate matter (PM2.5) in vitro. Environ. Pollut. 2018, 237, 592–600. [Google Scholar] [CrossRef]
- Fujii, T.; Hayashi, S.; Hogg, J.C.; Vincent, R.; Van Eeden, S.F. Particulate matter induces cytokine expression in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2001, 25, 265–271. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med. 2003, 9, 169–176. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation.; cellular dysfunction.; and disease progression. J. Cell Mol. Med. 2006, 10, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Piao, M.J.; Ahn, M.J.; Kang, K.A.; Ryu, Y.S.; Hyun, Y.J.; Shilnikova, K.; Zhen, A.X.; Jeong, J.W.; Choi, Y.H.; Kang, H.K.; et al. Particulate matter 2.5 damages skin cells by inducing oxidative stress, subcellular organelle dysfunction, and apoptosis. Arch. Toxicol. 2018, 92, 2077–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aztatzi-Aguilar, O.; Valdés-Arzate, A.; Debray-García, Y.; Calderón-Aranda, E.; Uribe-Ramirez, M.; Acosta-Saavedra, L.; Gonsebatt, M.E.; Maciel-Ruiz, J.A.; Petrosyan, P.; Mugica-Alvarez, V.; et al. Exposure to ambient particulate matter induces oxidative stress in lung and aorta in a size- and time-dependent manner in rats. Toxicol. Res. Appl. 2018, 2. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, J.L.; Pereira, M.F. The role of surface chemistry in catalysis with carbons. Catal. Today 2010, 150, 2–7. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Longhin, E.; Holme, J.A.; Gualtieri, M.; Camatini, M.; Øvrevik, J. Milan winter fine particulate matter (wPM2.5) induces IL-6 and IL-8 synthesis in human bronchial BEAS-2B cells, but specifically impairs IL-8 release. Toxicol. Vitr. 2018, 52, 365–373. [Google Scholar] [CrossRef]
- Hong, Z.; Guo, Z.; Zhang, R.; Xu, J.; Dong, W.; Zhuang, G.; Deng, C. Airborne Fine Particulate Matter Induces Oxidative Stress and Inflammation in Human Nasal Epithelial Cells. Tohoku J. Exp. Med. 2016, 239, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Rangasamy, T.; Guo, J.; Mitzner, W.A.; Roman, J.; Singh, A.; Fryer, A.D.; Yamamoto, M.; Kensler, T.W.; Tuder, R.M.; Georas, S.N.; et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 2005, 202, 47–59. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, G.; Yang, Z. Mitochondrial superoxide mediates PM2.5-induced cytotoxicity in human pulmonary lymphatic endothelial cells. Environ. Pollut. 2020, 263. [Google Scholar] [CrossRef]
- World Health Organization. Ambient Air Pollution: A Global Assessment of Exposure and Burden of Disease. World Health Organization. 2016. Available online: http://apps.who.int/iris/bitstream/10665/250141/1/9789241511353-eng.pdf (accessed on 6 August 2020).
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: An analysis of data from the Global Burden of Diseases Study 2015. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef] [Green Version]
- Pope, C.A.; Bhatnagar, A.; McCracken, J.P.; Abplanalp, W.; Conklin, D.J.; O’Toole, T. Exposure to Fine Particulate Air Pollution Is Associated With Endothelial Injury and Systemic Inflammation. Circ. Res. 2016, 119, 1204–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, S.; Bisig, C.; Petri-Fink, A.; Rothen-Rutishauser, B. Diesel exhaust: Current knowledge of adverse effects and underlying cellular mechanisms. Arch. Toxicol. 2016, 90, 1541–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, S.; Heeb, N.V.; Czerwinski, J.; Comte, P.; Mayer, A.; Petri-Fink, A.; Rothen-Rutishauser, B. Test-methods on the test-bench: A comparison of complete exhaust and exhaust particle extracts for genotoxicity/mutagenicity assessment. Environ. Sci. Technol. 2014, 48, 5237–5244. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zanobetti, A.; Kloog, I.; Coull, B.A.; Koutrakis, P.; Melly, S.J.; Schwartz, J.D. Low-Concentration PM2.5 and Mortality: Estimating Acute and Chronic Effects in a Population-Based Study. Environ. Health Perspect. 