1. Introduction
The musculoskeletal system consists principally of bones, muscles, tendons, ligaments, and articular cartilage. These are arranged throughout the body and physically and mechanically interact to assure the integrated motor activity and the musculoskeletal metabolism/homeostasis [
1]. In particular, the musculoskeletal system’s primary function includes supporting the body, allowing movement, and protecting vital organs. In this system, bones relate to other bones and muscles via connective tissue such as tendons and ligaments. Bones provide stability and muscles keep bones in place and allow the motor activity. Moreover, the skeletal portion of the system has the role to store calcium and phosphorus and contains the hematopoietic system [
2].
There are several diseases and disorders that may affect this system, and often they are very difficult to diagnose due to the close relation of the musculoskeletal system to other internal systems. Musculoskeletal conditions range from those that arise in an acute manner and are short-lived, such as fractures, to lifelong pathologies associated with chronic pain and disability such as genetic diseases (achondroplasia, osteogenesis imperfecta, muscular dystrophy, and myopathies) [
3,
4], metabolic diseases such as osteoporosis and osteopenia [
5], tumors of the bones and the muscle such as osteosarcoma and rhabdomyosarcoma [
6], joint diseases such as osteoarthritis and rheumatoid arthritis [
7], and aging in general characterized by bone and muscle weakness (osteopenia and sarcopenia) [
8,
9]. All these diseases comprise dysregulation of the physiology of all the tissues among the musculoskeletal system; therefore, a better understanding of the molecular mechanisms responsible for the crosstalk among these tissues is needed. In particular, the condition related to musculoskeletal aging is one of the major public health interests and is typical of the demographic changes in the population. It is associated with a high risk of fractures and joint and muscle hypofunctionality with consequent loss of autonomy in elderly people, and this condition is therefore correlated with high morbidity and healthcare rates [
10,
11]. Sarcopenia and osteoporosis are linked and commonly associated with aging, often leading to a frailty syndrome, a pathological condition characterized by a general decline, which includes multifactorial causes [
12,
13].
During the last decade, bone and muscle were increasingly recognized as interacting tissues, not only because of their proximity and their integrated function for motor activity, but also for the fact to be recognized as endocrine target tissues and to be endocrine organs themselves [
14]. In fact, the two tissues interact with each other by paracrine and endocrine signals and modulate their mutual development and function since intrauterine life to oldness [
15]. The muscle–bone crosstalk is supported by preclinical and clinical data, showing the presence of many tissue-specific factors released by osteoblasts and osteocytes, including prostaglandin E
2, osteocalcin, and IGF-1, which have a potential impact on skeletal muscle cells. Moreover, a growing number of muscle-released factors with bone-modulating properties have been identified. These include insulin-like growth factor-1 (IGF-1), fibroblast growth factor-2, IL-6, IL-15, myostatin, osteoglycin, irisin, and osteoactivin, which have been initially implicated in the pathogenesis of sarcopenia but appear to also be regulators of bone remodeling and thus potentially relevant for osteoporosis [
16,
17,
18,
19,
20,
21,
22]. However, despite this recent effort, limited data exist about the molecular basis controlling the muscle-to-bone crosstalk.
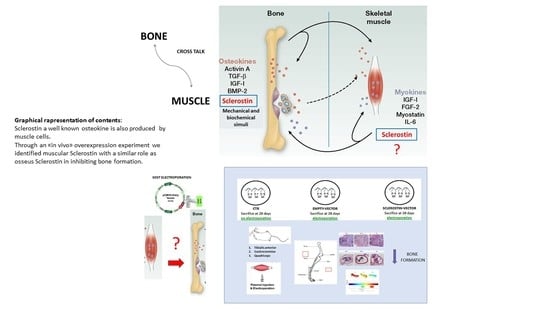
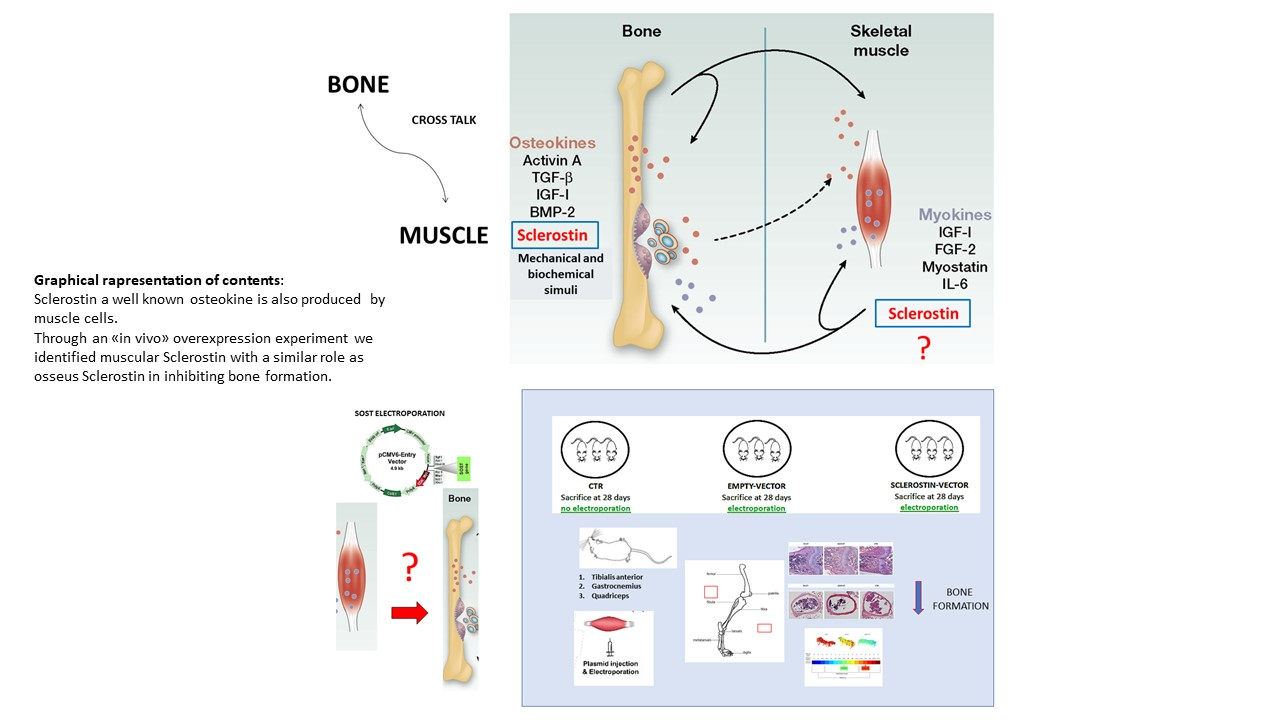
In our study, for the first time, we clearly identified a new putative myokine, sclerostin, that until recently had been described as a potent inhibitor of bone formation produced mainly by osteocytes [
23,
24]. Our data demonstrate the expression of sclerostin both in muscle cells in vitro and in muscles collected from variously aged mice and differing in metabolic and load-bearing features. Moreover, through a functional approach, we investigated the role of muscle-originated sclerostin in bone formation. In conclusion, this study highlights, for the first time, skeletal muscle as a new source of sclerostin. Sclerostin produced at the muscular level may act in combination with bone sclerostin in severe conditions of increased bone fragility. This discovery is particularly relevant for the development of treatments for pathophysiological conditions characterized by simultaneous bone and muscle loss, such as aging-associated osteoporosis/sarcopenia or forced immobilization. It is of note that this new finding could also lead to new paths in the knowledge of the mechanisms underlying genetic diseases such as neuromuscular diseases or cancers where both muscle and bone tissues are severely affected. Targeting pathways that regulate bone and muscle systemically and/or through the manipulation of more localized pathways that facilitate communication between the two tissues are the privileged directions for the identification of new molecules, which could simultaneously prevent, reduce, or restore bone and muscle age-related deterioration.
