
An Alternative Cell Therapy for Cancers: Induced Pluripotent Stem Cell (iPSC)-Derived Natural Killer Cells

 ,
,
Abstract
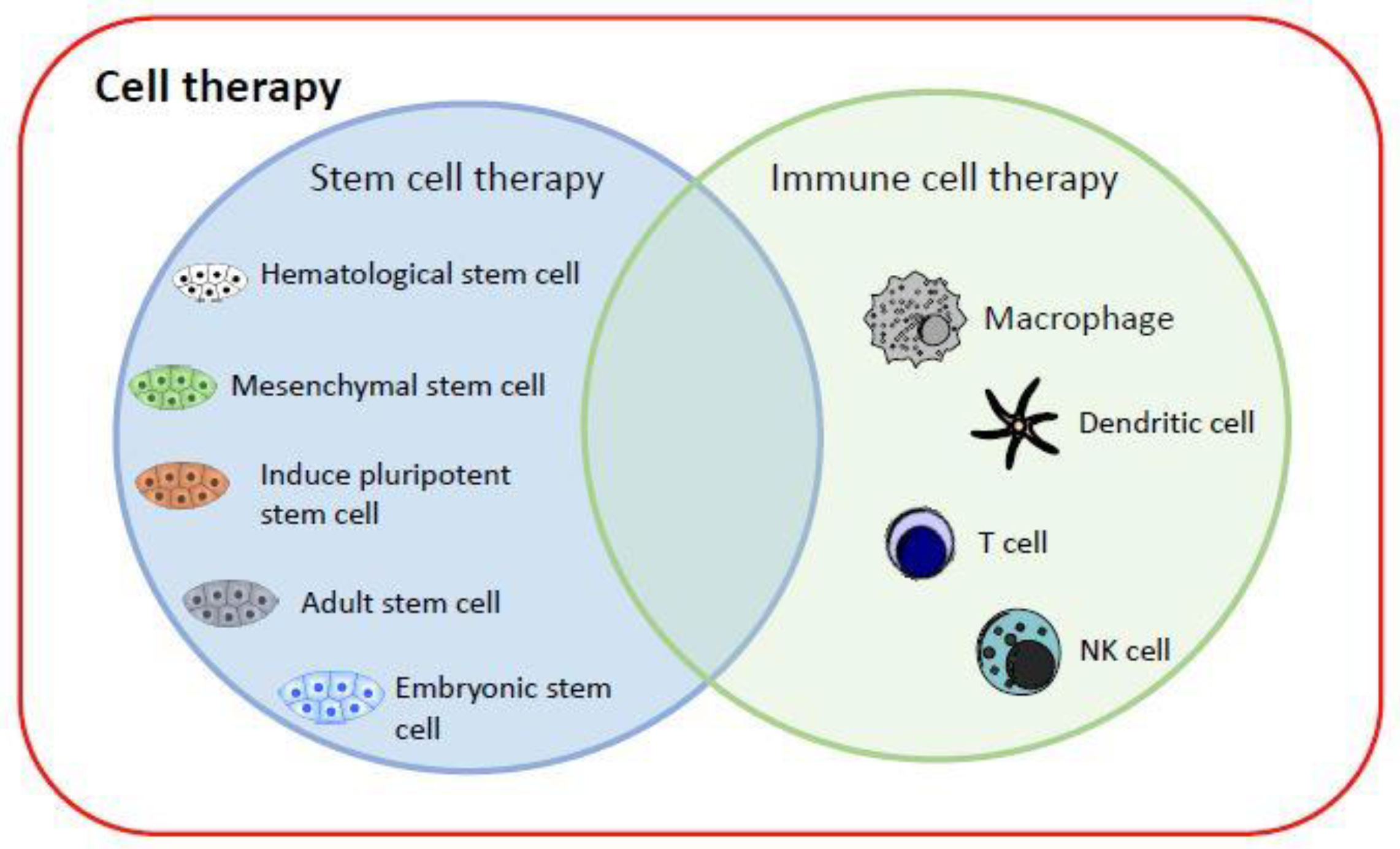
:1. Introduction
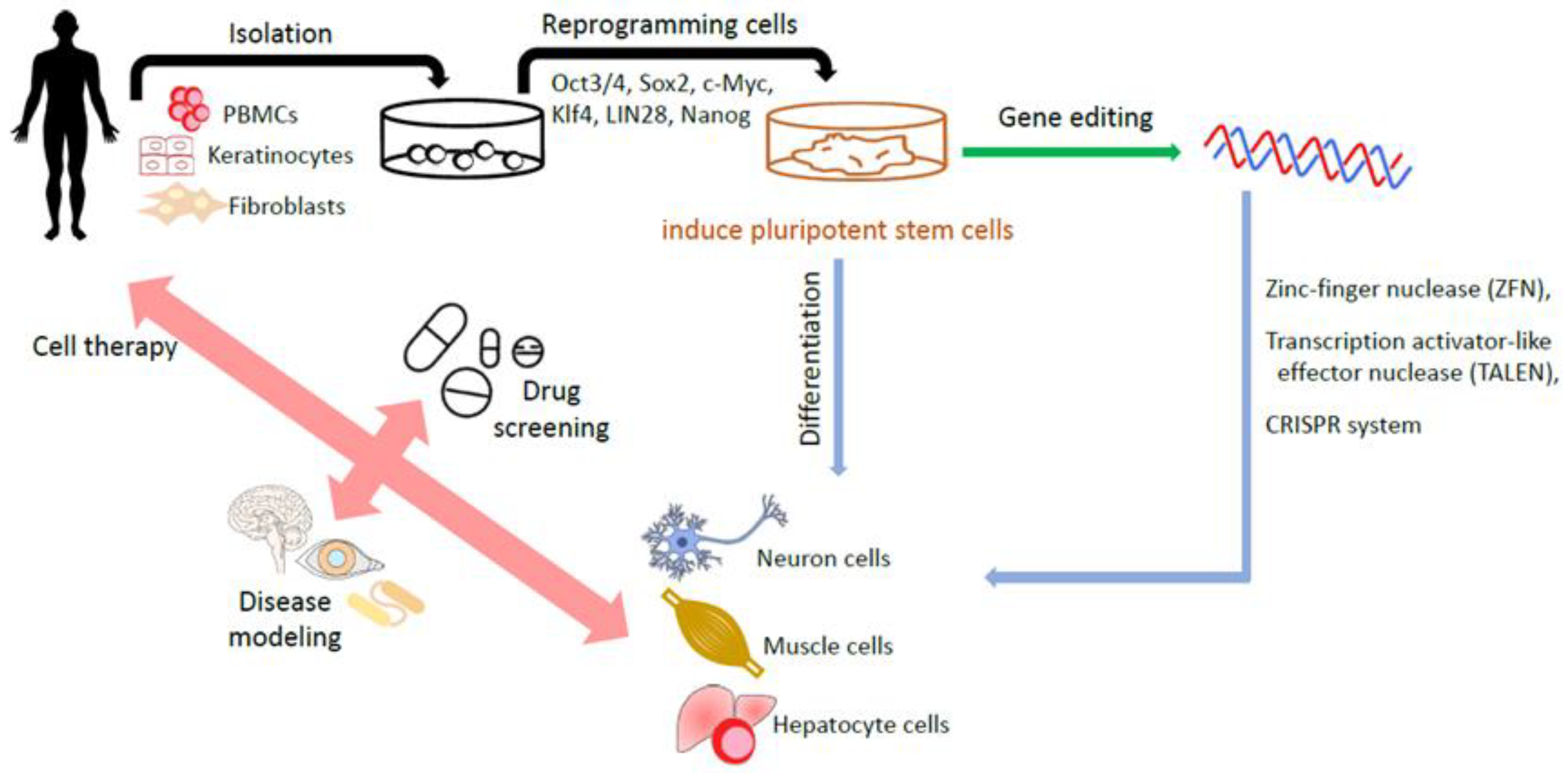
2. Induced Pluripotent Stem Cells (iPSCs)
2.1. Strategies for Generating iPSCs
2.2. Clinical Application of iPSC-Derived Products
- iPSC-DCs: In the study by Senju S et al., a method was developed to generate DCs from human iPSCs. These iPSC-DCs have the characteristics of original DCs, including the capability of T-cell stimulation, processing and presenting antigens, and producing cytokines [93]. Kitadani J et al. successfully established iPSC-DCs from the fibroblasts of healthy donors, as well as mouse iPSC-DCs from the iPS cell line iPS-MEF-Ng-20D-17 [94], which were derived from C57/BL6 MEFs. They demonstrated the therapeutic potential of mouse iPSC-DCs, in which the carcinoembryonic antigen (CEA) was transduced and expressed in a subcutaneous tumor model using CEA transgenic mice. These findings indicate that genetically modified iPSC-DCs, inducing the expression of CEA, are a promising strategy for the treatment of gastrointestinal cancer.
- iPSC-T: Adoptive immunotherapy with antigen-specific cytotoxic T lymphocytes (CTLs) represents a potential therapeutic strategy that can reduce tumor development and provide a survival advantage for patients undergoing cancer therapies. Maeda T et al. developed a simple method to generate antigen-specific CD8αβ T cells from the iPSCs of healthy volunteers and demonstrated their therapeutic potential against leukemia [95]. Wilms’ tumor antigen 1 (WT1)-specific CTLs regenerated by this method demonstrated antigen-specific cytotoxic activity in vitro and showed comparable potential to primary CTLs in producing IFNγ and TNFα [94]. When applied in vivo in a xenograft model, these CTLs prolonged the survival of mice bearing WT1-expressing leukemic cells [94]. Recent advances show the potential of the chimeric antigen receptor (CAR)-transduced T-cell immunotherapy for the treatment of a wide variety of diseases. Themeli M et al. demonstrated in clinical trials that CD19 CAR-modified T cells efficiently induce a complete remission in patients with acute or chronic lymphoblastic leukemias and eradicate B-cell malignancies in mice [96].
- iPSC-NK: The multiple dosing of allogeneic iPSC-NK cell therapy succeeded in treating solid tumors, such as ovarian cancer [97,98]. Hermanson D et al. established iPSCs derived from umbilical cord blood CD34+ cells, UCBiPS7, and derived iPSC-NK cells via spin embryoid bodies [98]. iPSC-NK cells were applied to treat NOD/SCID/γc−/− (NSG) mice, which were inoculated with ovarian cancer cells (MA148), and the median survival improved from 73 to 98 days. Moreover, such iPSC-NK cells were found in the peritoneal cavity of mice and were able to markedly inhibit tumor growth [97], indicating the therapeutic potential of iPSC-NK cells for treating solid tumors.