2016, 124, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Gioda, A.; Fuentes-Mattei, E.; Jimenez-Velez, B. Evaluation of cytokine expression in BEAS cells exposed to fine particulate matter (PM2.5) from specialized indoor environments. Int. J. Environ. Health Res. 2011, 21, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Bauer, R.N.; Diaz-Sanchez, D.; Jaspers, I. Effects of air pollutants on innate immunity: The role of Toll-like receptors and nucleotide-binding oligomerization domain-like receptors. J. Allergy Clin. Immunol. 2012, 129, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Lafferty, E.I.; Qureshi, S.T.; Schnare, M. The role of toll-like receptors in acute and chronic lung inflammation. J. Inflamm. 2010, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Bach, N.S.; Låg, M.; Øvrevik, J. Toll like receptor-3 priming alters diesel exhaust particle-induced cytokine responses in human bronchial epithelial cells. Toxicol. Lett. 2014, 228, 42–47. [Google Scholar] [CrossRef]
- Totlandsdal, A.I.; Refsnes, M.; Låg, M. Mechanisms involved in ultrafine carbon black-induced release of IL-6 from primary rat epithelial lung cells. Toxicol. Vitr. 2010, 24, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Watkins, L.R.; Hansen, M.K.; Nguyen, K.T.; Lee, J.E.; Maier, S.F. Dynamic regulation of the proinflammatory cytokine, interleukin-1beta: Molecular biology for non-molecular biologists. Life Sci. 1999, 65, 449–481. [Google Scholar] [CrossRef]
- Monn, C.; Becker, S. Cytotoxicity and induction of proinflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse particles (PM10-2.5) in outdoor and indoor air. Toxicol. Appl. Pharmacol. 1999, 155, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Pavilonis, B.T.; Anthony, T.R.; O’Shaughnessy, P.T.; Humann, M.J.; Merchant, J.A.; Moore, G.; Thorne, P.S.; Weisel, C.P.; Sanderson, W.T. Indoor and outdoor particulate matter and endotoxin concentrations in an intensely agricultural county. J. Expo. Sci. Environ. Epidemiol. 2013, 23, 299–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamara, M.; Thornburg, J.; Semmens, E.; Ward, T.; Noonan, C. Coarse particulate matter and airborne endotoxin within wood stove homes. Indoor Air 2013, 23, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, L.K.; Thompson-Figueroa, J.; Leclair, T.; Sullivan, M.J.; Poynter, M.E.; Irvin, C.G.; Bates, J.H. Tumor necrosis factor-alpha overexpression in lung disease: A single cause behind a complex phenotype. Am. J. Respir. Crit. Care Med. 2005, 171, 1363–1370. [Google Scholar] [CrossRef] [Green Version]
- Pease, J.E.; Sabroe, I. The role of interleukin-8 and its receptors in inflammatory lung disease: Implications for therapy. Am. J. Respir. Med. 2002, 1, 19–25. [Google Scholar] [CrossRef]
- Cooper, D.M.; Loxham, M. Particulate matter and the airway epithelium: The special case of the underground? Eur. Respir. Rev. 2019, 28, 190066. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Rui, W.; Zhang, F.; Ding, W. PM2.5 induces Nrf2-mediated defense mechanisms against oxidative stress by activating PIK3/AKT signaling pathway in human lung alveolar epithelial A549 cells. Cell Biol. Toxicol. 2013, 29, 143–157. [Google Scholar] [CrossRef]
- Leclercq, B.; Kluza, J.; Antherieu, S.; Sotty, J.; Alleman, L.Y.; Perdrix, E.; Loyens, A.; Coddeville, P.; Guidice, J.-M.L.; Marchetti, P.; et al. Air pollution-derived PM2.5 impairs mitochondrial function in healthy and chronic obstructive pulmonary diseased human bronchial epithelial cells. Environ. Pollut. 2018, 243, 1434–1449. [Google Scholar] [CrossRef]
- Cho, C.C.; Hsieh, W.Y.; Tsai, C.H.; Chen, C.Y.; Chang, H.F.; Lin, C.S. In Vitro and In Vivo Experimental Studies of PM2.5 on Disease Progression. Int. J. Environ. Res. Public Health 2018, 15, 1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.S.; Kim, S.R.; Lee, Y.C. Impact of oxidative stress on lung diseases. Respirology 2009, 14, 27–38. [Google Scholar] [CrossRef]
