
Computationally Assisted Lead Optimization of Novel Potent and Selective MAO-B Inhibitors


 , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. General
2.2. Synthesis of Derivatives
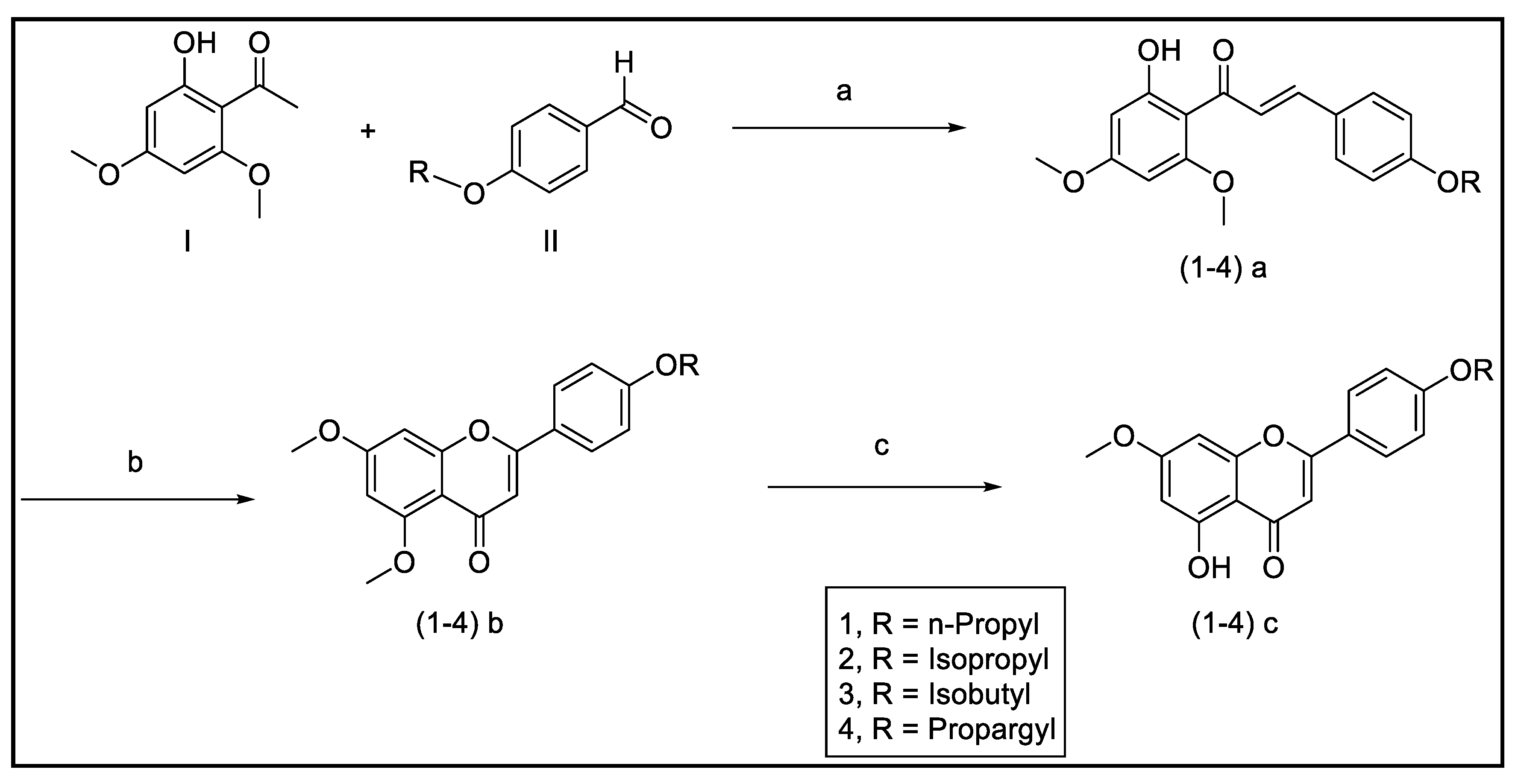
2.2.1. Procedure for Preparation of Compounds 1–4
Synthesis of (E) 4-Propyloxy-4′,6′-Dimethoxy-2′-Hydroxychalcone (1a)
Synthesis of 5,7-Dimethoxy-4′-Propyloxyflavone (1b)
Synthesis of 7-Methoxy-4′-Propyloxy-5-Hydroxyflavone (1c)
Synthesis of (E) 4-Isopropyloxy-4′,6′-Dimethoxy-2′-Hydroxychalcone (2a)
Synthesis of 5,7-Dimethoxy-4′-Isopropyloxyflavone (2b)
Synthesis of 7-Methoxy-4′-Isopropyloxy-5-Hydroxyflavone (2c)
Synthesis of (E) 4-Isobutyloxy-4′,6′-Dimethoxy-2′-Hydroxychalcone (3a)
Synthesis of 5,7-Dimethoxy-4′-Isobutyloxyflavone (3b)
Synthesis of 7-Methoxy-4′-Isobutyloxy-5-Hydroxyflavone (3c)
Synthesis of (E) 4-Propargyloxy-4′,6′-Dimethoxy-2′-Hydroxychalcone (4a)
Synthesis of 5,7-Dimethoxy-4′-Propargyloxyflavone (4b)
Synthesis of 7-Methoxy, 4′-Propargyloxy-5-Hydroxyflavone (4c)
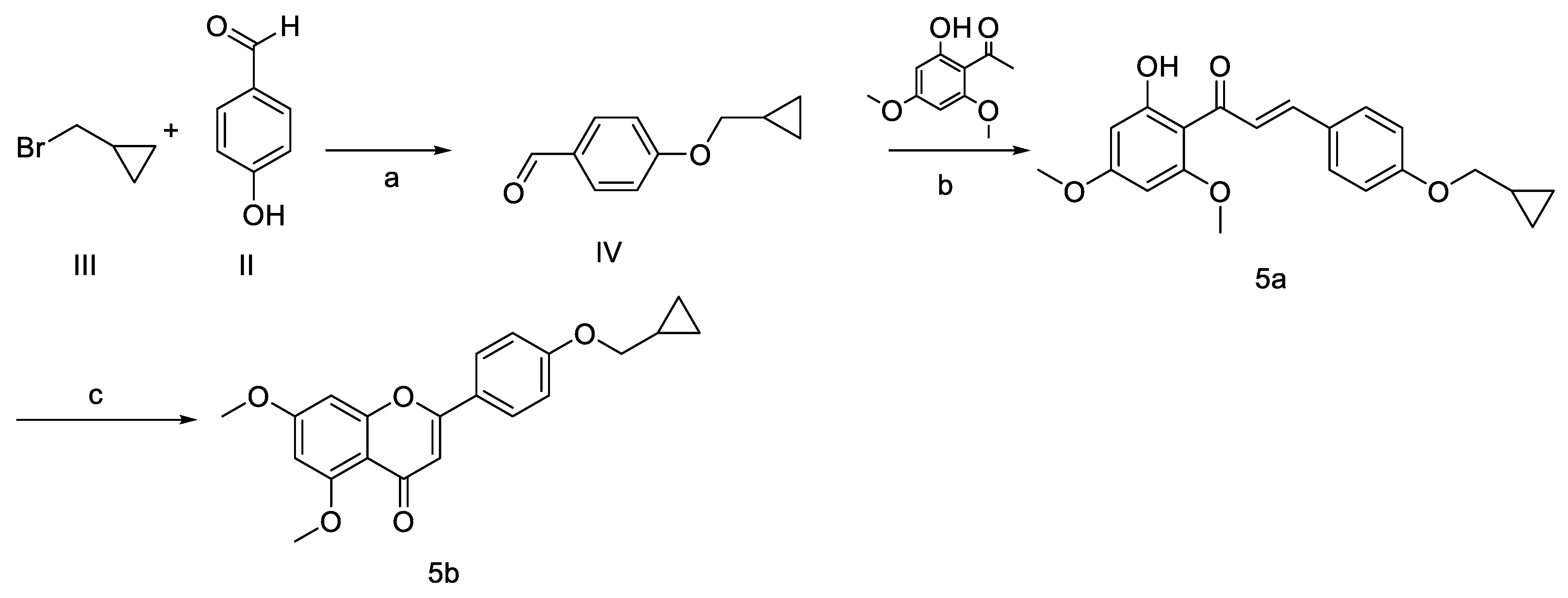
2.2.2. Procedure for Preparation of Compounds 5
Synthesis of 4-(Cyclopropyl Methoxy)Benzaldehyde (Compound IV)
Synthesis of (E) 4-(Cyclopropyl Methoxy)-4′,6′-Dimethoxy-2′-Hydroxychalcone (5a)
Synthesis of 5,7-Dimethoxy-4′(Cyclopropyl Methoxy)Flavone (5b)
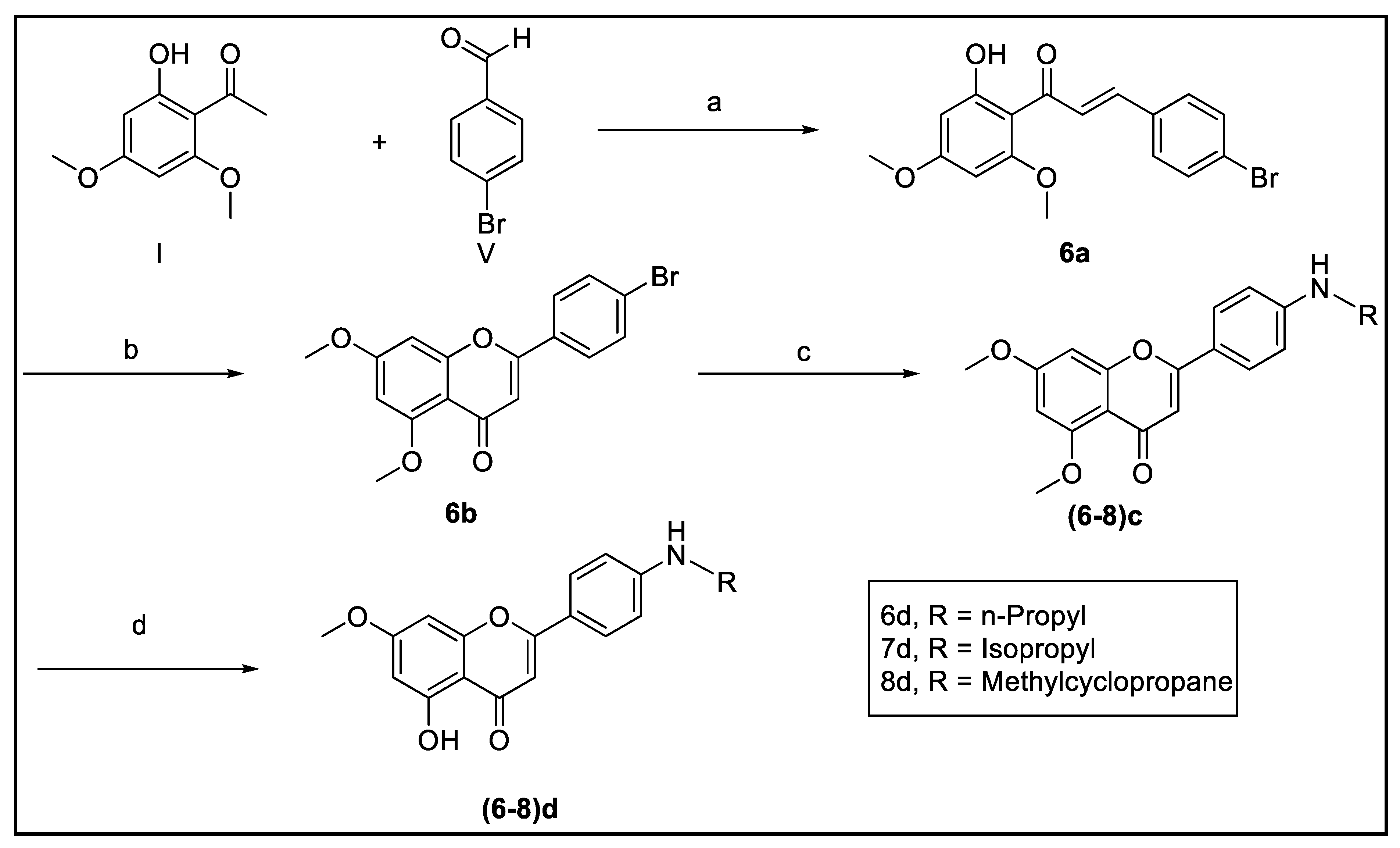
2.2.3. Procedure for Preparation of Compounds 6–8
Synthesis of (E) 4-Bromo-4′,6′-Dimethoxy-2′-Hydroxychalcone (6a)
Synthesis of 5,7-Dimethoxy-4′-Bromoflavone (6b)
Synthesis of 5,7-Dimethoxy-4′-Aminopropylflavone (6c)
Synthesis of 7-Methoxy-4′-Aminopropyl-5-Hydroxyflavone (6d)
Synthesis of 5,7-Dimethoxy-4′-Aminoisopropylflavone (7c)
Synthesis of 7-Methoxy-4′-Aminoisopropyl-5-Hydroxyflavone (7d)
Synthesis of 5,7-Dimethoxy-4′-(Cyclopropyl Methylamino)Flavone (8c)
Synthesis of 7-Methoxy-4′-(Cyclopropyl Methylamino)-5-Hydroxyflavone (8d)
2.3. Evaluation of Pan Assay Interference Compounds (PAINS)
2.4. Monoamine Oxidase Inhibition Assay and Determination of IC50 Values for Synthesized Compounds
