
Role of PARP in TNBC: Mechanism of Inhibition, Clinical Applications, and Resistance



Abstract
:1. Introduction
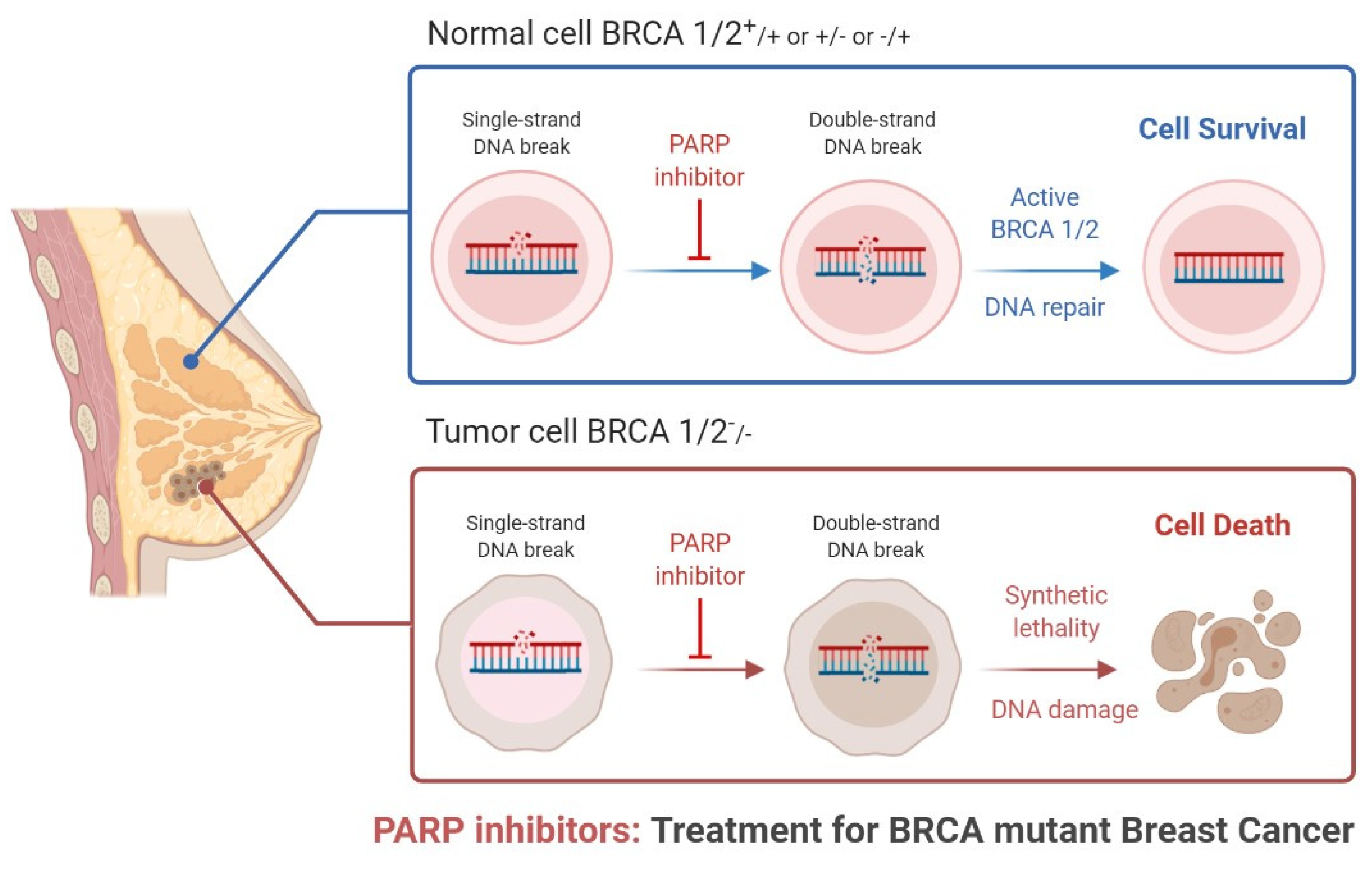