2. Materials and Methods
2.1. Cell Cultures
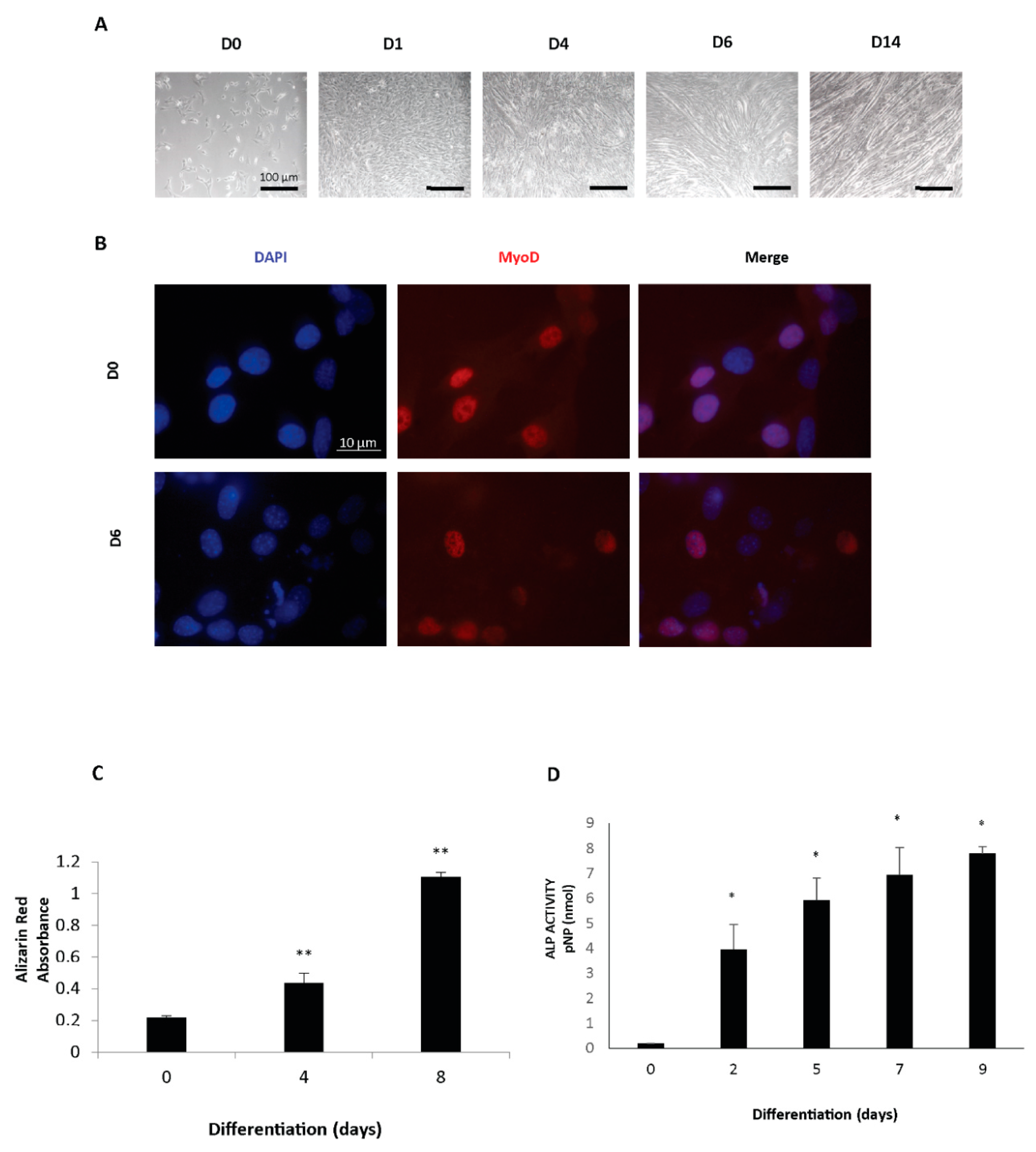
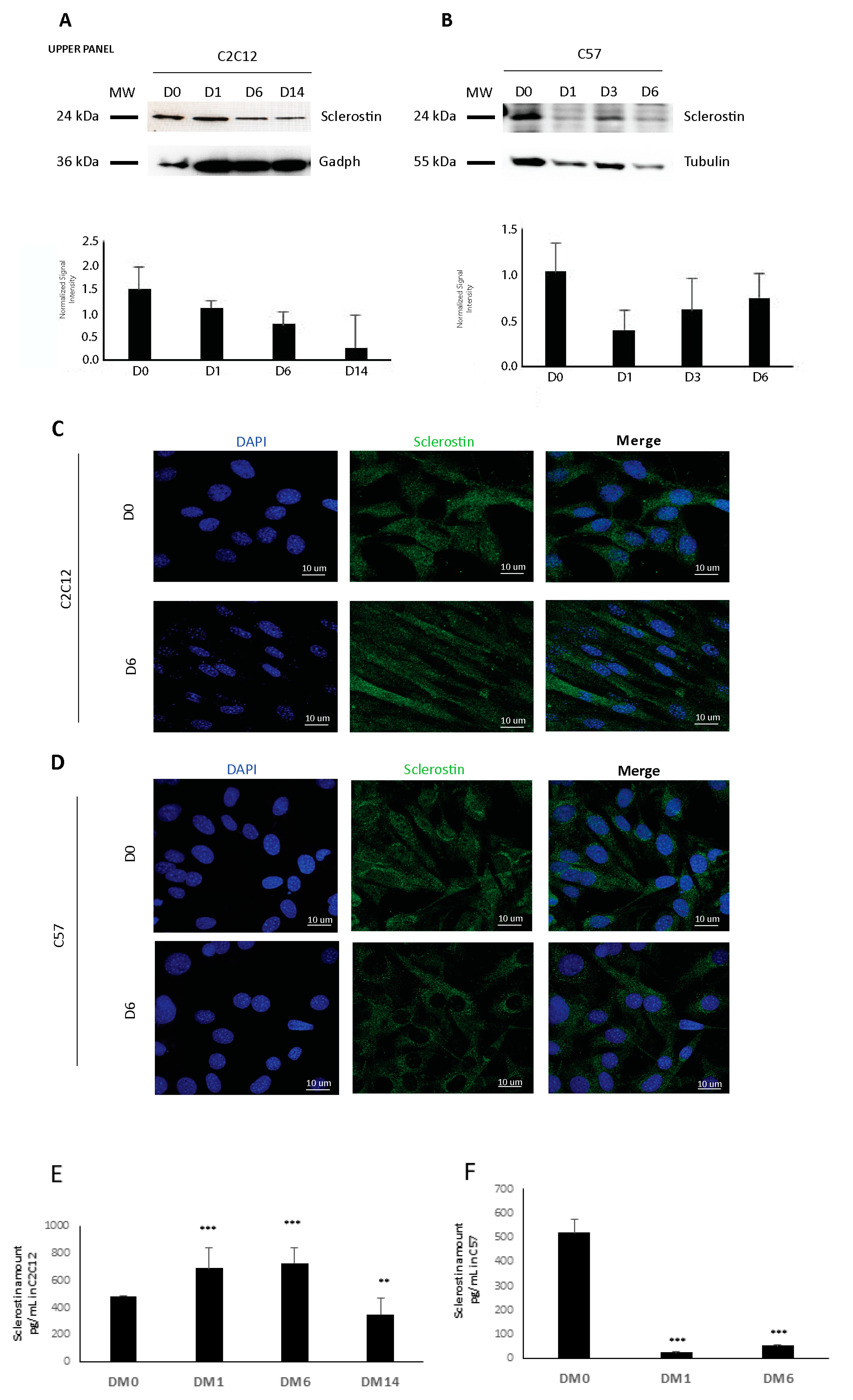
C2C12 myoblasts (CRL-1772; ATCC) were cultured in a growth medium (GM) composed of DMEM containing 10% FBS, 1% penicillin/streptomycin (P/S), and 2 mM L-glutamine in a 5% CO2 atmosphere at 37 °C. To induce myogenic differentiation, the cells were plated on tissue culture plates and grown to 95% confluence before switching to the differentiation medium (DM) (DMEM with 5 μg/mL insulin). The cells were replenished with fresh DM every 48 h to induce differentiation until 14 days.
Osteoblastic cell line 2T3 provided by Lynda Bonewald (Indiana University School of Medicine, Indianapolis, IN, USA) was cultured in a complete medium composed of a MEM supplemented with 10% FBS, 2 mM L-glutamine, and 1% P/S. For osteogenic differentiation, a complete medium supplemented with 50 μg/mL ascorbic acid and 4 mM β-glycerophosphate was used. The differentiation medium was changed every two days of culture.
C57 primary myoblasts were isolated from limb muscles of young (14–30 days old) mice previously injured with a 30-gauge needle and dissociated to isolate pure populations of myoblasts as described previously [