2.5. Enzyme Kinetics, Mechanism Studies, Analysis of Reversibility, and Binding Assays of Acacetin 7-O-Methyl Ether Analogs
2.6. Molecular Modeling Studies
3. Results and Discussion
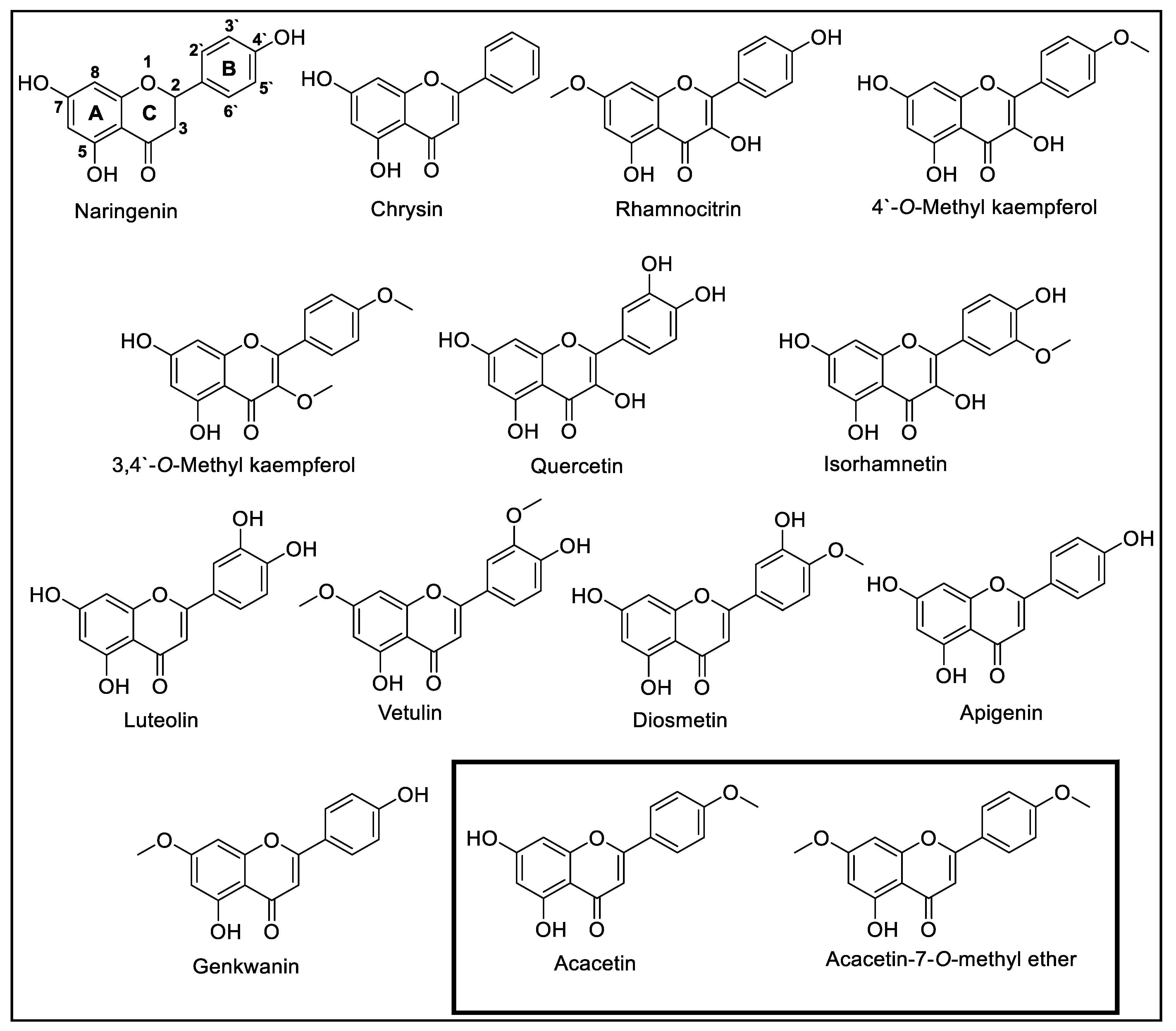
3.1. Comparison of Acacetin and Acacetin 7-O-Methyl Ether with Known Flavonoids
3.2. In Silico Optimization and Design
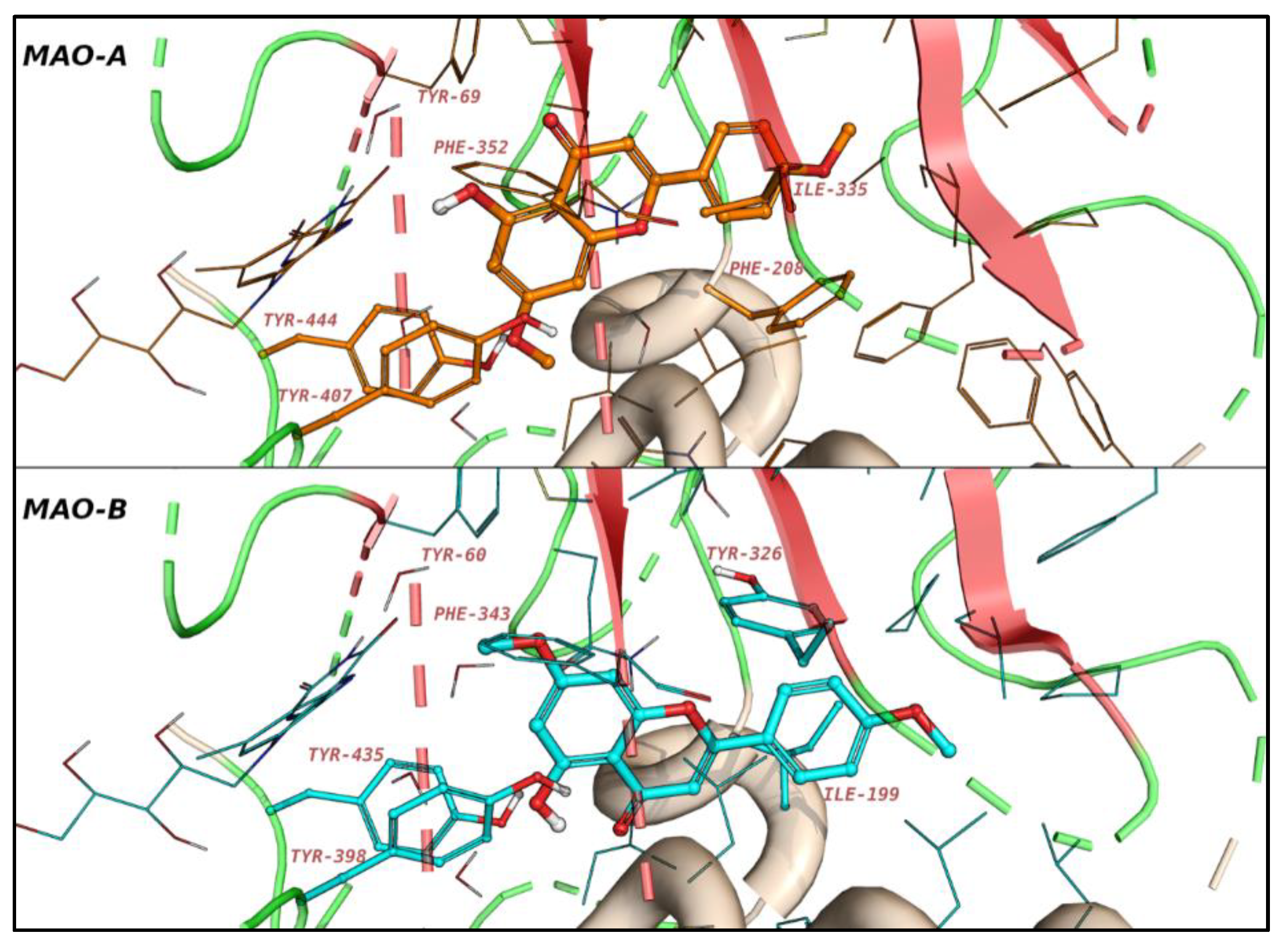
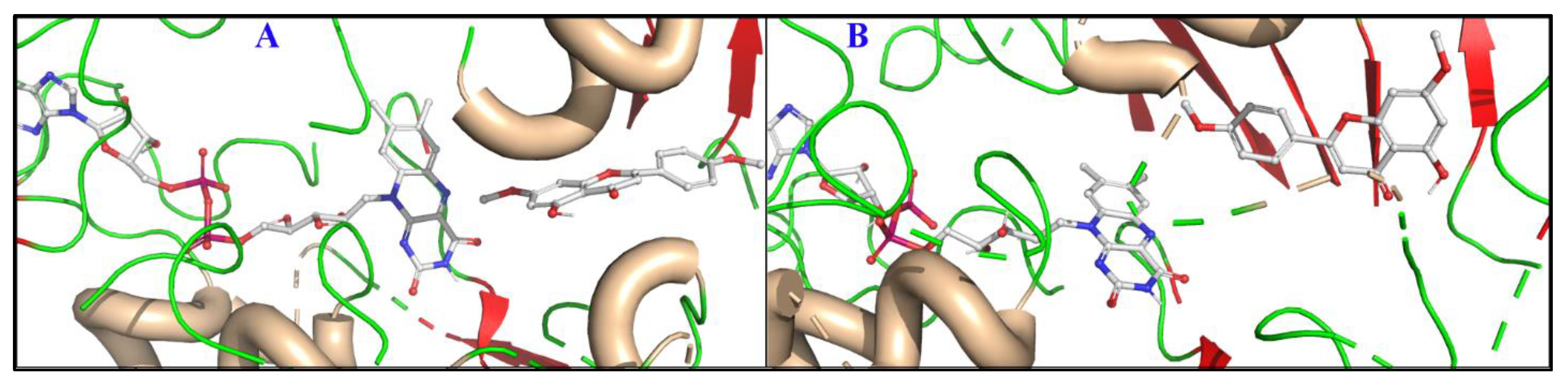
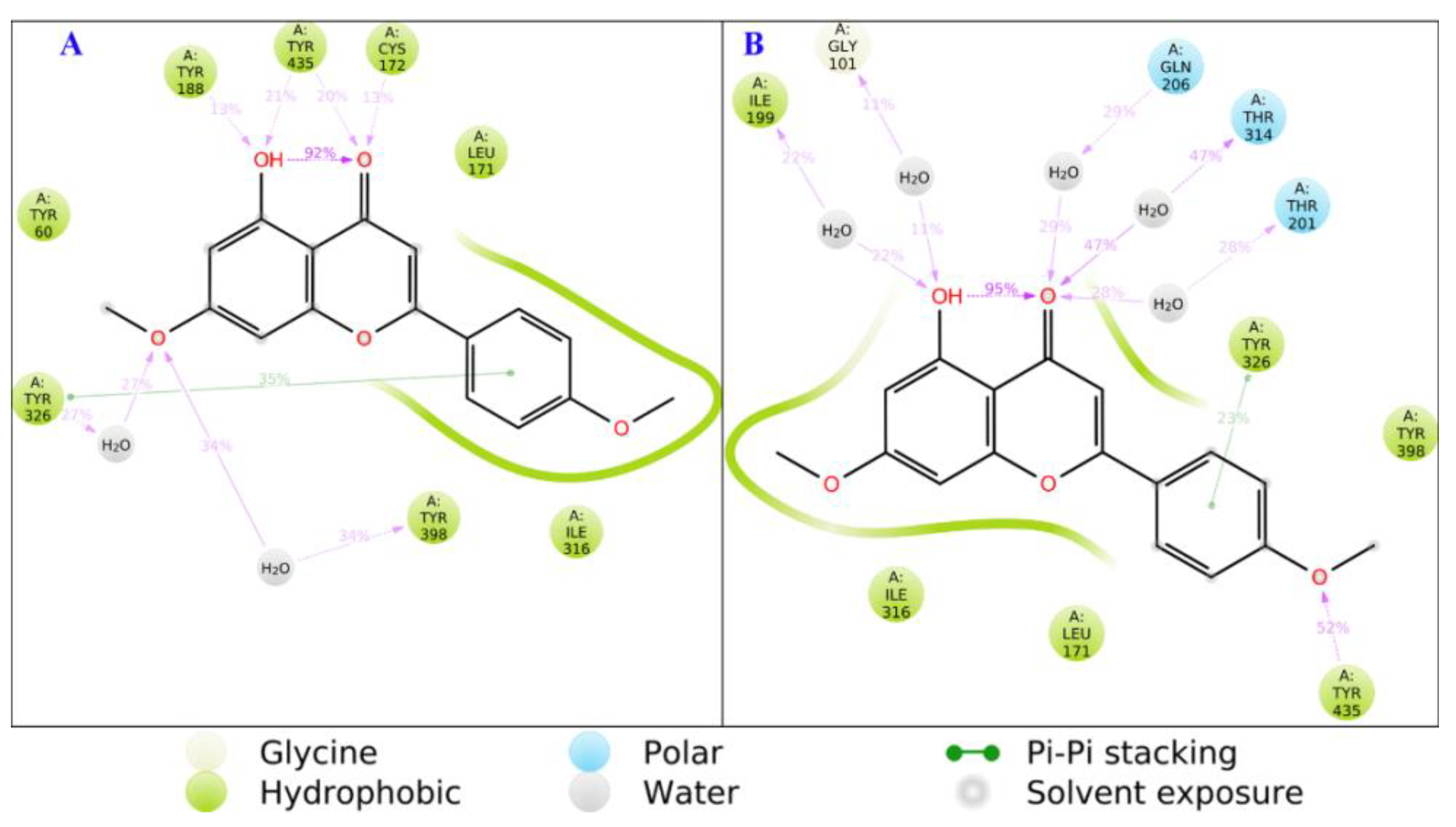
3.2.1. Molecular Dynamics (MD) Simulations of the Binding Modes of Acacetin and Acacetin 7-O-Methyl Ether
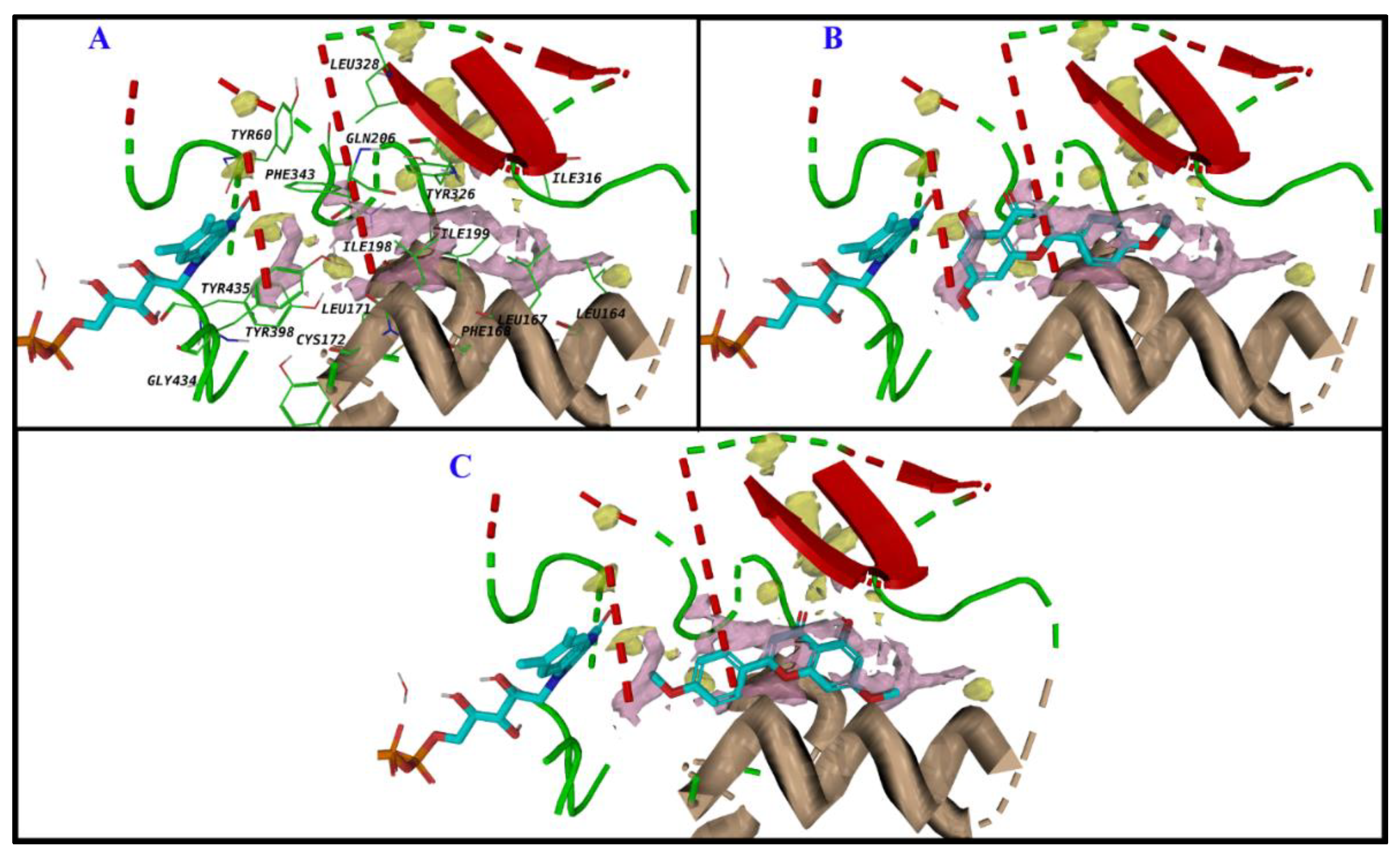
3.2.2. Active Site Hydration of MAO-B: MD Simulations, Thermodynamics, and Ligand Designs
3.3. Chemistry
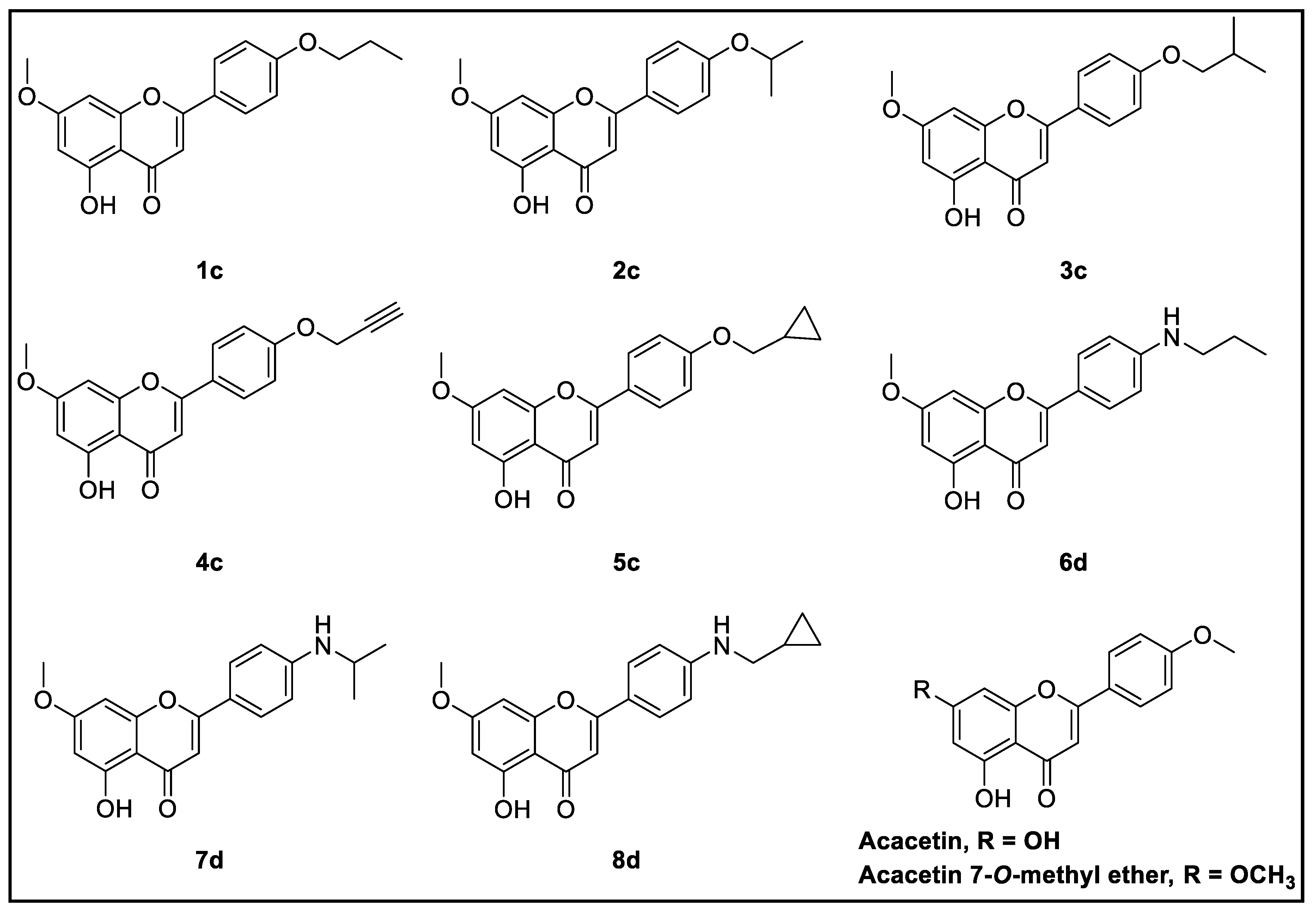
3.3.1. Synthesis of Modified Flavonoids 1–5
3.3.2. Synthesis of Modified Flavonoids 6–8
3.4. Biological Assays
3.4.1. Determination of Inhibitory Effects of Modified Flavonoids and Intermediates on MAO-A and -B
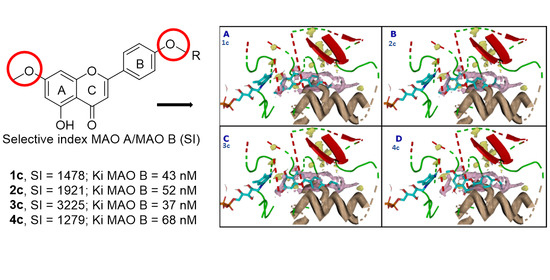
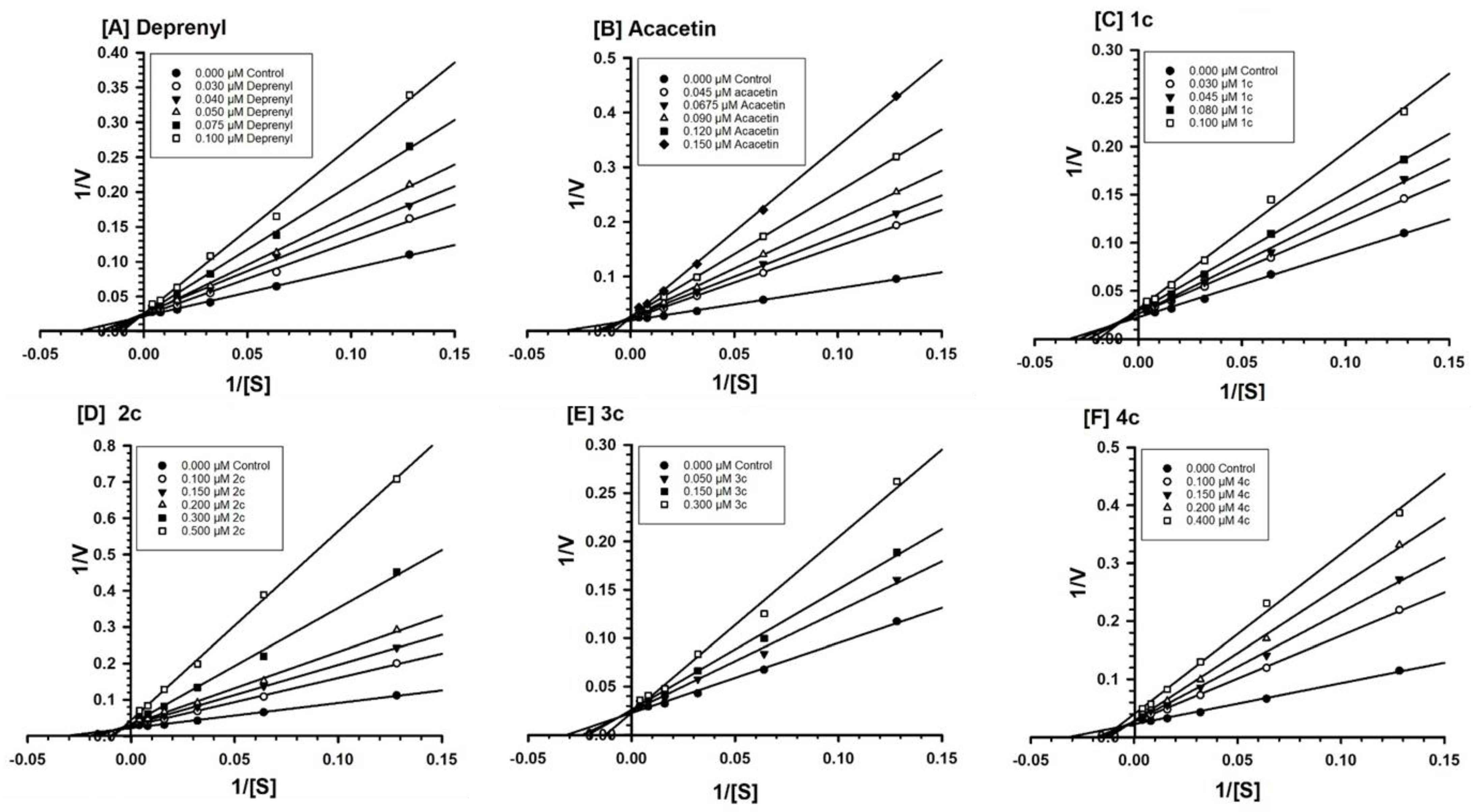
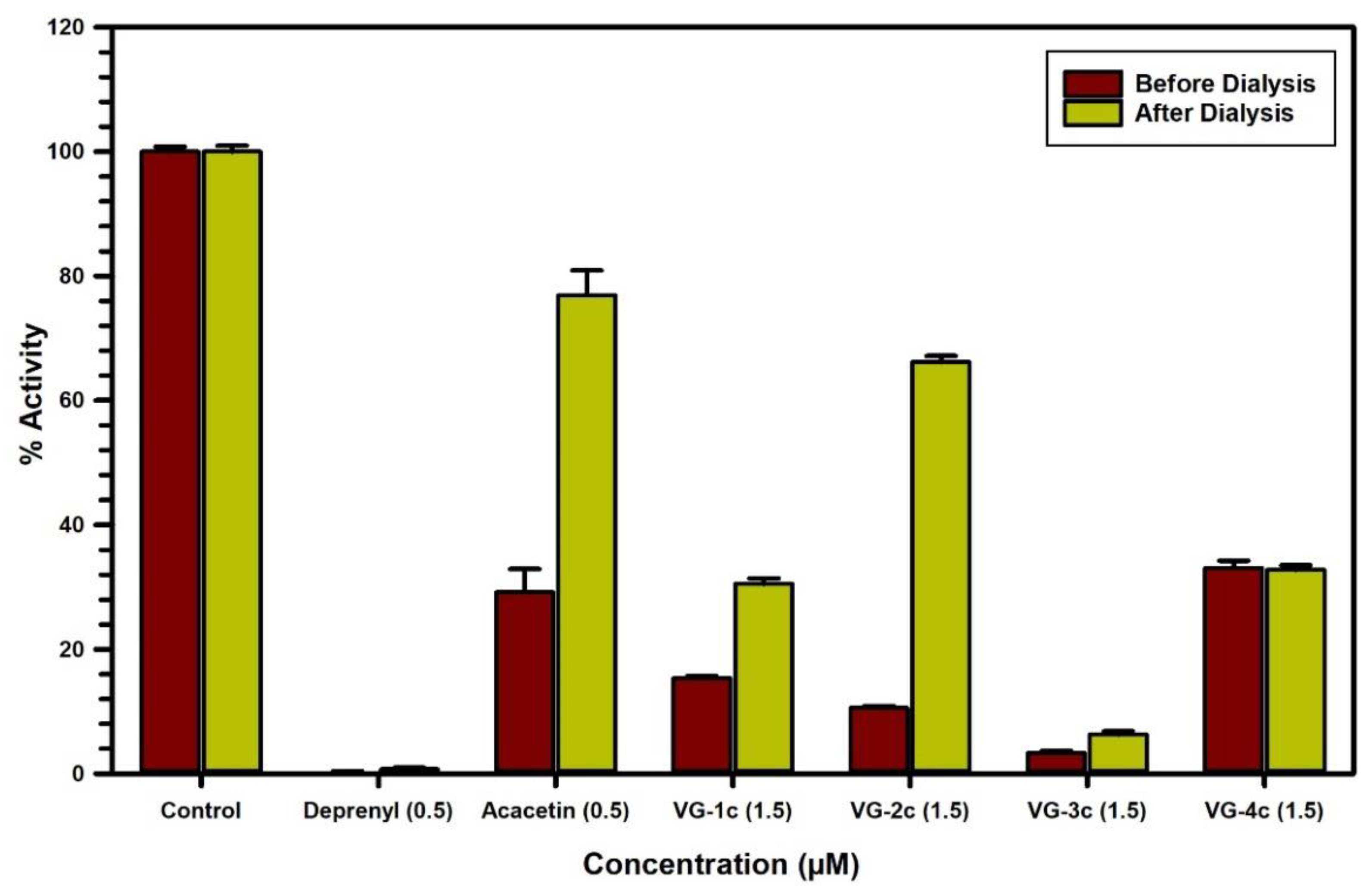
3.4.2. Evaluation of MAO-B Inhibition Kinetics and Analysis of Binding and Time-Dependent Inhibition of Modified Flavonoids (1–4) c
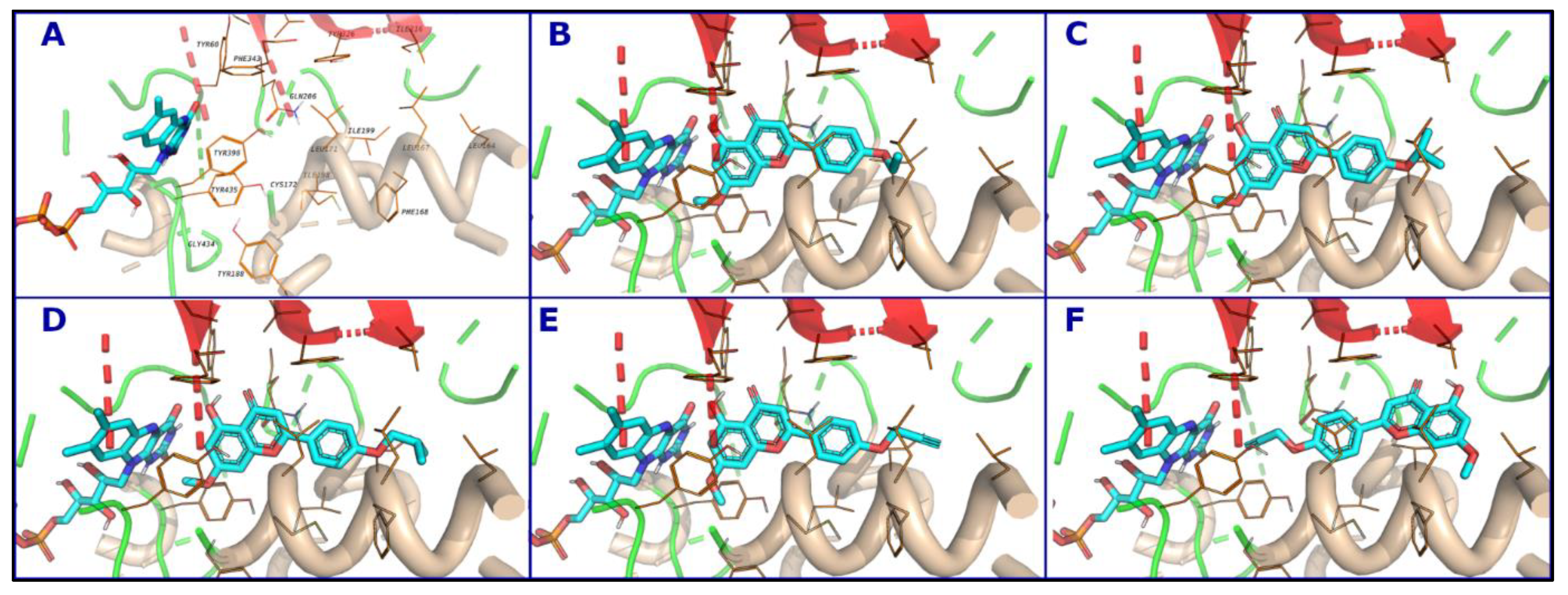
3.5. Computational Analysis of Enzyme–Inhibitor Interactions for Modified Flavonoids (1–4) c
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scott, L.; Dawson, V.L.; Dawson, T.M. Trumping Neurodegeneration: Targeting Common Pathways Regulated by Autosomal Recessive Parkinson’s Disease Genes. Exp. Neurol. 2017, 298, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D.L. The Prevalence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative Stress and Parkinson’s Disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Sarrafchi, A.; Bahmani, M.; Shirzad, H.; Rafieian-Kopaei, M. Oxidative Stress and Parkinson’s Disease: New Hopes in Treatment with Herbal Antioxidants. Curr. Pharm. Des. 2016, 22, 238–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.; Rathitharan, G.; Meyer, J.H.; Furukawa, Y.; Ang, L.-C.; Boileau, I.; Guttman, M.; Hornykiewicz, O.; Kish, S.J. Brain Monoamine Oxidase B and A in Human Parkinsonian Dopamine Deficiency Disorders. Brain 2017, 140, 2460–2474. [Google Scholar] [CrossRef] [PubMed]