2.3. Generation of iPSC-Derived NK Cells
- PB-iPSCs with OP9: On day 0, iPSCs derived from peripheral blood cells (PB-iPSCs) were cocultured with OP9 cells (a bone marrow stromal cell line) in αMEM with 20% fetal bovine serum (FBS). On day 12, the modified OP9 cell line expressing Notch ligand Delta-like-1 (OP9-DLL1) replaced OP9 cells and was cocultured with the above iPSCs (mainly CD34+) in the presence of the stem cell factor (SCF) and Flt3L, together with IL-7 and IL-15. On day 26, a small population of CD45+CD56+ cells appeared; the CD45+CD56+ cells became the dominant population, with a purity of 99% on day 40. A yield of 7.93 × 106 CD45+CD56+ cells was obtained on day 40 and increased to 15 × 106 cells on day 47 [104].
- FB-iPSCs with OP9: On day 0, the iPSC cell line from primate skin fibroblasts (FB-iPSCs) was cocultured with OP9 in αMEM containing 20% FBS and supplemented with a basic fibroblast growth factor (bFGF), activin A, vascular endothelial growth factor (VEGF) and CHIR99021. On days 6 and 8, SCF, thrombopoietin (TPO), IL-3 and IL-6 were added to the above culture medium for the differentiation of mesodermal cells to hematopoietic stem cells. On day 10, the floating cells were harvested and cocultured with the modified OP9 cell line expressing Notch ligand Delta-like-4 (OP9-DLL4) in αMEM containing 20% FBS, in addition to IL-7, FLT3L and IL-2 for up to 4 weeks. On day 38, a yield of 1.0–3.5 × 106 iPSC NK cells expressing perforin and IFNγ was obtained [105].
- CB-iPSC: On day 0, UCBiPS7 iPSCs derived from umbilical cord blood CD34+ cells were seeded in round-bottomed plates for the development of embryoid bodies in a BPEL culture medium (bovine serum albumin, polyvinyl alcohol, essential lipids) containing SCF, VEGF and bone morphogenic protein 4 (BMP-4). During days 8–12, the formed embryoid bodies (EBs) containing CD34+CD43+ cells were directly transferred into flat-bottomed plates, and BPELs were cultured in the presence of IL-3, IL-7, IL-15, SCF and FLT3L. After 28~32 days, iPSC-NK cells were obtained and expanded in RPMI-1640 containing 10% FBS, 1% penicillin/streptomycin and 50 units/mL IL-2 and stimulated with irradiated (10,000 cGy) artificial antigen-presenting cells (aAPCs) (2:1 v/v) upon initiation of culture. The culture medium was changed twice weekly, and iPSC-NK cells could be restimulated with aAPCs every 7 days. The purity of the expanded NK cells almost reached 97% [97].