25]. Primary cultures were plated on 5 μg/mL laminin-1/collagen-coated dishes and amplified in a growth medium (GM) consisting of Ham’s F-12 with 20% FBS, 2.5 ng/mL basic fibroblast growth factor, 2 mM L-glutamine, and 1% P/S.
To induce differentiation, myoblast cultures were seeded on coated dishes at 60% confluence and the growth medium was switched to a differentiation medium (DM) consisting of DMEM with 2% horse serum, L-glutamine, and P/S. The timepoints analyzed were days 1, 3, and 6 of differentiation. All tissue culture reagents were from Thermo Fisher, Waltham, MA, USA.
2.2. Animal Procedures
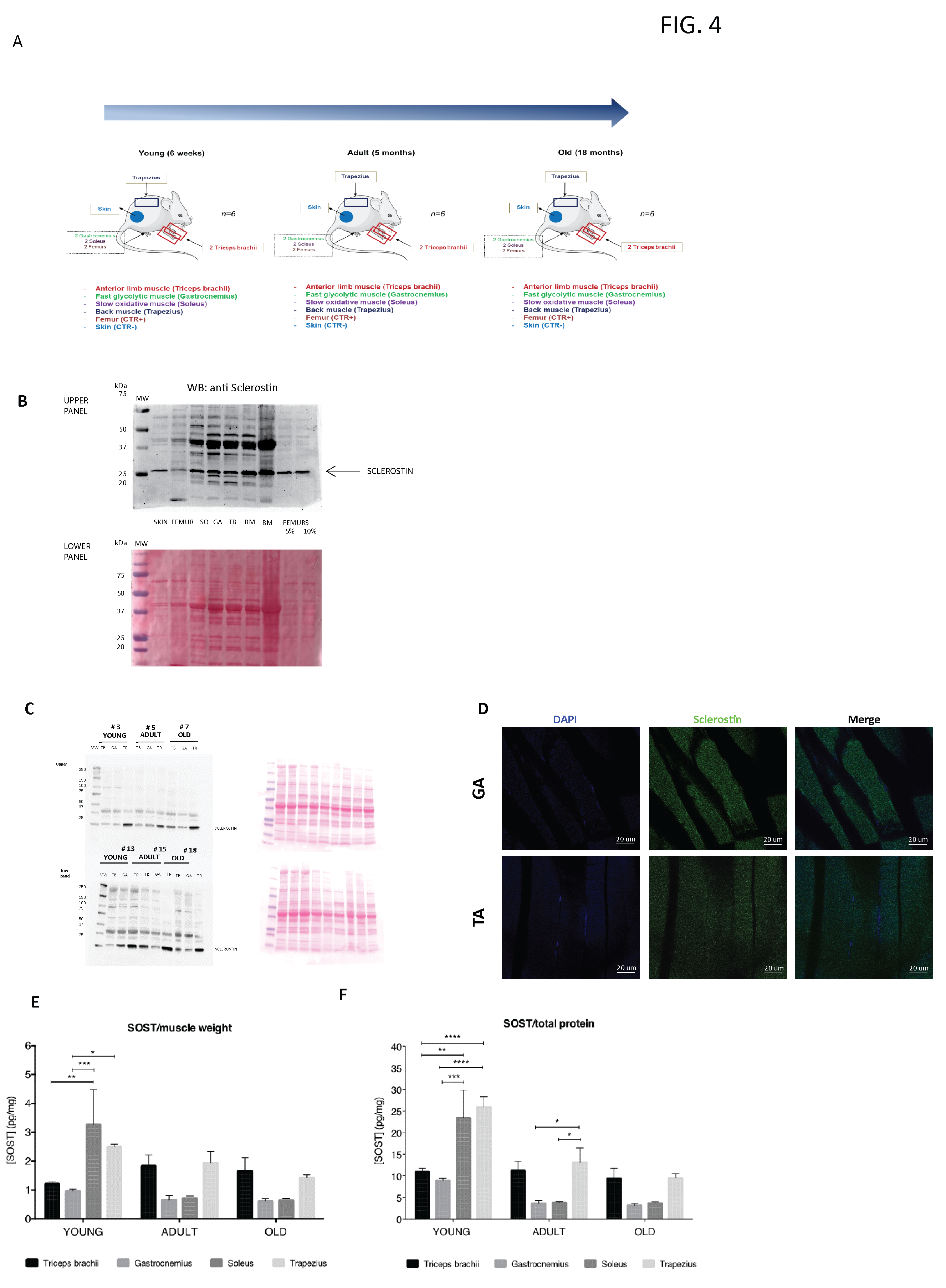
In vivo experiments of sclerostin characterization took place in the conventional animal facility of Bordeaux University (agreement number 063-917 obtained 7 May 2018). Eighteen male C57BL/6 mice at the age of 6 weeks (young; n = 6), 5 months (adult; n = 6) and 18 months (old; n = 6) were purchased from Janvier Labs (Saint-Berthevin, France). Animal housing and caretaking were provided by the animal facility in accordance with the national guidelines. All animal procedures were performed in accordance with the European Guidelines for Care and Use of Laboratory Animals.