- Szökő, É.; Tábi, T.; Riederer, P.; Vécsei, L.; Magyar, K. Pharmacological Aspects of the Neuroprotective Effects of Irreversible MAO-B Inhibitors, Selegiline and Rasagiline, in Parkinson’s Disease. J. Neural Transm. 2018, 125, 1735–1749. [Google Scholar] [CrossRef]
- Liguori, C.; Stefani, A.; Mercuri, N.B.; Pierantozzi, M. Effective Treatment of Restless Legs Syndrome by Safinamide in Parkinson’s Disease Patients. Sleep Med. 2018, 41, 113–114. [Google Scholar] [CrossRef]
- Guglielmi, P.; Carradori, S.; Ammazzalorso, A.; Secci, D. Novel Approaches to the Discovery of Selective Human Monoamine Oxidase-B Inhibitors: Is There Room for Improvement? Expert Opin. Drug Discov. 2019, 14, 995–1035. [Google Scholar] [CrossRef]
- Chaurasiya, N.D.; Gogineni, V.; Elokely, K.M.; León, F.; Núñez, M.J.; Klein, M.L.; Walker, L.A.; Cutler, S.J.; Tekwani, B.L. Isolation of Acacetin from Calea urticifolia with Inhibitory Properties against Human Monoamine Oxidase-A and -B. J. Nat. Prod. 2016, 79, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Semwal, R.B.; Semwal, D.K.; Combrinck, S.; Trill, J.; Gibbons, S.; Viljoen, A. Acacetin–A Simple Flavone Exhibiting Diverse Pharmacological Activities. Phytochem. Lett. 2019, 32, 56–65. [Google Scholar] [CrossRef]
- Chaurasiya, N.D.; Zhao, J.; Pandey, P.; Doerksen, R.J.; Muhammad, I.; Tekwani, B.L. Selective Inhibition of Human Monoamine Oxidase B by Acacetin 7-Methyl Ether Isolated from Turnera diffusa (Damiana). Molecules 2019, 24, 810. [Google Scholar] [CrossRef] [Green Version]
- Amawi, H.; Ashby, C.R., Jr.; Tiwari, A.K. Cancer Chemoprevention Through Dietary Flavonoids: What’s Limiting? Chin. J. Cancer 2017, 36, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.-F.; Wang, Z.-Z.; Chen, K.-Z.; Xu, T.-F.; Hao, G.-F. Computational Fragment-Based Design Facilitates Discovery of Potent and Selective Monoamine Oxidase-B (MAO-B) Inhibitor. J. Med. Chem. 2020, 63, 15021–15036. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, C.; Bertozzi, C.; Georg, G.I.; Kiessling, L.; Lindsley, C.; Liotta, D.; Merz, K.M., Jr.; Schepartz, A.; Wang, S. The Ecstasy and Agony of Assay Interference Compounds. J. Med. Chem. 2017, 60, 2165–2168. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15–Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Chaurasiya, N.D.; León, F.; Ding, Y.; Gómez-Betancur, I.; Benjumea, D.; Walker, L.A.; Cutler, S.J.; Tekwani, B.L. Interactions of Desmethoxyyangonin, a Secondary Metabolite from Renealmia alpinia, with Human Monoamine Oxidase-A and Oxidase-B. Evid. Based Complement. Altern. Med. 2017, 2017, 4018724. [Google Scholar] [CrossRef] [Green Version]
- Parikh, S.; Hanscom, S.; Gagne, P.; Crespi, C.; Patten, C. A Fluorescent-Based, High-Throughput Assay for Detecting Inhibitors of Human Monoamine Oxidase A and B. BD Biosci. Discov. Labware 2002, S02T081R2. [Google Scholar] [CrossRef]
- Small-Molecule Drug Discovery Suite 2017-4; Schrödinger, LLC.: New York, NY, USA, 2017.
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the Comprehensive, Rapid, and Accurate Prediction of the Favorable Tautomeric States of Drug-Like Molecules in Aqueous Solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for pKa Prediction and Protonation State Generation for Drug-Like Molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-4: Epik; Schrödinger, LLC.: New York, NY, USA, 2017.
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Schrödinger Release 2017-4: Glide; Schrödinger, LLC.: New York, NY, USA, 2017.
- Arul Murugan, N.; Chiotis, K.; Rodriguez-Vieitez, E.; Lemoine, L.; Ågren, H.; Nordberg, A. Cross-interaction of tau PET tracers with monoamine oxidase B: Evidence from in silico modelling and in vivo imagining. Eur. J. Nucl. Med. Mol. Imagining 2019, 46, 1369–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arul Murugan, N.; Zaleśny, R. Multiscale modeling of two-photon probes for Parkinson’s diagnostics based on monoamine oxidase B biomarker. J. Chem. Inf. Model. 2020, 60, 3854–3863. [Google Scholar] [CrossRef]