- CB-iPSC: On day 0, the iPSC cell lines 409B7 (B7) and CB-A11 (A11), derived from cord blood mononuclear cells, were seeded in iMatrix 511-coated plates (Osaka, Japan) for the development of EBs in an Essential 8 culture medium supplemented with CHIR99021, BMP-4, and VEGF. On day 2, the formed EBs containing CD34+ cells were cultured in an Essential 6 culture medium containing SB431542, SCF, and VEGF. During days 4–12, the formed EBs appeared to contain CD34+, CD43+ and CD45+ hemoangiogenic progenitor cells (HPCs) and were cultured in a Stem Line II medium together with SCF and Flt3L. From day 12 onwards, the cells were cultured in DMEM containing 20% human AB serum or a Stem Line II, in addition to SCF, Flt3L, IL-7 and IL-15. On day 48, the purity of iPSC-NK cells reached 63.10 ± 7.01%~78.23 ± 5.66% [106].
3. Natural Killer Cells (NK cells)
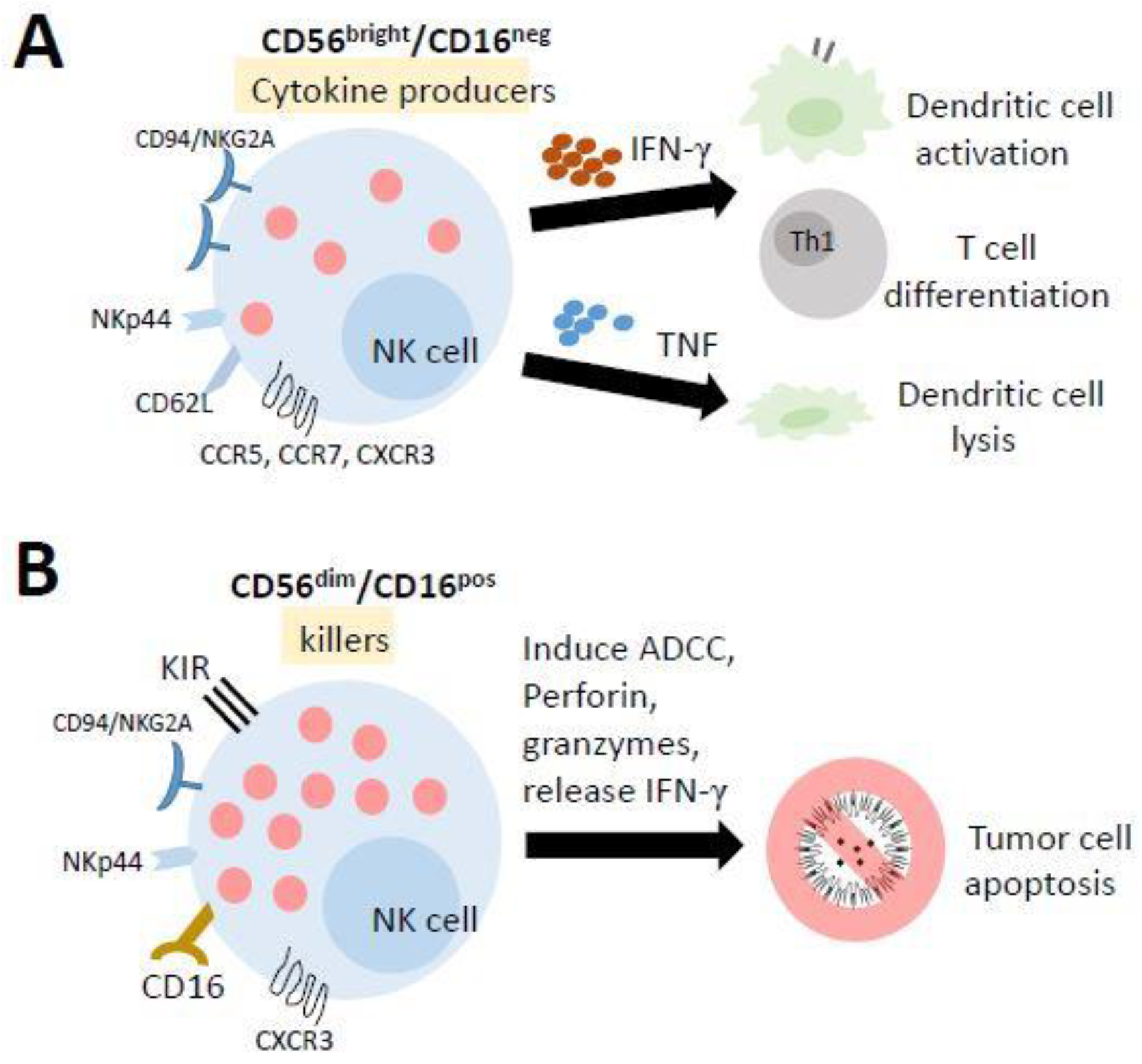
3.1. The Physiological Conditions in NK Cells
3.2. NK Cell Education
3.3. NK Cell-Based Therapy in Tumors
4. Conclusions
Funding
Conflicts of Interest
References
- Chan, M.K.S.; Wong, M.B.F.; Nallenthiran, L.; Klokol, D. Live cell therapy: Historical aspects, mechanisms of action, safety and success stories. J. Stem Cell Res. Ther. 2019, 5, 5. [Google Scholar]
- Kumar, A.; D’Souza, S.S.; Thakur, A.S. Understanding the Journey of Human Hematopoietic Stem Cell Development. Stem Cells Int. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrzewski, W.; Dobrzyński, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 1–22. [Google Scholar] [CrossRef]
- Mukhopadhyay, M. Macrophages enter CAR immunotherapy. Nat. Methods 2020, 17, 559. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Mamonkin, M.; Brenner, M.K.; Heslop, H.E. Taking T-Cell Oncotherapy Off-the-Shelf. Trends Immunol. 2021, 42, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Sadeghzadeh, M.; Bornehdeli, S.; Mohahammadrezakhani, H.; Abolghasemi, M.; Poursaei, E.; Asadi, M.; Zafari, V.; Aghebati-Maleki, L.; Shanehbandi, D. Dendritic cell therapy in cancer treatment; the state-of-the-art. Life Sci. 2020, 254, 117580. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Chu, D.T.; Nguyen, T.T.; Tien, N.L.B.; Tran, D.K.; Jeong, J.H.; Anh, P.G.; Thanh, V.V.; Truong, D.T.; Dinh, T.C. Recent progress of stem cell therapy in cancer treatment: Molecular mechanisms and potential applications. Cells 2020, 9, 563. [Google Scholar] [CrossRef] [Green Version]
- Nianias, A.; Themeli, M. Induced pluripotent stem cell (ipsc)-derived lymphocytes for adoptive cell immunotherapy: Recent advances and challenges. Curr. Hematol. Malig. Rep. 2019, 14, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Ronson, A.; Tvito, A.; Rowe, J.M. Treatment of Relapsed/Refractory Acute Lymphoblastic Leukemia in Adults. Curr. Oncol. Rep. 2016, 18, 36. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.J. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin. Immunopathol 2019, 41, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Jiang, J.; Wu, C. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2020, 472, 175–180. [Google Scholar] [CrossRef]
- Mehta, R.S.; Randolph, B.; Daher, M.; Rezvani, K. NK cell therapy for hematologic malignancies. Int. J. Hematol. 2018, 107, 262–270. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, N.; Ishikawa, T.; Kokura, S.; Okayama, T.; Oka, K.; Ideno, M.; Sakai, F.; Kato, A.; Tanabe, M.; Enoki, T.; et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J. Transl. Med. 2015, 13, 277. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; DeFor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.-H.; Leung, W. NKAML: A Pilot Study to Determine the Safety and Feasibility of Haploidentical Natural Killer Cell Transplantation in Childhood Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliopoulou, E.G.; Kountourakis, P.; Karamouzis, M.V.; Doufexis, D.; Ardavanis, A.; Baxevanis, C.N.; Rigatos, G.; Papamichail, M.; Perez, S.A. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol. Immunother. 2010, 59, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Basar, R.; Daher, M.; Rezvani, K. Next-generation cell therapies: The emerging role of CAR-NK cells. Blood Adv. 2020, 4, 5868–5876. [Google Scholar] [CrossRef]
- Hodgins, J.J.; Khan, S.T.; Park, M.M.; Auer, R.C.; Ardolino, M. Killers 2.0: NK cell therapies at the forefront of cancer control. J. Clin. Investig. 2019, 129, 3499–3510. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, J.; Chen, T.; Wang, Y.; Xin, S.; Li, J.; Pei, G.; Kang, J. Yamanaka factors critically regulate the developmental signaling network in mouse embryonic stem cells. Cell Res. 2008, 18, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nat. Cell Biol. 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, I.; Maeda, T.; Shimada, H.; Kawai, K.; Okada, Y.; Igarashi, H.; Oiwa, R.; Iwasaki, T.; Aoki, M.; Kimura, T.; et al. Generating induced pluripotent stem cells from common marmoset (Callithrix jacchus) fetal liver cells using defined factors, including Lin28. Genes Cells 2010, 15, 959–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Jiang, J.; Kraus, P.; Heng, J.-C.D.; Chan, Y.-S.; Yaw, L.-P.; Zhang, W.; Loh, Y.-H.; Han, J.; Vega, V.B.; et al. Reprogramming of fibroblasts into induced pluripotent stem cells with orphan nuclear receptor Esrrb. Nat. Cell Biol. 2009, 11, 197–203. [Google Scholar] [CrossRef]