Mice were kept under a 12 h–12 h light/dark cycle and the temperature was controlled. After the first week of adaptation, animals were euthanized by cervical dislocation and the following muscles were dissected: Gastrocnemius, Soleus, spinotrapezius, and triceps brachii.
Each selected muscle and its contralateral were withdrawn: one was snap-frozen for molecular biology assays and the other was fixed in 4% paraformaldehyde (PFA) for histology.
2.3. In Vivo Gene Transfer and Electroporation Procedures
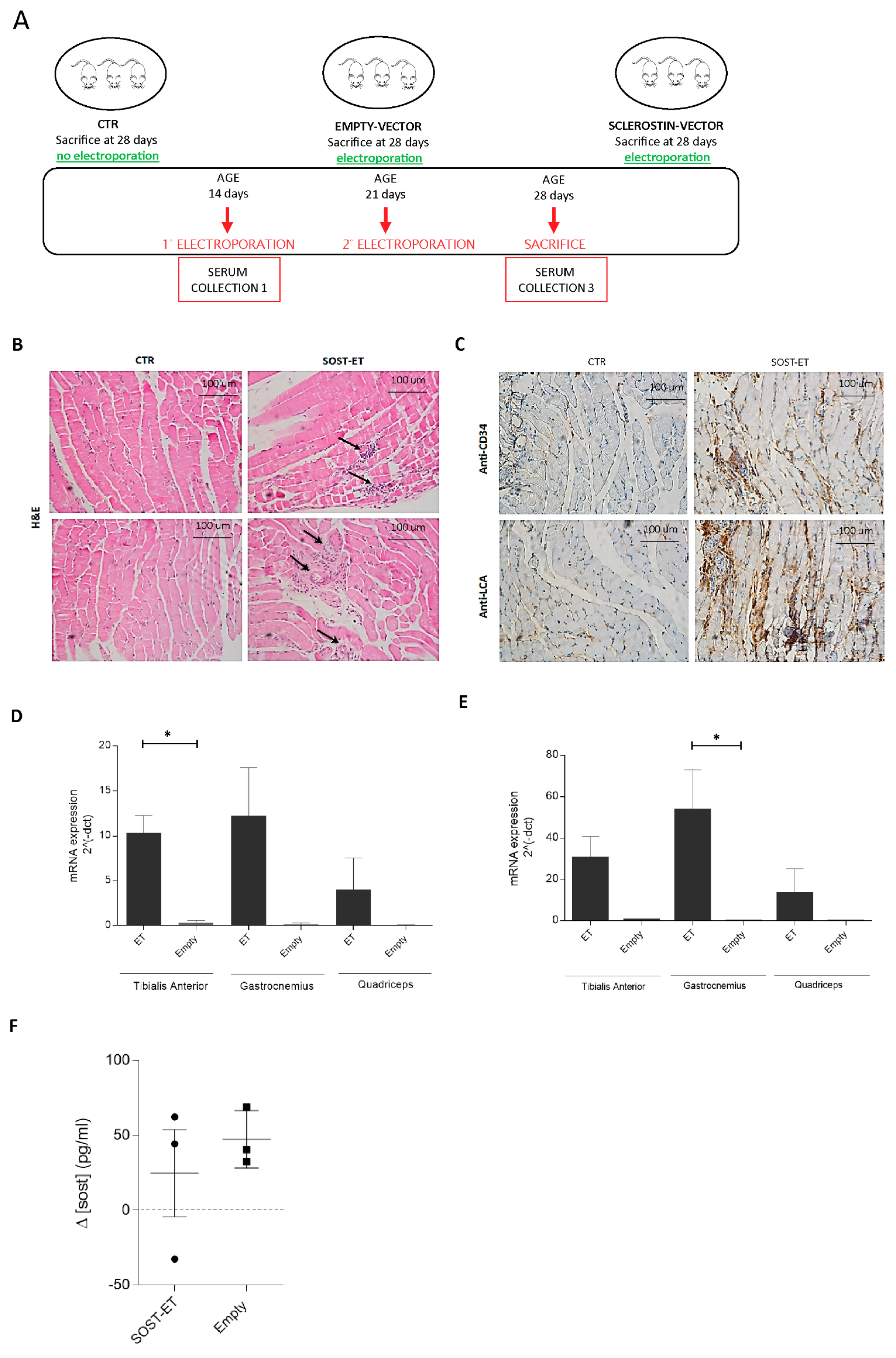
Eight male C57BL/6 mice (Charles River) all belonging to the same progeny were housed in the institutional animal facility (Department of Cellular, Computational and Integrative Biology (CIBIO), University of Trento, Trento, Italy) and maintained on standard chow ad libitum. Three animals (n = 3) were electroporated with the SOST vector, three (n = 3)—with the empty vector, and two (n = 2) were non-electroporated (CTRs). The pCMV6 expression plasmid (MR222588, Origene, Rockville, MD, USA) containing the SOST gene tagged with Myc-DDK was amplified in Escherichia coli.
Eleven days after birth, mice were anesthetized by inhaled isoflurane and plasmid DNA was injected into the tibialis anterior (TA), quadriceps femoris (QF), and gastrocnemius (GA) muscles with a 0.3 mL insulin syringe through a 31-gauge needle in a constant volume of ~40 μL. Each animal received ~240 γ of plasmid DNA (80 γ/site). A pair of stainless steel plates was then applied over the muscle to encompass the injection area. In all the cases, the distance between the electrodes was 4–4.5 mm. Current was delivered 5 min after DNA injection as a constant current, square wave pulse with a digital stimulator (Panlab 3100, Biological Instruments, Harvard Apparatus, Holliston, MA, USA). The characteristics of the electric field applied were 200 V/cm, 20 ms amplitude, 1 Hz, eight consecutive pulses. The animals were then kept warm until recovery.
The electroporations were performed at days 11 and 17 of age; the animals were then sacrificed at 25 days of age. At each timepoint, animal sera were collected.