- Arul Murugan, N.; Muvva, C.; Jeyarajpandian, C.; Jeyakanthan, J.; Subramanian, V. Performance of force-field and machine learning-based scoring functions in ranking MAO-B protein-inhibitor complexes in relevance to developing Parkinson’s therapeutics. Int. J. Mol. Sci. 2020, 21, 7648. [Google Scholar] [CrossRef]
- Schrödinger Release 2017-4: LigPrep; Schrödinger, LLC.: New York, NY, USA, 2017.
- Elokely, K.M.; Doerksen, R.J. Docking Challenge: Protein Sampling and Molecular Docking Performance. J. Chem. Inf. Model. 2013, 53, 1934–1945. [Google Scholar] [CrossRef] [Green Version]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC ’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar] [CrossRef] [Green Version]
- Desmond Molecular Dynamics System, D.E. Shaw Research, New York, NY, 2021. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY. 2021. Available online: https://www.schrodinger.com/citations (accessed on 12 December 2017).
- SZMAP 1.2.1.4; OpenEye Scientific Software: Santa Fe, NM, USA. Available online: http://www.eyesopen.com (accessed on 12 December 2017).
- Bayden, A.S.; Moustakas, D.T.; Joseph-McCarthy, D.; Lamb, M.L. Evaluating Free Energies of Binding and Conservation of Crystallographic Waters Using SZMAP. J. Chem. Inf. Model. 2015, 55, 1552–1565. [Google Scholar] [CrossRef] [PubMed]
- Elokely, K.; Velisetty, P.; Delemotte, L.; Palovcak, E.; Klein, M.L.; Rohacs, T.; Carnevale, V. Understanding TRPV1 Activation by Ligands: Insights from the Binding Modes of Capsaicin and Resiniferatoxin. Proc. Natl. Acad. Sci. USA. 2016, 113, E137–E145. [Google Scholar] [CrossRef] [Green Version]
- Olsen, H.T.; Stafford, G.I.; van Staden, J.; Christensen, S.B.; Jäger, A.K. Isolation of the MAO-Inhibitor Naringenin from Mentha aquatica L. J. Ethnopharmacol. 2008, 117, 500–502. [Google Scholar] [CrossRef]
- Larit, F.; Elokely, K.M.; Chaurasiya, N.D.; Benyahia, S.; Nael, M.A.; León, F.; Abu-Darwish, M.S.; Efferth, T.; Wang, Y.-H.; Belouahem-Abed, D.; et al. Inhibition of Human Monoamine Oxidase A and B by Flavonoids Isolated from Two Algerian Medicinal Plants. Phytomedicine 2018, 40, 27–36. [Google Scholar] [CrossRef]