- Loh, Y.-H.; Agarwal, S.; Park, I.-H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, G.Q. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 5476–5479. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, A.; Montserrat, N.; Aasen, T.; Gonzalez, F.; Rodríguez-Pizà, I.; Vassena, R.; Raya, A.; Boue, S.; Barrero, M.; Corbella, B.A.; et al. Generation of Induced Pluripotent Stem Cells from Human Cord Blood Using OCT4 and SOX2. Cell Stem Cell 2009, 5, 353–357. [Google Scholar] [CrossRef] [Green Version]
- Eminli, S.; Foudi, A.; Stadtfeld, M.; Maherali, N.; Ahfeldt, T.; Mostoslavsky, G.; Hock, H.; Hochedlinger, K. Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nat. Genet. 2009, 41, 968–976. [Google Scholar] [CrossRef] [Green Version]
- Aoki, T.; Ohnishi, H.; Oda, Y.; Tadokoro, M.; Sasao, M.; Kato, H.; Hattori, K.; Ohgushi, H. Generation of Induced Pluripotent Stem Cells from Human Adipose-Derived Stem Cells Without c-MYC. Tissue Eng. Part A 2010, 16, 2197–2206. [Google Scholar] [CrossRef]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2007, 26, 101–106. [Google Scholar] [CrossRef]
- Buganim, Y.; Markoulaki, S.; van Wietmarschen, N.; Hoke, H.; Wu, T.; Ganz, K.; Akhtar-Zaidi, B.; He, Y.; Abraham, B.J.; Porubsky, D.; et al. The Developmental Potential of iPSCs Is Greatly Influenced by Reprogramming Factor Selection. Cell Stem Cell 2014, 15, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Utikal, J.; Maherali, N.; Kulalert, W.; Hochedlinger, K. Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J. Cell Sci. 2009, 122, 3502–3510. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.B.; Greber, B.; Arauzo-Bravo, M.J.; Meyer, J.; Park, K.I.; Zaehres, H.; Scholer, H.R. Direct reprogramming of human neural stem cells by oct4. Nature 2009, 461, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ye, Z.; Kim, Y.; Sharkis, S.; Jang, Y.Y. Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology 2010, 51, 1810–1819. [Google Scholar] [CrossRef]
- Sugii, S.; Kida, Y.; Kawamura, T.; Suzuki, J.; Vassena, R.; Yin, Y.Q.; Lutz, M.K.; Berggren, W.T.; Izpisua Belmonte, J.C.; Evans, R.M. Human and mouse adipose-derived cells support feeder-independent induction of pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 3558–3563. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.B.; Zaehres, H.; Wu, G.; Gentile, L.; Ko, K.; Sebastiano, V.; Araúzo-Bravo, M.J.; Ruau, D.; Han, D.W.; Zenke, M.; et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nat. Cell Biol. 2008, 454, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-Specific Induced Pluripotent Stem Cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Omole, A.E.; Fakoya, A.O.J. Ten years of progress and promise of induced pluripotent stem cells: Historical origins, characteristics, mechanisms, limitations, and potential applications. PeerJ 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzino, A. Concise review: The sox2-oct4 connection: Critical players in a much larger interdependent network integrated at multiple levels. Stem Cells 2013, 31, 1033–1039. [Google Scholar] [CrossRef] [Green Version]
- Avilion, A.A.; Nicolis, S.K.; Pevny, L.H.; Perez, L.; Vivian, N.; Lovell-Badge, R. Multipotent cell lineages in early mouse development depend on sox2 function. Genes. Dev. 2003, 17, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Kopp, J.L.; Ormsbee, B.D.; Desler, M.; Rizzino, A. Small increases in the level of sox2 trigger the differentiation of mouse embryonic stem cells. Stem Cells 2008, 26, 903–911. [Google Scholar] [CrossRef]
- Shi, G.; Jin, Y. Role of oct4 in maintaining and regaining stem cell pluripotency. Stem Cell Res. Ther. 2010, 1, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, J.; Zevnik, B.; Anastassiadis, K.; Niwa, H.; Klewe-Nebenius, D.; Chambers, I.; Schöler, H.; Smith, A. Formation of Pluripotent Stem Cells in the Mammalian Embryo Depends on the POU Transcription Factor Oct4. Cell 1998, 95, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Matz, P.; Adjaye, J. Episomal-based generation of an iPS cell line from human fetal foreskin fibroblasts. Stem Cell Res. 2016, 16, 67–69. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, K.; Kawabata, K.; Inamura, M.; Takayama, K.; Furukawa, N.; Sakurai, F.; Katayama, K.; Hayakawa, T.; Furue, M.K.; Mizuguchi, H. Adenovirus Vector-Mediated Efficient Transduction into Human Embryonic and Induced Pluripotent Stem Cells. Cell. Reprogramming 2010, 12, 501–507. [Google Scholar] [CrossRef]
- Chen, I.P.; Fukuda, K.; Fusaki, N.; Iida, A.; Hasegawa, M.; Lichtler, A.; Reichenberger, E.J. Induced pluripotent stem cell reprogramming by integration-free sendai virus vectors from peripheral blood of patients with craniometaphyseal dysplasia. Cell Reprogram 2013, 15, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Woltjen, K.; Michael, I.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nat. Cell Biol. 2009, 458, 766–770. [Google Scholar] [CrossRef]
- Yu, J.; Chau, K.F.; Vodyanik, M.A.; Jiang, J.; Jiang, Y. Efficient Feeder-Free Episomal Reprogramming with Small Molecules. PLoS ONE 2011, 6, e17557. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of Induced Pluripotent Stem Cells Using Recombinant Proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef] [Green Version]