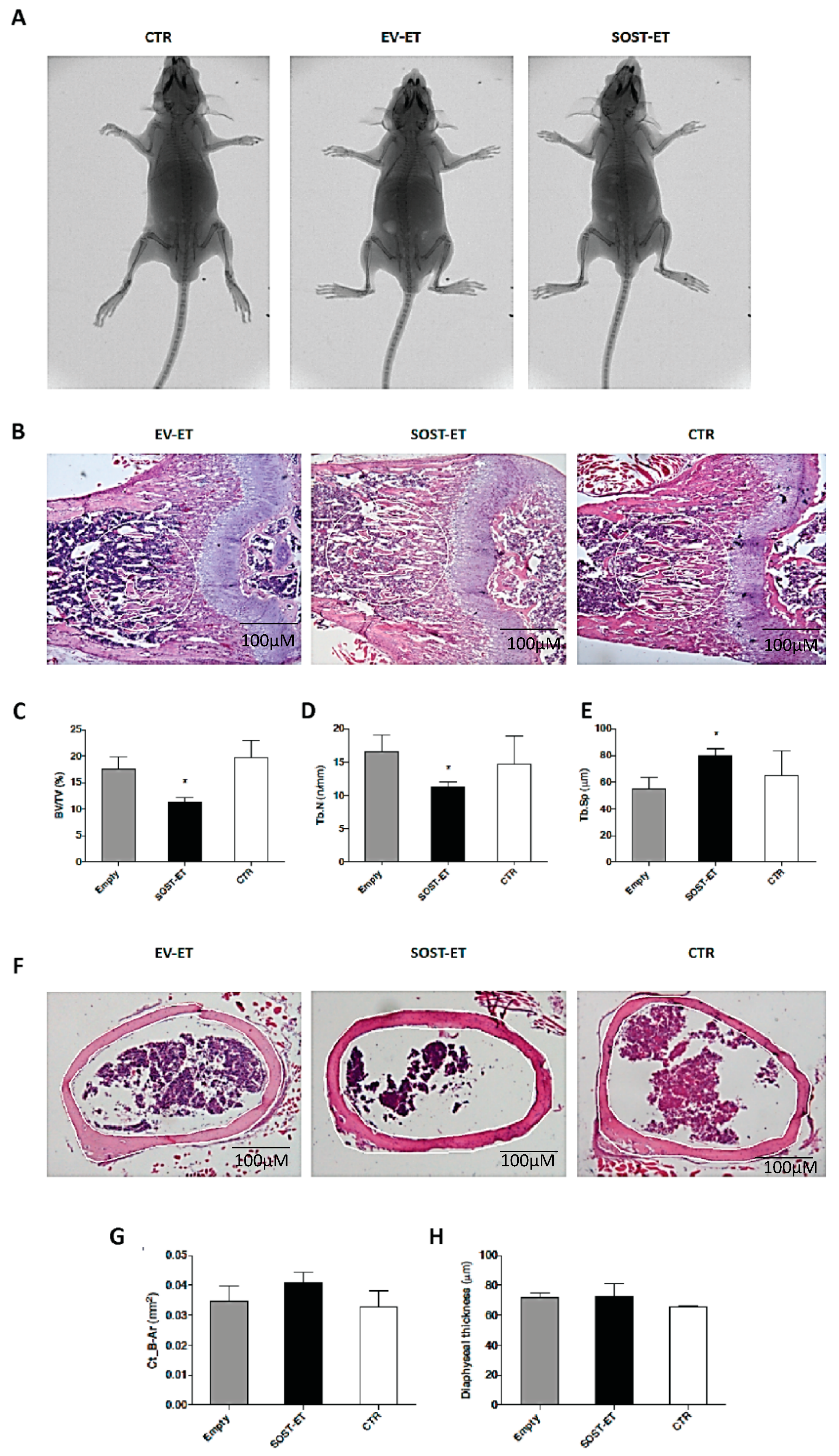
Shortly after sacrifice, the animals were X-ray scanned using an In-Vivo Xtreme-Bruker instrument. Animal studies were approved by the institutional Animal Use and Welfare Committee and the National Ministry of Health (protocol No. 62/2020-PR, granted on the 29 January 2020).
2.4. Western Blot
At the scheduled timepoints, the cells were lysed with the lysis buffer (10 mM Tris-Cl, pH 7.4–7.6, 1% NP-40, 150 mM NaCl, 10% glycerol, 10 mM EDTA, 20 mM NaF, 5 mM sodium pyrophosphate, 1 mM Na3VO4, and freshly added protease inhibitor cocktail) at 4 °C for 20 min.
Proteins from animal tissues were extracted by addition of the AT buffer sonicated 3 times for 10 s each and incubated for 4 h (muscle) or 24 h (bone) at 4 °C with agitation.
Proteins were resolved by SDS-PAGE, then transferred to 0.2 μm nitrocellulose membranes and probed with the following specific primary antibodies: sclerostin (ABCAM ab63097), tubulin (Sigma Aldrich, St. Louis, MO, USA, T5168), GAPDH (SIGMA G9545). Then, the membranes were washed with Tris-buffered saline Tween (TBST) three times and incubated with specific HRP-conjugated secondary antibodies. Finally, the membranes were treated with enhanced chemiluminescence (ECL) reagents (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. Images were visualized using Image LabTM Software (Bio-Rad, Hercules, CA, USA).
2.5. RNA Extraction and Quantitative Real-Time PCR
Total RNA was extracted with an RNeasy kit (Qiagen, Hilden, Germany) or TRIzol (Thermo Fisher Scientific, Waltham, MA, USA) and quantified by spectrophotometry with a NanoDrop 2000 device (Thermo Fisher Scientific, Waltham, MA, USA). Then, 500 ng of the total RNA were reverse-transcribed using an iScriptTM cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Levels of mRNA were quantitatively determined on a CFX96 TouchTM Real-Time PCR Detection System using an SsoFast™ EvaGreen Supermix according to the manufacturer’s instructions (Bio-Rad, Hercules, CA, USA). PCR primer sequences were as follows: Runx2 forward primer 5′-TTAATCCACAAGGACAGA-3′, Runx2 reverse primer 3′-GTAAGACTGGTCATAGGA-5′; Wisp2 forward primer 5′-TGTGACCAGGCAGTGATG-3′, Wisp2 reverse primer 5′-AGTGACAAGGGCAGAAAGT-3′; Hprt forward primer 5′-GGCATTGCTCTCTCAATGACAA-3′, Hprt reverse primer 5′-ATGTAGGCCATGAGGTCCAC-3′.
For the “In vivo SOST overexpression” experiment (electroporation procedure), the animal tissues (muscles) were probed with sclerostin forward primer 5′-AACAACCAGACCATGAAC-3′, sclerostin reverse primer 5′-TACTCGGACACATCTTTG-3′ (to detect the endogenous sclerostin); sclerostin Myc-Tag forward primer 5′-CGGAGCTGGAGAACGCCT-3′, sclerostin Myc-Tag reverse primer 5′-AATCCAGGATATCATTTGCTGCC-3′ (to detect the exogenous Myc-tagged sclerostin); Hprt forward primer 5′-TCA GAC CGC TTT TTG CCG CGA-3′, Hprt reverse primer 5′-ATC GCT AAT CAC GAC GCT GGG AC-3′.