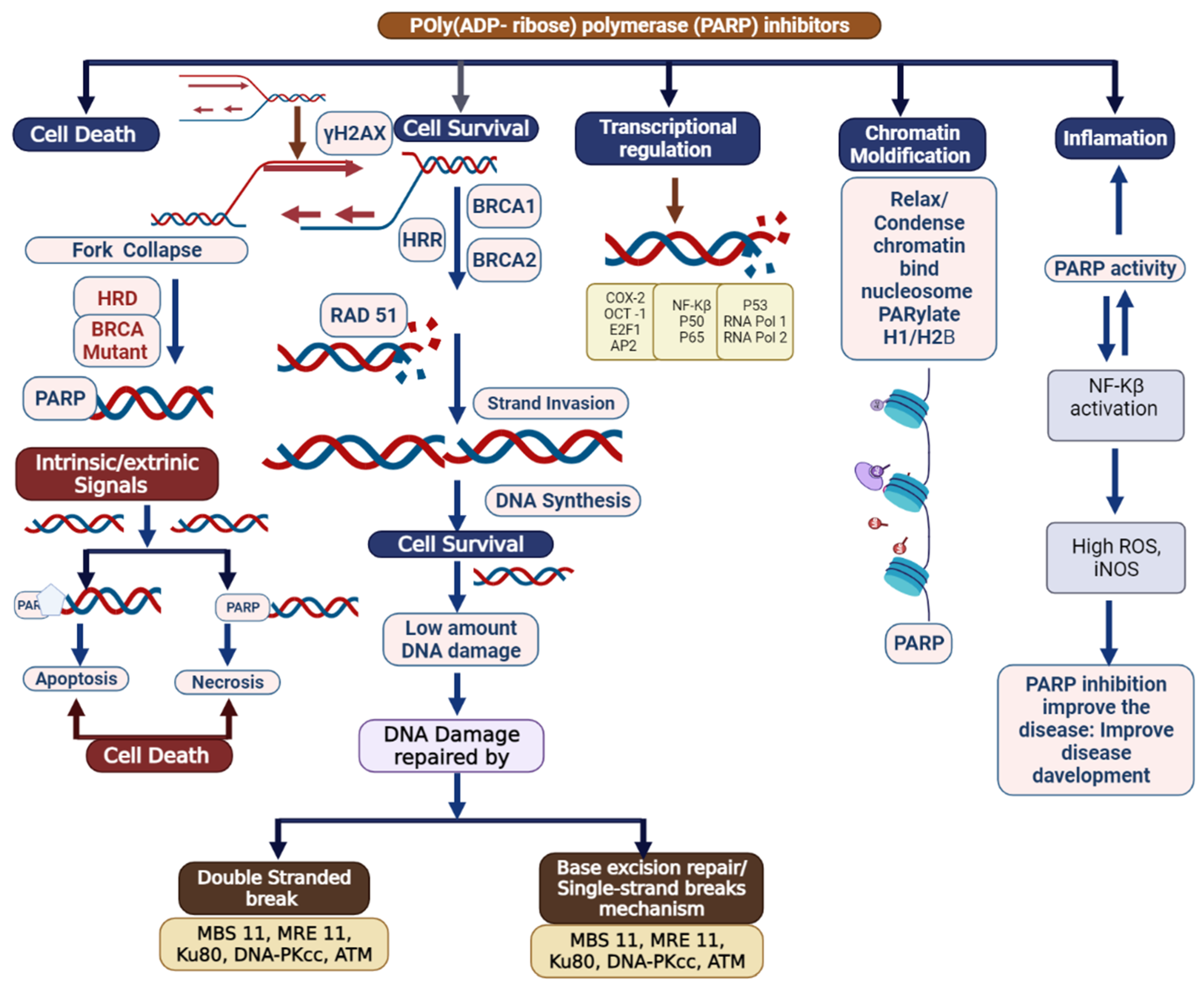
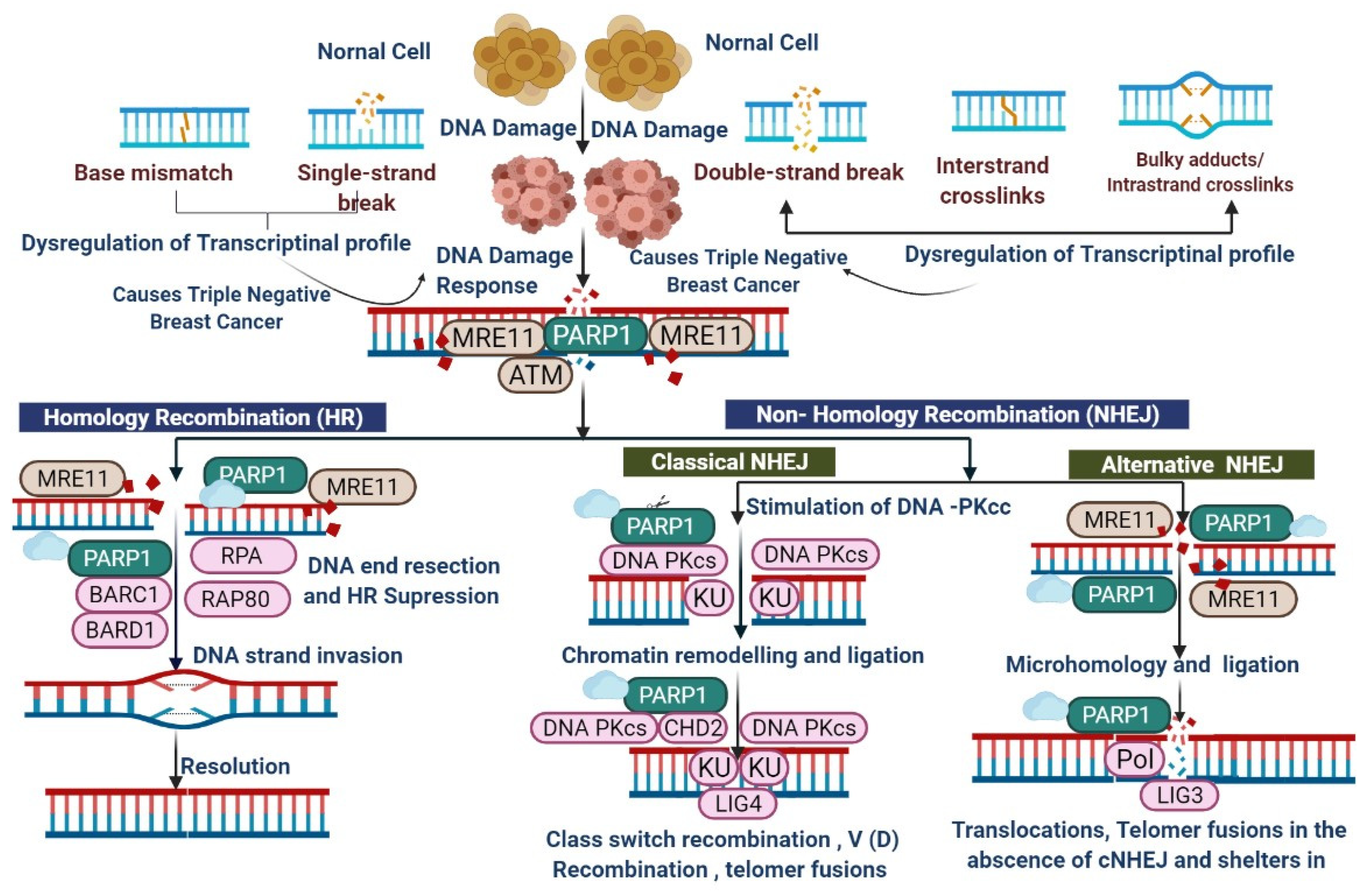