- Kogut, I.; McCarthy, S.M.; Pavlova, M.; Astling, D.P.; Chen, X.; Jakimenko, A.; Jones, K.L.; Getahun, A.; Cambier, J.C.; Pasmooij, A.M.G.; et al. High-efficiency RNA-based reprogramming of human primary fibroblasts. Nat. Commun. 2018, 9, 745. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.S.; Li, Z.; Rana, T.M. Micrornas modulate ips cell generation. RNA 2011, 17, 1451–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si-Tayeb, K.; Noto, F.K.; Sepac, A.; Sedlic, F.; Bosnjak, Z.J.; Lough, J.W.; Duncan, S.A. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev. Biol. 2010, 10, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narsinh, K.H.; Jia, F.; Robbins, R.C.; Kay, M.A.; Longaker, M.T.; Wu, J.C. Generation of adult human induced pluripotent stem cells using nonviral minicircle DNA vectors. Nat. Protoc. 2010, 6, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Woltjen, K.; Hämäläinen, R.; Kibschull, M.; Mileikovsky, M.; Nagy, A. Transgene-Free Production of Pluripotent Stem Cells Using piggyBac Transposons. Cardiovasc. Dev. 2011, 767, 87–103. [Google Scholar] [CrossRef]
- Zhou, Y.-Y.; Zeng, F. Integration-free Methods for Generating Induced Pluripotent Stem Cells. Genom. Proteom. Bioinform. 2013, 11, 284–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haridhasapavalan, K.K.; Borgohain, M.P.; Dey, C.; Saha, B.; Narayan, G.; Kumar, S.; Thummer, R.P. An insight into non-integrative gene delivery approaches to generate transgene-free induced pluripotent stem cells. Gene 2019, 686, 146–159. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Stoddard-Bennett, T.; Pera, R.R. Treatment of Parkinson’s Disease through Personalized Medicine and Induced Pluripotent Stem Cells. Cells 2019, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Kouroupi, G.; Antoniou, N.; Prodromidou, K.; Taoufik, E.; Matsas, R. Patient-Derived Induced Pluripotent Stem Cell-Based Models in Parkinson’s Disease for Drug Identification. Int. J. Mol. Sci. 2020, 21, 7113. [Google Scholar] [CrossRef]
- Kondo, T.; Imamura, K.; Funayama, M.; Tsukita, K.; Miyake, M.; Ohta, A.; Woltjen, K.; Nakagawa, M.; Asada, T.; Arai, T.; et al. Ipsc-based compound screening and in vitro trials identify a synergistic anti-amyloid beta combination for alzheimer’s disease. Cell Rep. 2017, 21, 2304–2312. [Google Scholar] [CrossRef] [Green Version]
- Yahata, N.; Asai, M.; Kitaoka, S.; Takahashi, K.; Asaka, I.; Hioki, H.; Kaneko, T.; Maruyama, K.; Saido, T.C.; Nakahata, T.; et al. Anti-abeta drug screening platform using human ips cell-derived neurons for the treatment of alzheimer’s disease. PLoS ONE 2011, 6, e25788. [Google Scholar] [CrossRef] [PubMed]
- Pomeshchik, Y.; Puttonen, K.; Kidin, I.; Ruponen, M.; Lehtonen, S.; Malm, T.; Åkesson, E.; Hovatta, O.; Koistinaho, J. Transplanted Human Induced Pluripotent Stem Cell-Derived Neural Progenitor Cells Do Not Promote Functional Recovery of Pharmacologically Immunosuppressed Mice with Contusion Spinal Cord Injury. Cell Transplant. 2015, 24, 1799–1812. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, O.; Miura, K.; Okada, Y.; Fujiyoshi, K.; Mukaino, M.; Nagoshi, N.; Kitamura, K.; Kumagai, G.; Nishino, M.; Tomisato, S.; et al. Therapeutic potential of appropriately evaluated safe-induced pluripotent stem cells for spinal cord injury. Proc. Natl. Acad. Sci. USA 2010, 107, 12704–12709. [Google Scholar] [CrossRef] [Green Version]
- Kimbrel, E.A.; Lanza, R. Next-generation stem cells—ushering in a new era of cell-based therapies. Nat. Rev. Drug Discov. 2020, 19, 463–479. [Google Scholar] [CrossRef] [PubMed]
- Oji, A.; Noda, T.; Fujihara, Y.; Miyata, H.; Kim, Y.J.; Muto, M.; Nozawa, K.; Matsumura, T.; Isotani, A.; Ikawa, M. CRISPR/Cas9 mediated genome editing in ES cells and its application for chimeric analysis in mice. Sci. Rep. 2016, 6, 31666. [Google Scholar] [CrossRef] [PubMed]
- Ferreccio, A.; Mathieu, J.; Detraux, D.; Logeshwaran, S.; Cavanaugh, C.; Sopher, B.; Fischer, K.; Bello, T.; Hussein, A.M.; Levy, S.; et al. Inducible CRISPR genome editing platform in naive human embryonic stem cells reveals JARID2 function in self-renewal. Cell Cycle 2018, 17, 535–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grobarczyk, B.; Franco, B.; Hanon, K.; Malgrange, B. Generation of isogenic human ips cell line precisely corrected by genome editing using the crispr/cas9 system. Stem Cell Rev. Rep. 2015, 11, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, Y.; Yan, W.; Smith, C.; Ye, Z.; Wang, J.; Gao, Y.; Mendelsohn, L.; Cheng, L. Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient ipscs after genome editing of the sickle point mutation. Stem Cells 2015, 33, 1470–1479. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Maeder, M.L.; Gersbach, C.A. Genome-editing Technologies for Gene and Cell Therapy. Mol. Ther. 2016, 24, 430–446. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F. CRISPR/Cas9: Prospects and Challenges. Hum. Gene Ther. 2015, 26, 409–410. [Google Scholar] [CrossRef] [Green Version]
- Bak, R.O.; Dever, D.P.; Porteus, M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018, 13, 358–376. [Google Scholar] [CrossRef] [Green Version]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. Crispr-cas9 gene editing for sickle cell disease and beta-thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ren, S.; Yu, S.; Pan, H.; Li, T.; Ge, S.; Zhang, J.; Xia, N. Methods favoring homology-directed repair choice in response to crispr/cas9 induced-double strand breaks. Int. J. Mol. Sci. 2020, 21, 6461. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; de Haas, M.; Holland, H.A.; Akhtar, W.; van Steensel, B. Kinetics and fidelity of the repair of cas9-induced double-strand DNA breaks. Mol. Cell 2018, 70, 801–813.e806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradi, S.; Mahdizadeh, H.; Šarić, T.; Kim, J.; Harati, J.; Shahsavarani, H.; Greber, B.; Iv, J.B.M. Research and therapy with induced pluripotent stem cells (iPSCs): Social, legal, and ethical considerations. Stem Cell Res. Ther. 2019, 10, 341. [Google Scholar] [CrossRef] [Green Version]