Relative gene expression values were calculated by applying the 2−∆∆Ct method [
26]: using this method, we obtained the fold changes in gene expression normalized to the internal control gene Hprt.
2.6. Alizarin Red Staining and Mineralization
Matrix mineralization was evaluated by Alizarin Red S (ARS) staining. The cells were fixed in 4% paraformaldehyde for 20 min at room temperature. Next, the cell layer was stained with 40 mM Alizarin S (Sigma Aldrich, St. Louis, MO, USA - A5533) at pH ~ 4.2 for 5 min under gentle agitation. Cell preparations were washed with PBS to eliminate non-specific staining. To assess the Alizarin Red S staining, 10% cetylpyridinium chloride was added into each well and the cells were incubated for 20 min. Optical density was measured by spectrophotometry at 570 nm.
2.7. Alkaline Phosphatase (ALP) Assay
Alkaline phosphatase (ALP) activity was measured with a Alkaline Phosphatase Assay Kit (colorimetric) (Abcam). The staining procedures were performed according to the manufacturer’s suggested protocols. The ALP activity was assessed using p-nitrophenol phosphate (pNPP) as the substrate at pH 10.2 by evaluating the optical density of the yellow substance at 405 nm using an iMarkTM Microplate reader (Bio-Rad, Hercules, CA, USA).
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
Sclerostin levels in blood serum samples, animal tissues, and cell lysates were determined with a quantitative sandwich ELISA. Mouse SOST (EM1815, FineTest, Wuhan, Hubei, China) ELISA kits were used according to the standard protocols provided by the manufacturer. All samples were assayed in duplicate.
2.9. Immunofluorescence
C2C12 and C57 cells grown on glass coverslips were fixed in 4% paraformaldehyde at 4 °C for 20 min, blocked in 4% (w/v) bovine serum albumin (BSA) PBS and incubated with primary anti-sclerostin (ab63097, Abcam, Cambridge, United Kingdom) anti-MyoD antibody (Santa Cruz Biotechnology, Dallas, USA) overnight at 4 °C. Coverslips were next washed with PBS and incubated with fluorescence-labeled secondary antibodies (Thermo Scientific, Waltham, MA, USA) for 1 h at room temperature. Slides were mounted with a 10% DABCO (1,4-diazabicyclo[2.2.2]octane) solution and were observed using a Nikon A1 confocal laser scanning microscope. The confocal serial sections were processed with ImageJ software to obtain three-dimensional projections and image rendering was performed using Adobe Photoshop CS 8.0 software (Adobe Systems, San Jose, CA, USA). All the images shown in this paper are representative of at least 3 independent experiments carried out under the same conditions.
2.10. Histological Staining and Immunohistochemistry
Animal tissues (muscle, bone) were fixed with 4% PFA overnight. Next, the samples were processed for paraffin embedding (Leica EG1150 H) following the procedure described here: 70% ethanol for 30 min, three times in 95% ethanol (1 h each), three times in 100% ethanol (1 h each), three times in toluene (1 h each), and two times in paraffin (1 h each). After paraffin embedding, 5–7 μm sections were obtained from each sample by means of a Leica RM2155 microtome (Leica Inc. Wetzlar, Germany).
Hematoxylin and eosin (H&E) staining was performed on murine muscles and bone tissues. Briefly, two slides per muscle were deparaffinized by means of xylene (two times for 5 min each), hydrated through a graded series of alcohol (100%, 95%, 80%, and 70%) for 5 min each, and rinsed; first with tap water and then with deionized water for 1 min each. After the dehydration procedure, the slides were stained with a Carazzi hematoxylin solution (C0203, DiaPath, Bergamo, Italy), rinsed with tap water, stained with alcoholic 0.5% eosin Y (C0353, DiaPath, Bergamo, Italy), and then rinsed with deionized water for 1 min each.
Histomorphometric analysis was conducted on the right and left femurs. Briefly, both femurs were removed from each animal, deprived of soft tissues, fixed in PBS—4% paraformaldehyde (pH 7.4), dehydrated in graded ethanol, and paraffin-embedded. The femurs were cut sagittally at the distal metaphyseal level and transversally cut at the mid-diaphyseal region to obtain serial 5 μm-thick sections for histomorphometric analysis (ImageJ software, 1.52v, NIH). The sections were stained with H&E to measure the following static histomorphometric parameters: trabecular bone volume (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular separation (Tb.Sp) of the distal femoral metaphyses, as well as the cortical bone area (Ct-B-Ar) of femoral mid-diaphyses. The areas in which the measurements were performed were selected as previously described.
For immunohistochemistry staining, from the paraffin block of each case, we cut a 4 µm-thick slides for immunohistochemical analysis against CD34 (pre-diluted QBEnd/10 Mouse Monoclonal Antibody clone; Ventana Medical Systems, Tucson, AZ, USA) and CD45/LCA (pre-diluted 2B11&PD7/26 Mouse Monoclonal Antibody clone; Cell Marque™- a Sigma-Aldrich®, St. Louis, MO, USA) using a Bench Mark ULTRA automated stainer (Ventana Medical Systems, Tucson, AZ, USA).
For both antibodies, microwave oven irradiation was used as the immunohistochemical antigen unmasking treatment. In addition, a pre-diluted ULTRA Cell Conditioning Solution (ULTRA CC1—Ventana) was used as a pre-treatment step in the processing of tissue samples for immunohistochemistry reaction. Sample sections of the vascular tumor and lymphoid tissue were used as the internal control for CD34 and LCA/CD45 markers, respectively. The sections were blocked for non-specific binding with 3% normal bovine serum and incubated with primary antibodies for 1 h at room temperature. For immunofluorescence, the sections were then incubated with secondary antibodies at 1:200 for 30 min at room temperature (DABCO). The sections were mounted with a VECTASHIELD HardSet mounting medium. All the stained sections were evaluated under a Nikon Eclipse Ni microscope (Nikon) equipped with a DS-Fi2 camera (Nikon) and processed by means of NIS-Elements D 5.11.00 software (Nikon, Rochester, New York, NY, USA).