- Baek, S.C.; Park, M.H.; Ryu, H.W.; Lee, J.P.; Kang, M.-G.; Park, D.; Park, C.M.; Oh, S.-R.; Kim, H. Rhamnocitrin Isolated from Prunus padus var. seoulensis: A Potent and Selective Reversible Inhibitor of Human Monoamine Oxidase A. Bioorg. Chem. 2019, 83, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, N.D.; Midiwo, J.; Pandey, P.; Bwire, R.N.; Doerksen, R.J.; Muhammad, I.; Tekwani, B.L. Selective Interactions of O-Methylated Flavonoid Natural Products with Human Monoamine Oxidase-A and -B. Molecules 2020, 25, 5358. [Google Scholar] [CrossRef] [PubMed]
- Carradori, S.; Gidaro, M.C.; Petzer, A.; Costa, G.; Guglielmi, P.; Chimenti, P.; Alcaro, S.; Petzer, J.P. Inhibition of Human Monoamine Oxidase: Biological and Molecular Modeling Studies on Selected Natural Flavonoids. J. Agric. Food Chem. 2016, 64, 9004–9011. [Google Scholar] [CrossRef]
- Park, S.E.; Paudel, P.; Wagle, A.; Seong, S.H.; Kim, H.R.; Fauzi, F.M.; Jung, H.A.; Choi, J.S. Luteolin, a Potent Human Monoamine Oxidase-A Inhibitor and Dopamine D4 and Vasopressin V1A Receptor Antagonist. J. Agric. Food Chem. 2020, 68, 10719–10729. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, N.D.; Ibrahim, M.A.; Muhammad, I.; Walker, L.A.; Tekwani, B.L. Monoamine Oxidase Inhibitory Constituents of Propolis: Kinetics and Mechanism of Inhibition of Recombinant Human MAO-A and MAO-B. Molecules 2014, 19, 18936–18952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Mrazek, A.A.; Wang, X.; Ding, C.; Ding, Y.; Porro, L.J.; Liu, H.; Chao, C.; Hellmich, M.R.; Zhou, J. Design, Synthesis, and Characterization of Novel Apigenin Analogues that Suppress Pancreatic Stellate Cell Proliferation In vitro and Associated Pancreatic Fibrosis In vivo. Bioorg. Med. Chem. 2014, 22, 3393–3404. [Google Scholar] [CrossRef] [Green Version]
- Pandey, P.; Chaurasiya, N.D.; Tekwani, B.L.; Doerksen, R.J. Interactions of endocannabinoid virodhamine and related analogs with human monoamine oxidase-A and-B. Biochem. Pharmacol. 2018, 155, 82–91. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flavonoids | MAO-A (IC50, μM) * | MAO-B (IC50, μM) * | Selectivity Index MAO-A/B |