- Bocci, V.; Valacchi, G.; Corradeschi, F.; Aldinucci, C.; Silvestri, S.; Paccagnini, E.; Gerli, R. Studies on the biological effects of ozone: 7. Generation of reactive oxygen species (ROS) after exposure of human blood to ozone. J. Biol. Regul. Homeost. Agents 1998, 12, 67–75. [Google Scholar] [PubMed]
- Bell, M.L.; Son, J.Y.; Peng, R.D.; Wang, Y.; Dominici, F. Ambient PM2.5 and Risk of Hospital Admissions: Do Risks Differ for Men and Women? Epidemiology 2015, 26, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Yin, P.; Meng, X.; Liu, C.; Wang, L.; Xu, X.; Ross, J.A.; Tse, L.A.; Zhao, Z.; Kan, H.; et al. Fine Particulate Air Pollution and Daily Mortality. A Nationwide Analysis in 272 Chinese Cities. Am. J. Respir. Crit. Care Med. 2017, 196, 73–81. [Google Scholar] [CrossRef]
- Gerlofs-Nijland, M.E.; Totlandsdal, A.I.; Tzamkiozis, T.; Leseman, D.L.; Samaras, Z.; Låg, M.; Schwarze, P.; Ntziachristos, L.; Cassee, F.R. Cell toxicity and oxidative potential of engine exhaust particles: Impact of using particulate filter or biodiesel fuel blend. Environ. Sci. Technol. 2013, 47, 5931–5938. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Sioutas, C.; Cho, A.; Schmitz, D.; Misra, C.; Sempf, J.; Wang, M.; Oberley, T.; Froines, J.; Nel, A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ. Health Perspect. 2003, 111, 455–460. [Google Scholar] [CrossRef]
- Zhu, X.M.; Wang, Q.; Xing, W.W.; Long, M.H.; Fu, W.L.; Xia, W.R.; Jin, C.; Guo, N.; Xu, D.Q.; Xu, D.G. PM2.5 induces autophagy-mediated cell death via NOS2 signaling in human bronchial epithelium cells. Int. J. Biol. Sci. 2018, 14, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Longhin, E.; Holme, J.A.; Gutzkow, K.B.; Arlt, V.M.; Kucab, J.E.; Camatini, M.; Gualtieri, M. Cell cycle alterations induced by urban PM2.5 in bronchial epithelial cells: Characterization of the process and possible mechanisms involved. Part. Fibre Toxicol. 2013, 10, 63. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Nethery, R.C.; Sabath, B.M.; Braun, D.; Dominici, F. Exposure to air pollution and COVID-19 mortality in the United States: A nationwide cross-sectional study. Preprint. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soot | Treatment | Measured Atomic 1 % | |||
|---|---|---|---|---|---|
| C | O | N | S | ||
| S1 | None | 97.2 | 1.3 | -- | 0.9 |
| S2 | HNO3 | 67.8 | 31.5 | 1.3 | -- |
| S3 | Ozone | 90 | 9.4 | -- | 0.6 |
| S4 | HNO3 + 300 °C | 86.5 | 13.2 | -- | 0.2 |
| Soot | Treatment | Oxygen Groups % | |||
|---|---|---|---|---|---|
| C-O | C=O | O-C=O | Total O | ||
| S1 | None | -- | -- | -- | 1.3 |
| S2 | HNO3 | 10.2 | 4.9 | 9.4 | 34.0 |
| S3 | Ozone | 2.2 | 1.4 | 2.4 | 8.3 |
| S4 | HNO3 + 300 °C | 7.3 | 3.8 | 0.5 | 12.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Housseiny, H.; Singh, M.; Emile, S.; Nicoleau, M.; Wal, R.L.V.; Silveyra, P. Identification of Toxicity Parameters Associated with Combustion Produced Soot Surface Chemistry and Particle Structure by in Vitro Assays. Biomedicines 2020, 8, 345. https://doi.org/10.3390/biomedicines8090345
Al Housseiny H, Singh M, Emile S, Nicoleau M, Wal RLV, Silveyra P. Identification of Toxicity Parameters Associated with Combustion Produced Soot Surface Chemistry and Particle Structure by in Vitro Assays. Biomedicines. 2020; 8(9):345. https://doi.org/10.3390/biomedicines8090345
Chicago/Turabian StyleAl Housseiny, Heba, Madhu Singh, Shaneeka Emile, Marvin Nicoleau, Randy L. Vander Wal, and Patricia Silveyra. 2020. "Identification of Toxicity Parameters Associated with Combustion Produced Soot Surface Chemistry and Particle Structure by in Vitro Assays" Biomedicines 8, no. 9: 345. https://doi.org/10.3390/biomedicines8090345
APA StyleAl Housseiny, H., Singh, M., Emile, S., Nicoleau, M., Wal, R. L. V., & Silveyra, P. (2020). Identification of Toxicity Parameters Associated with Combustion Produced Soot Surface Chemistry and Particle Structure by in Vitro Assays. Biomedicines, 8(9), 345. https://doi.org/10.3390/biomedicines8090345