2.11. Micro-CT Measurements
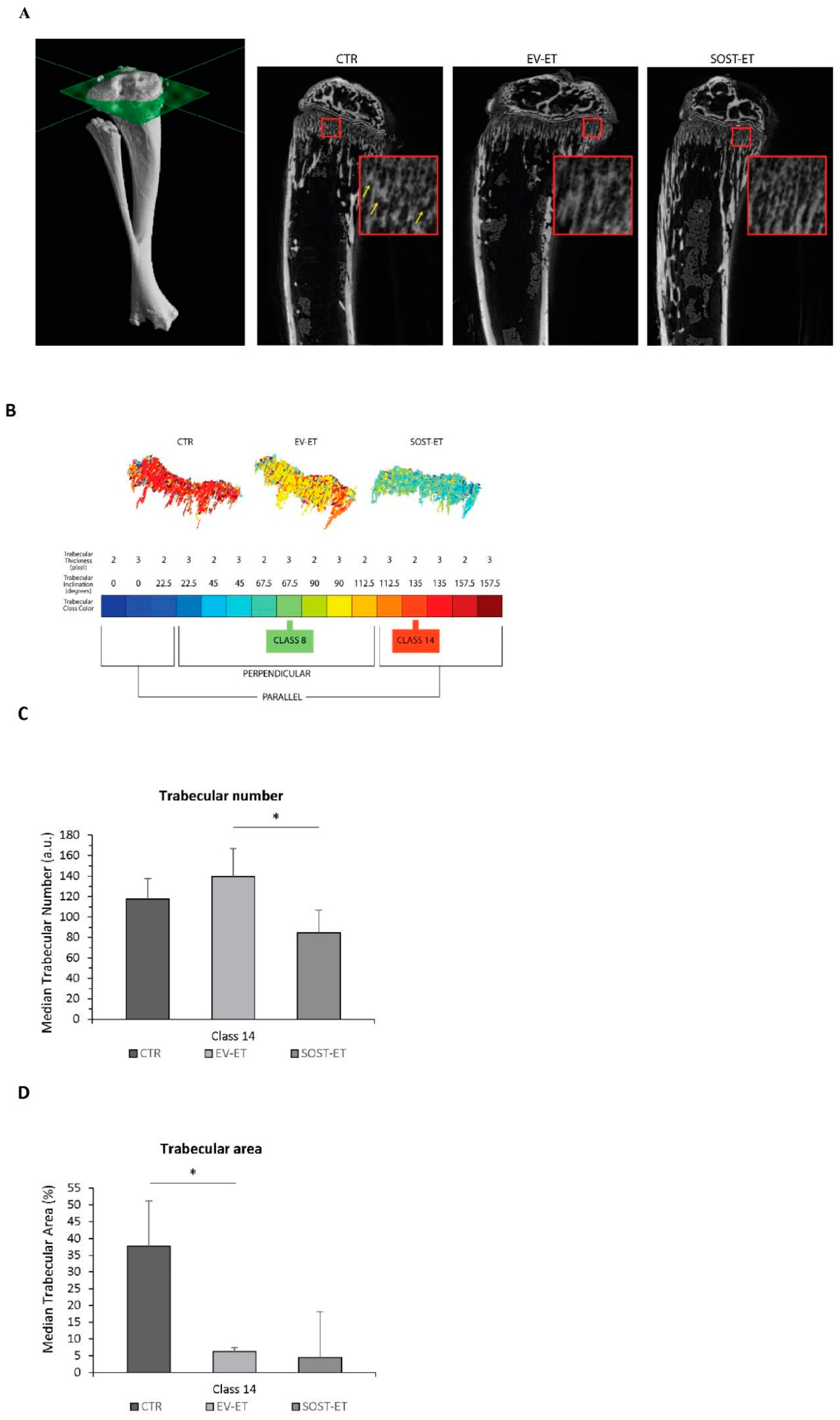
Micro-CT scanning was carried out on both left and right tibiae of C57BL/6 mice after electroporation and bone harvesting. Micro-CT scanning and analysis were performed using a KEVEX PXS10 130 kV–0.5 mA X-ray tube and a Photonic Science VHR1 CCD (charge-coupled device) camera with a fiber optic plate with a scintillator (FOS) 4000 × 2600 (9 μm pixel). The isometric voxel size was 4.5 μm and the X-ray tube potential was 70 kV.
For each tibia, axial sections (from distal epiphysis to middle diaphysis) were acquired and stacked in a single DICOM (Digital Imaging and Communications in Medicine) file. The acquisition volume was reconstructed from each DICOM file and the coronal sections were then extracted. The most informative (i.e., with less background and more signal, that is, imaged bone) coronal section was selected and analyzed together with the previous five and the subsequent five for a total of 11 coronal sections per sample. For each section, the epiphysis, metaphysis and metaphyseal plate were segmented separately and automatically; in each of the 11 sections, the metaphysis was further segmented through the subdivision of metaphyseal trabeculae into 16 different types, or classes. To this purpose, a Gabor filter bank was used, as it is a well-established approach to analyze objects’ thickness and orientation in grey-level images in computer vision applications [
27]. In the case of our classification, the trabecular thickness can have a value of 2 or 3 pixels (therefore, it is related to a range of about 10–15 µm), while the angle of trabecular inclination with respect to the metaphyseal plate w quantized by multiples of 22.5 degrees. Thus, the classes of metaphyseal trabeculae were defined by a unique combination of trabecular thickness and orientation as follows in
Table 1.
Each trabecular class identified was analyzed separately considering the trabecular number, thickness, shape, and area. The statistical analysis was performed pooling the left and right tibiae of the same experimental group (untreated (CTR), empty vector-treated (EV-ET), and sclerostin vector-treated (SOST-EV)). First, the three groups of tibiae were compared to each other in pairs (i.e., CTR against EV-ET, CTR against SOST-ET, and EV-ET against SOST-ET, separately). Then, for each pair of groups, their individual tibiae were compared in pairs by considering all the possible pairs of inter-group comparison in order to verify whether what is observed from the comparison between the groups can also be valid for the individual tibiae that compose them. As an example, if the CTR group is composed of tibiae 1A and 1B and the EV-ET group is composed of tibiae 2A and 2B, after pair-comparing the whole groups (CTR vs. EV-ET), the single samples that compose them are pair-compared in all possible combinations (1A vs. 2A, 1A vs. 2B, 1B vs. 2A, 1B vs. 2B).
2.12. Statistical Analyses
All the data were analyzed using Prism version 6.0e (GraphPad Software, Graph Pad Holdings, CA, USA). In vitro data are presented as the means ± standard deviation (SD) from three independent experiments. Unless otherwise stated, the data for animal experiments are expressed as the means ± standard deviation, and statistical analyses were performed using one-way ANOVA followed by Dunnett’s, Sidak, or Tukey’s multiple comparison tests. Differences between groups were considered statistically significant at p < 0.05.
4. Discussion
Since the discovery of myostatin in 1997, the first myokine that has been identified, secretome-based analysis of the human myocyte culture medium has revealed over 600 myokines to date [
33,
34,
35,
36,
37]. However, the majority of these myokines are still not sufficiently characterized and need to be extensively investigated to gain insight into the pathogenesis of musculoskeletal diseases.