|---|---|---|---|
| Naringenin [38] | 955 ± 129 | 288 ± 18 | 3.32 |
| Chrysin [39] | 0.25 ± 0.04 | 1.04 ± 0.17 | 0.24 |
| Rhamnocitrin [40] | 0.051 ± 0.001 | 2.97 ± 2.97 | 0.02 |
| 4′-O-Methyl kaempferol [38] | 1.350 ± 0.198 | >100 | 0.01 |
| 3,4′-Di-O-methyl kaempferol [41] | 0.033 ± 0.042 | 9.667 ± 2.39 | 0.03 |
| Quercetin [39] | 1.52 ± 0.09 | 28.39 ± 5.41 | 0.05 |
| Isorhamnetin [42] | 6.42 ± 7.69 | 21.2 ± 4.99 | 0.30 |
| Luteolin [43] | 8.57 ± 0.47 | >100 | 0.08 |
| Vetulin [11] | 18.79 ± 0.29 | 0.44 ± 0.01 | 42.70 |
| Diosmetin [42] | 5.74 ± 0.57 | 1.58 ± 0.88 | 3.63 |
| Apigenin [44] | 0.64 ± 0.11 | 1.12 ± 0.27 | 0.30 |
| Genkwanin [40] | 0.14 ± 0.01 | 0.35 ± 0.03 | 0.40 |
| Acacetin [9] | 0.121 ± 0.001 | 0.049 ± 0.0007 | 4.28 |
| Acacetin 7-O-methyl ether [11] | >100 | 0.198 ± 0.001 | >505.05 |
| Synthesized Analogs | MAO-A (IC50, μM) * | MAO-B (IC50, μM) * | Selectivity Index MAO-A/B |
|---|---|---|---|
| 1a | 0.515 ± 0.024 | 0.40 ± 0.024 | 1.3 |
| 2a | 0.117 ± 0.05 | 0.049 ± 0.0035 | 2.4 |
| 3a | 0.42 ± 0.002 | 0.22 ± 0.0211 | 1.9 |
| 4a | 9.42 ± 1.78 | 8.33 ± 0.7871 | 1.1 |
| 5a | 1.30 ± 0.09 | 0.90 ± 0.035 | 1.4 |
| 1b | >100 | 1.70 ± 0.076 | >58.8 |
| 2b | >100 | 2.64 ± 0.0880 | >37.9 |
| 3b | >100 | 7.23 ± 2.1438 | >13.8 |
| 4b | >100 | >100 | >1 |
| 4a | 9.42 ± 1.78 | 8.33 ± 0.7871 | 1.1 |
| 4b | >100 | >100 | >1 |
| 5b | >100 | 22.82 ± 2.098 | >4.4 |
| 1c | 48.78 ± 2.01 | 0.033 ± 0.0042 | 1478.2 |
| 2c | 30.74 ± 0.023 | 0.016 ± 0.0070 | 1921.3 |
| 3c | >100 | 0.031 ± 0.0070 | >3225.8 |
| 4c | 62.70 ± 5.21 | 0.049 ± 0.0014 | 1279.6 |
| 6a | 12.349 ± 0.249 | >100 | >0.1 |
| 6b | 87.830 ± 5.449 | 28.407 ± 2.639 | 3.1 |
| 6c | >100 | 22.097 ± 2.479 | >4.5 |
| 7c | 37.492 ± 0.476 | 9.447 ± 0.113 | 4.0 |
| 8c | 39.095 ± 5.144 | 12.727 ± 0.290 | 3.1 |
| 6d | 85.484 ± 1.585 | 1.554 ± 0.137 | 55.0 |
| 7d | 40.500 ± 2.374 | 0.417 ± 0.012 | 97.1 |
| 8d | 39.114 ± 0.555 | 2.185 ± 0.088 | 17.9 |
| Acacetin | 0.105 ± 0.0014 | 0.042 ± 0.0021 | 2.5 |
| Acacetin 7-O-methyl ether [11] | >100 | 0.198 ± 0.001 | >505.05 |