- Kanemura, H.; Go, M.J.; Shikamura, M.; Nishishita, N.; Sakai, N.; Kamao, H.; Mandai, M.; Morinaga, C.; Takahashi, M.; Kawamata, S. Tumorigenicity studies of induced pluripotent stem cell (ipsc)-derived retinal pigment epithelium (rpe) for the treatment of age-related macular degeneration. PLoS ONE 2014, 9, e85336. [Google Scholar] [CrossRef]
- Sugita, S. Retinal regeneration with iPS cells—Clinical trials for retinal degenerative disorders. Jpn. J. Clin. Immunol. 2015, 38, 7. [Google Scholar] [CrossRef] [Green Version]
- Sugita, S.; Iwasaki, Y.; Makabe, K.; Kamao, H.; Mandai, M.; Shiina, T.; Ogasawara, K.; Hirami, Y.; Kurimoto, Y.; Takahashi, M. Successful Transplantation of Retinal Pigment Epithelial Cells from MHC Homozygote iPSCs in MHC-Matched Models. Stem Cell Rep. 2016, 7, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Koga, K.; Wang, B.; Kaneko, S. Current status and future perspectives of HLA-edited induced pluripotent stem cells. Inflamm. Regen. 2020, 40, 23. [Google Scholar] [CrossRef]
- Umekage, M.; Sato, Y.; Takasu, N. Overview: An iPS cell stock at CiRA. Inflamm. Regen. 2019, 39, 17. [Google Scholar] [CrossRef] [Green Version]
- Deinsberger, J.; Reisinger, D.; Weber, B. Global trends in clinical trials involving pluripotent stem cells: A systematic multi-database analysis. NPJ Regen. Med. 2020, 5, 15. [Google Scholar] [CrossRef]
- Nagoshi, N.; Tsuji, O.; Nakamura, M.; Okano, H. Cell therapy for spinal cord injury using induced pluripotent stem cells. Regen. Ther. 2019, 11, 75–80. [Google Scholar] [CrossRef]
- Tewarie, R.S.N.; Hurtado, A.; Bartels, R.H.; Grotenhuis, A.; Martin Oudega, P. Stem cell–based therapies for spinal cord injury. J. Spinal. Cord. Med. 2009, 32, 10. [Google Scholar]
- Kobayashi, Y.; Okada, Y.; Itakura, G.; Iwai, H.; Nishimura, S.; Yasuda, A.; Nori, S.; Hikishima, K.; Konomi, T.; Fujiyoshi, K.; et al. Pre-evaluated safe human iPSC-derived neural stem cells promote functional recovery after spinal cord injury in common marmoset without tumorigenicity. PLoS ONE 2012, 7, e52787. [Google Scholar]
- Nori, S.; Okada, Y.; Yasuda, A.; Tsuji, O.; Takahashi, Y.; Kobayashi, Y.; Fujiyoshi, K.; Koike, M.; Uchiyama, Y.; Ikeda, E.; et al. Grafted human-induced pluripotent stem-cell-derived neurospheres promote motor functional recovery after spinal cord injury in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 16825–16830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, O.; Sugai, K.; Yamaguchi, R.; Tashiro, S.; Nagoshi, N.; Kohyama, J.; Iida, T.; Ohkubo, T.; Itakura, G.; Isoda, M.; et al. Concise Review: Laying the Groundwork for a First-In-Human Study of an Induced Pluripotent Stem Cell-Based Intervention for Spinal Cord Injury. Stem Cells 2019, 37, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Eintracht, J.; Toms, M.; Moosajee, M. The Use of Induced Pluripotent Stem Cells as a Model for Developmental Eye Disorders. Front. Cell. Neurosci. 2020, 14, 265. [Google Scholar] [CrossRef]
- Senju, S.; Matsunaga, Y.; Fukushima, S.; Hirata, S.; Motomura, Y.; Fukuma, D.; Matsuyoshi, H.; Nishimura, Y. Immunotherapy with pluripotent stem cell-derived dendritic cells. Semin. Immunopathol 2011, 33, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Kitadani, J.; Ojima, T.; Iwamoto, H.; Tabata, H.; Nakamori, M.; Nakamura, M.; Hayata, K.; Katsuda, M.; Miyajima, M.; Yamaue, H. Cancer vaccine therapy using carcinoembryonic antigen—expressing dendritic cells generated from induced pluripotent stem cells. Sci. Rep. 2018, 8, 4569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, T.; Nagano, S.; Ichise, H.; Kataoka, K.; Yamada, D.; Ogawa, S.; Koseki, H.; Kitawaki, T.; Kadowaki, N.; Takaori-Kondo, A.; et al. Regeneration of cd8alphabeta t cells from t-cell-derived ipsc imparts potent tumor antigen-specific cytotoxicity. Cancer Res. 2016, 76, 6839–6850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, D.L.; Bendzick, L.; Pribyl, L.; McCullar, V.; Vogel, R.I.; Miller, J.S.; Geller, M.A.; Kaufman, D.S. Induced pluripotent stem cell-derived natural killer cells for treatment of ovarian cancer. Stem Cells 2016, 34, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, E.S.; Davis, R.; Stanley, E.G.; Elefanty, A.G. A protocol describing the use of a recombinant protein-based, animal product-free medium (apel) for human embryonic stem cell differentiation as spin embryoid bodies. Nat. Protoc. 2008, 3, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Iriguchi, S.; Yasui, Y.; Kawai, Y.; Arima, S.; Kunitomo, M.; Sato, T.; Ueda, T.; Minagawa, A.; Mishima, Y.; Yanagawa, N.; et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat. Commun. 2021, 12, 430. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Shankar, K.; Capitini, C.M.; Saha, K. Genome engineering of induced pluripotent stem cells to manufacture natural killer cell therapies. Stem Cell Res. Ther. 2020, 11, 234. [Google Scholar] [CrossRef]