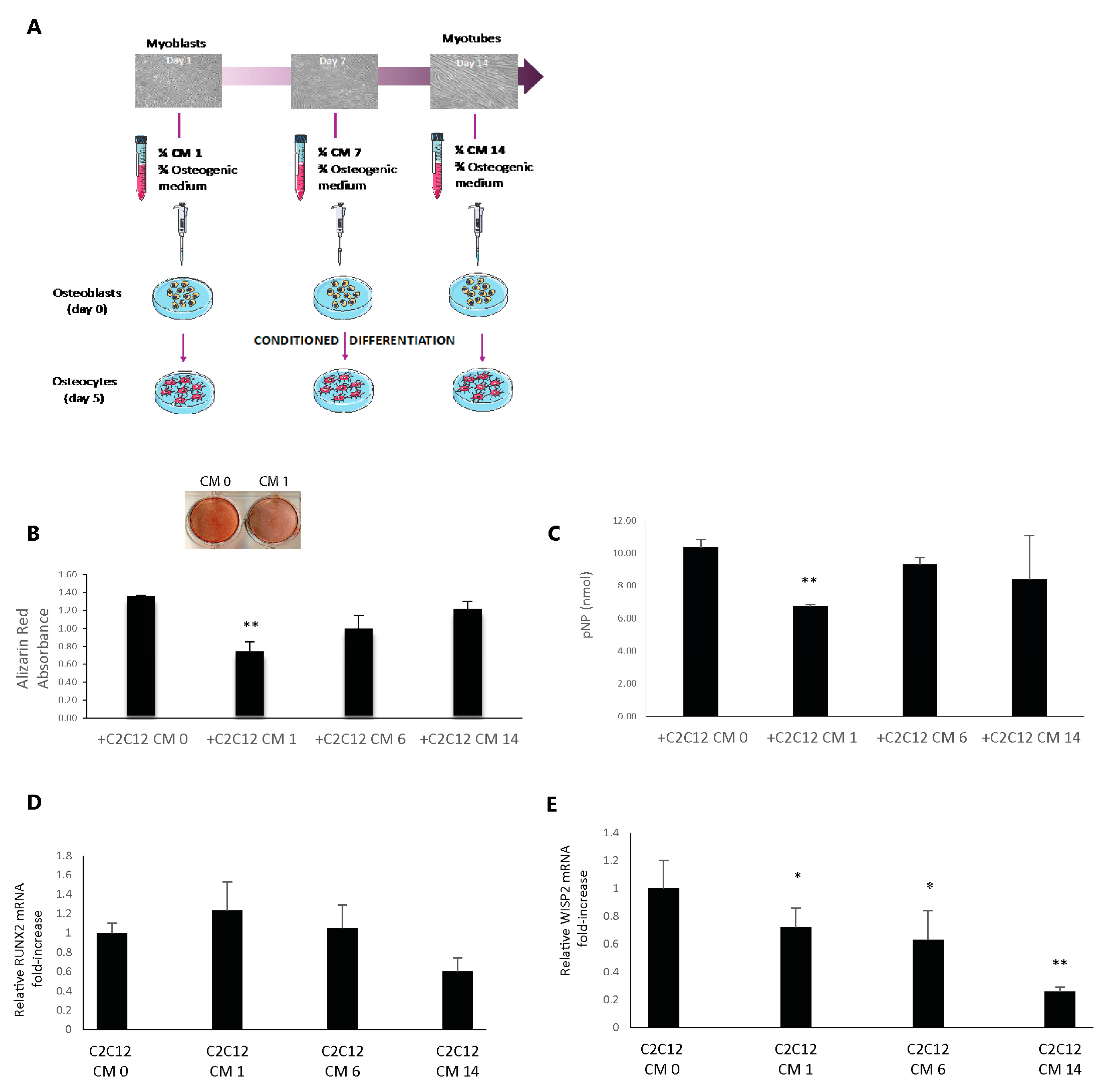
In this study, we proposed to model in vitro the muscle-to-bone communication with osteogenic (2T3) and myogenic (C2C12) cell lines. We found that the conditioned medium from differentiating C2C12 cells significantly altered the functional maturation of osteoblasts. Therefore, we hypothesized the presence of muscle cell-secreted factors that may exert an inhibitory action on osteogenesis. Recently, our group described an osteocyte-produced factor, Wisp2, that is an autocrine/paracrine activator of canonical Wnt signaling pathway with a potent role in accelerating the osteogenesis process [
28]. Since sclerostin, an inhibitor of the Wnt/β-catenin pathway that impairs bone formation, has become an attractive therapeutic target for treating osteoporosis, we wondered if this molecule could also be produced by muscle cells. Despite being considered an osteocyte-specific protein,
SOST mRNA expression was found in other organs, including skeletal muscle [
29], but no one had ever characterized its actual synthesis by muscle in detail. Thus, we analyzed SOST/sclerostin protein expression in C2C12 and C57 muscle cells and its secretion into the culture medium, discovering that it is produced at all these levels. We then focused on the characterization of sclerostin in vivo, highlighting that it is widely expressed in young, adult, and old mouse muscles and that its expression undergoes changes in muscles with different metabolic profiles.
In order to determine whether muscular sclerostin was similar to that of osseous sclerostin in the ability to modulate bone remodeling, a transient gain-of-function experiment was performed via plasmid electroporation in muscles of young mice characterized by a growing skeleton. This experiment allowed outlining that muscle-released sclerostin is able to negatively influence the skeletal system. This hypothesis was confirmed by (i) the static histomorphometry performed in the trabecular bone of the femur distal metaphysis (see above); (ii) the micro-CT analysis performed on tibiae, which revealed that SOST overexpression induced a decrease in the number and extension of parallel trabeculae. Diversely, the increased amount of perpendicular trabeculae observed in SOST-ET animals has an explanation related to skeletal homeostasis. As regards the mechanical meaning of trabecular orientation, it is well established that trabecular structures are aligned in an organized manner associated with the direction of load distribution so that trabeculae are mainly oriented according to the principal applied mechanical load directions [
38,
39]; in line with this evidence, our results on electroporation of the plasmid containing the
SOST gene show a significant decrease under the effect of muscle-derived sclerostin of bony trabeculae less subjected to increasing load during growth (class 14), differently from the trabeculae (class 8) mostly subjected to increasing load during growth, on which the effect of growing is likely dominant compared to other conditioning factors. Thus, we argue that, particularly in dynamic situations (such as in the growing skeleton) and despite sophisticated analyses (such as micro-CT), the effect of downregulation on osteogenesis exerted by sclerostin of muscular origin can be masked by the effect exerted on the skeleton by the increase in body mass during somatic growth.
Taken together, our data indicate that the action of muscular sclerostin resembles the activity of the osseous one. Therefore, muscular sclerostin seems to behave as an inhibitory factor for osteogenesis that may act synergistically with bone sclerostin to block bone deposition. In suggesting the in vivo role of muscular sclerostin on bone tissue, the authors are aware of the need to confirm the role of muscular sclerostin after the end of the somatic growth before proposing the validation of this protein as a therapeutic target for the treatment of bone processes directly linked to the pathological condition or indirectly, due to a muscular weakness, such as the condition of muscular dystrophy, neuronal atrophy, or cachexia. In line with this target, further steps will be as follows: (i) detailed analyses of the biochemical structure of muscular sclerostin (comparing it with the osseous one) and (ii) investigations on the potential of anti-sclerostin compounds to also inhibit the muscular form.
In conclusion, this study highlights for the first time the presence of a new form of sclerostin produced at the muscular level that can act in combination with bone sclerostin in the phenomena of increased bone fragility. Targeting pathways that centrally regulate bone and muscle or pathways that facilitate communication between the two tissues are the privileged directions for the identification of new chemical molecules, which could prevent, reduce, or restore bone loss.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}