| Clorgyline | 0.0039 ± 0.0002 | 2.15 ± 0.212 | 0.002 |
| Deprenyl | 33.00 ± 1.411 | 0.046 ± 0.0014 | 717.4 |
| Safinamide [46] | 90.00 ± 2.470 | 0.060 ± 0.005 | 1500.0 |
| Harmine | 0.0031 ± 0.0003 | 39.000 ± 1.412 | 0.00008 |
| Compound | Monoamine Oxidase A | Monoamine Oxidase B | ||
|---|---|---|---|---|
| Ki (nM) * | Type of Inhibition | Ki (nM) * | Type of Inhibition | |
| 1c | - | - | 43 ± 3.8 | mixed/partially reversible |
| 2c | - | - | 52 ± 3.1 | mixed/reversible |
| 3c | - | - | 37 ± 9.5 | mixed/irreversible |
| 4c | - | - | 68 ± 7.1 | mixed/irreversible |
| Acacetin | 30 ± 1.8 | competitive/reversible | 21 ± 1.8 | competitive/reversible |
| Acacetin 7-O-methyl ether [11] | -- | 45 ± 3.0 | competitive/partially reversible | |
| Deprenyl | - | - | 43 ± 4.0 | mixed/irreversible |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gogineni, V.; Nael, M.A.; Chaurasiya, N.D.; Elokely, K.M.; McCurdy, C.R.; Rimoldi, J.M.; Cutler, S.J.; Tekwani, B.L.; León, F. Computationally Assisted Lead Optimization of Novel Potent and Selective MAO-B Inhibitors. Biomedicines 2021, 9, 1304. https://doi.org/10.3390/biomedicines9101304
Gogineni V, Nael MA, Chaurasiya ND, Elokely KM, McCurdy CR, Rimoldi JM, Cutler SJ, Tekwani BL, León F. Computationally Assisted Lead Optimization of Novel Potent and Selective MAO-B Inhibitors. Biomedicines. 2021; 9(10):1304. https://doi.org/10.3390/biomedicines9101304
Chicago/Turabian StyleGogineni, Vedanjali, Manal A. Nael, Narayan D. Chaurasiya, Khaled M. Elokely, Christopher R. McCurdy, John M. Rimoldi, Stephen J. Cutler, Babu L. Tekwani, and Francisco León. 2021. "Computationally Assisted Lead Optimization of Novel Potent and Selective MAO-B Inhibitors" Biomedicines 9, no. 10: 1304. https://doi.org/10.3390/biomedicines9101304
APA StyleGogineni, V., Nael, M. A., Chaurasiya, N. D., Elokely, K. M., McCurdy, C. R., Rimoldi, J. M., Cutler, S. J., Tekwani, B. L., & León, F. (2021). Computationally Assisted Lead Optimization of Novel Potent and Selective MAO-B Inhibitors. Biomedicines, 9(10), 1304. https://doi.org/10.3390/biomedicines9101304