- Morvan, M.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Bachanova, V.; Cooley, S.; DeFor, T.E.; Verneris, M.R.; Zhang, B.; McKenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lewis, D.; Hippen, K.; et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood 2014, 123, 3855–3863. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Tang, S.Y.; Toh, L.L.; Wang, S. Generation of “Off-the-Shelf” Natural Killer Cells from Peripheral Blood Cell-Derived Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 1796–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, S.S.; Maufort, J.; Kumar, A.; Zhang, J.; Smuga-Otto, K.; Thomson, J.A.; Slukvin, I.I. Gsk3beta inhibition promotes efficient myeloid and lymphoid hematopoiesis from non-human primate-induced pluripotent stem cells. Stem Cell Rep. 2016, 6, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubara, H.; Niwa, A.; Nakahata, T.; Saito, M.K. Induction of human pluripotent stem cell-derived natural killer cells for immunotherapy under chemically defined conditions. Biochem. Biophys. Res. Commun. 2019, 515, 1–8. [Google Scholar] [CrossRef]
- Oh, S.; Lee, J.-H.; Kwack, K.; Choi, S.-W. Natural Killer Cell Therapy: A New Treatment Paradigm for Solid Tumors. Cancers 2019, 11, 1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trabanelli, S.; Gomez-Cadena, A.; Salomé, B.; Michaud, K.; Mavilio, D.; Landis, B.N.; Jandus, P.; Jandus, C. Human innate lymphoid cells (ILCs): Toward a uniform immune-phenotyping. Cytom. Part B Clin. Cytom. 2018, 94, 392–399. [Google Scholar] [CrossRef]
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef]
- Zhu, L.; Kalimuthu, S.; Oh, J.M.; Gangadaran, P.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Enhancement of antitumor potency of extracellular vesicles derived from natural killer cells by il-15 priming. Biomaterials 2019, 190-191, 38–50. [Google Scholar] [CrossRef]
- O’Brien, K.L.; Finlay, D.K. Immunometabolism and natural killer cell responses. Nat. Rev. Immunol. 2019, 19, 282–290. [Google Scholar] [CrossRef]
- Anft, M.; Netter, P.; Urlaub, D.; Prager, I.; Schaffner, S.; Watzl, C. Nk cell detachment from target cells is regulated by successful cytotoxicity and influences cytokine production. Cell Mol. Immunol. 2020, 17, 347–355. [Google Scholar] [CrossRef]
- Michel, T.; Poli, A.; Cuapio, A.; Briquemont, B.; Iserentant, G.; Ollert, M.; Zimmer, J. Human CD56bright NK Cells: An Update. J. Immunol. 2016, 196, 2923–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Freud, A.G.; Caligiuri, M.A. Location and cellular stages of natural killer cell development. Trends Immunol. 2013, 34, 573–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, A.; Michel, T.; Theresine, M.; Andres, E.; Hentges, F.; Zimmer, J. Cd56bright natural killer (nk) cells: An important nk cell subset. Immunology 2009, 126, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Kaufman, D.S. Engineered human pluripotent stem cell-derived natural killer cells: The next frontier for cancer immunotherapy. Blood Sci. 2019, 1, 4–11. [Google Scholar] [CrossRef]
- Malhotra, A.; Shanker, A. NK cells: Immune cross-talk and therapeutic implications. Immunotherapy 2011, 3, 1143–1166. [Google Scholar] [CrossRef] [Green Version]
- Zamai, L.; Ponti, C.; Mirandola, P.; Gobbi, G.; Papa, S.; Galeotti, L.; Cocco, L.; Vitale, M. NK cells and cancer. J. Immunol. 2007, 178, 4011–4016. [Google Scholar] [CrossRef] [Green Version]
- Di Vito, C.; Mikulak, J.; Zaghi, E.; Pesce, S.; Marcenaro, E.; Mavilio, D. NK cells to cure cancer. Semin. Immunol. 2019, 41, 101272. [Google Scholar] [CrossRef]
- Nigro, C.L.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.J.; Wu, J.; Walcheck, B. Engineering anti-tumor monoclonal antibodies and fc receptors to enhance adcc by human nk cells. Cancers (Basel) 2021, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minetto, P.; Guolo, F.; Pesce, S.; Greppi, M.; Obino, V.; Ferretti, E.; Sivori, S.; Genova, C.; Lemoli, R.M.; Marcenaro, E. Harnessing nk cells for cancer treatment. Front. Immunol 2019, 10, 2836. [Google Scholar] [CrossRef] [Green Version]
- Barrow, A.D.; Martin, C.J.; Colonna, M. The Natural Cytotoxicity Receptors in Health and Disease. Front. Immunol. 2019, 10, 909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, M.J.; Swann, J.; Kelly, J.M.; Cretney, E.; Yokoyama, W.M.; Diefenbach, A.; Sayers, T.J.; Hayakawa, Y. Nkg2d recognition and perforin effector function mediate effective cytokine immunotherapy of cancer. J. Exp. Med. 2004, 200, 1325–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldenson, B.H.; Zhu, H.; Wang, Y.M.; Heragu, N.; Bernareggi, D.; Ruiz-Cisneros, A.; Bahena, A.; Ask, E.H.; Hoel, H.J.; Malmberg, K.J.; et al. Umbilical cord blood and ipsc-derived natural killer cells demonstrate key differences in cytotoxic activity and kir profiles. Front. Immunol. 2020, 11, 561553. [Google Scholar] [CrossRef]
- Konjević, G.; Vuletić, A.; Martinović, K.M.; Džodić, R. The Role of Activating and Inhibitory NK Cell Receptors in Antitumor Immune Response. Nat. Kill. Cells 2017, 49–65. [Google Scholar] [CrossRef] [Green Version]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Terunuma, H.; Deng, X.; Dewan, Z.; Fujimoto, S.; Yamamoto, N. Potential role of nk cells in the induction of immune responses: Implications for nk cell-based immunotherapy for cancers and viral infections. Int. Rev. Immunol. 2008, 27, 93–110. [Google Scholar] [CrossRef]
- Larsen, S.K.; Gao, Y.; Basse, P.H. Nk cells in the tumor microenvironment. Crit. Rev. Oncog. 2014, 19, 91–105. [Google Scholar] [CrossRef] [Green Version]
- Bald, T.; Krummel, M.F.; Smyth, M.J.; Barry, K.C. The NK cell–cancer cycle: Advances and new challenges in NK cell–based immunotherapies. Nat. Immunol. 2020, 21, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Matosevic, S. Chemokine networks modulating natural killer cell trafficking to solid tumors. Cytokine Growth Factor Rev. 2021, 59, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J. Role of chemokines in the biology of natural killer cells. J. Leukoc. Biol. 2002, 71, 173–183. [Google Scholar] [PubMed]
- Yang, H.; Zhang, Q.; Xu, M.; Wang, L.; Chen, X.; Feng, Y.; Li, Y.; Zhang, X.; Cui, W.; Jia, X. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol. Cancer 2020, 19, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiskala, M.; Leidenius, M.; Joensuu, K.; Heikkilä, P. High expression of CCL2 in tumor cells and abundant infiltration with CD14 positive macrophages predict early relapse in breast cancer. Virchows Arch. 2019, 474, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Terrén, I.; Orrantia, A.; Vitallé, J.; Zenarruzabeitia, O.; Borrego, F. NK Cell Metabolism and Tumor Microenvironment. Front. Immunol. 2019, 10, 2278. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Bluman, E.M.; Porter, M.M.; Mrozek, E.; Cooper, M.; VanDeusen, J.B.; Frankel, S.R.; Stock, W.; Caligiuri, M.A. Potential mechanisms of human natural killer cell expansion in vivo during low-dose IL-2 therapy. J. Clin. Investig. 2000, 106, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Felices, M.; Lenvik, A.J.; McElmurry, R.; Chu, S.; Hinderlie, P.; Bendzick, L.; Geller, M.A.; Tolar, J.; Blazar, B.R.; Miller, J.S. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight 2018, 3, 3. [Google Scholar] [CrossRef]
- Bierie, B.; Moses, H.L. Tumour microenvironment: Tgfbeta: The molecular jekyll and hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human ipsc-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 2018, 23, 181–192.e185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollino, D.; Webb, T.J. Chimeric antigen receptor-engineered natural killer and natural killer t cells for cancer immunotherapy. Transl. Res. 2017, 187, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Kumagai, A.; Iriguchi, S.; Yasui, Y.; Miyasaka, T.; Nakagoshi, K.; Nakane, K.; Saito, K.; Takahashi, M.; Sasaki, A.; et al. Non–clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti–glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. 2020, 111, 1478–1490. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Primary Differentiation | Lymphoid Commitment | Yield | Reference | |||||
|---|---|---|---|---|---|---|---|---|
| Medium | Cytokine | Culture Day | Medium | Cytokines | Culture Day | (per 1 × 106 iPS cells) | ||
| 1 | αMEM + 20% FBS | - | 12 | αMEM + 20% FBS | SCF, Flt3L, IL-7, IL-15 | 47 | 15.0 × 106 | [104] |
| 2 | αMEM + 20% FBS | bFGF, activin A, VEGF, CHIR99021 | 10 | αMEM + 20% FBS | Flt3L, IL-7, IL-2 | 38 | 1.0~3.5 × 106 | [105] |
| 3 | BPEL (APEL + 10% FBS) | SCF, VEGF, BMP-4 | 11 | BPEL | SCF, Flt3L, IL-3, IL-7, IL-15 | 28~32 | >97% | [98] |
| 4 | Essential 8 | BMP-4, CHIR99021, VEGF | 0–2 | DMEM + 20% human AB-serum or Stem line II | SCF, Flt3L, IL-7, IL-15 | 48 | 63.10 ± 7.01%~78.23 ± 5.66% | [106] |
| Essential 6 | SCF, SB431542, VEGF | 2–4 | ||||||
| Stem line II | SCF, Flt3L | 4–12 | ||||||
| Pre-Clinical Research | ||||
|---|---|---|---|---|
| Disease target | Strategies | Outcome | References | |
| Ovarian cancer | Multiple dose, IL-2 stimulated | The median survival improved from 73 to 98 days | [97] | |
| Cell line (K562, SKOV3, SW480, HCT-8, MCF7, SCC-25) | IL-2 stimulated | Efficiently killed all tested cancer cell lines (p < 0.5) | [105] | |
| Ovarian cancer | Targeting Mesothelin, engineered with chimeric, NKG2D-CAR-iPSC-NK | NKG2D-CAR-iPSC-NK cells displayed in vivo function similar to NKG2D-CAR-iPSC-T cells | [141] | |
| Hematological cancers, Hepatocellular carcinomas, Ovarian cancer | Tetravalent bispecific trifunctional antibody targeting GPC3, NKp46-CAR-iPSC-NK-EGFR | Effectively suppressed GPC3-expressing tumor growth in vitro and in vivo and confirmed the therapeutic quality and safety of the final product | [142] | |
| Clinical trial | ||||
| NCT number | Disease target | Phase | Start date | Affiliation |
| NCT03841110 | Advanced solid tumors Lymphoma, Gastric cancer, Colorectal cancer, Head and neck cancer, Squamous cell Carcinoma EGFR positive solid tumor, HER2-positive breast cancer, Hepatocellular, Small cell lung cancer, Renal cell carcinoma, Pancreas cancer, Melanoma, NSCLC, Urothelial carcinoma, Cervical cancer, Microsatellite instability, Merkel cell carcinoma | I | 15-Feb-19 | Fate Therapeutics |
| NCT04023071 | Acute myelogenous leukemia, B-cell lymphoma | I | 4-Oct-19 | Fate Therapeutics |
| NCT04614636 | Multiple myeloma Relapsed/Refractory acute Myeloid leukemia, Acute myelogenous leukemia | I | 4-Nov-20 | Fate Therapeutics |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, L.-J.; Liu, C.-L.; Kuo, M.-L.; Shen, C.-N.; Shen, C.-R. An Alternative Cell Therapy for Cancers: Induced Pluripotent Stem Cell (iPSC)-Derived Natural Killer Cells. Biomedicines 2021, 9, 1323. https://doi.org/10.3390/biomedicines9101323
Hsu L-J, Liu C-L, Kuo M-L, Shen C-N, Shen C-R. An Alternative Cell Therapy for Cancers: Induced Pluripotent Stem Cell (iPSC)-Derived Natural Killer Cells. Biomedicines. 2021; 9(10):1323. https://doi.org/10.3390/biomedicines9101323
Chicago/Turabian StyleHsu, Li-Jie, Chao-Lin Liu, Ming-Ling Kuo, Chia-Ning Shen, and Chia-Rui Shen. 2021. "An Alternative Cell Therapy for Cancers: Induced Pluripotent Stem Cell (iPSC)-Derived Natural Killer Cells" Biomedicines 9, no. 10: 1323. https://doi.org/10.3390/biomedicines9101323
APA StyleHsu, L. -J., Liu, C. -L., Kuo, M. -L., Shen, C. -N., & Shen, C. -R. (2021). An Alternative Cell Therapy for Cancers: Induced Pluripotent Stem Cell (iPSC)-Derived Natural Killer Cells. Biomedicines, 9(10), 1323. https://doi.org/10.3390/biomedicines9101323