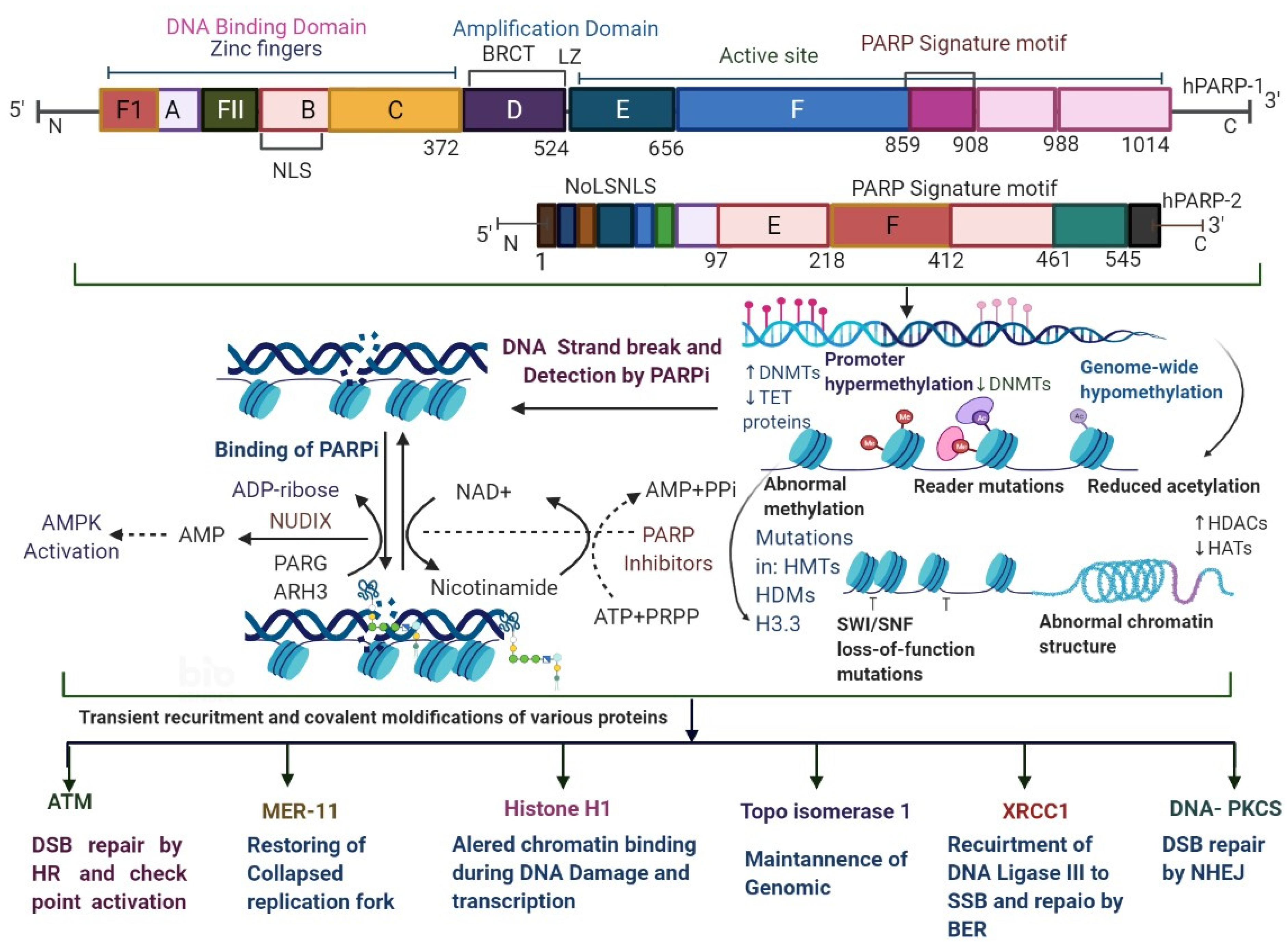
2. Mechanism of Poly (ADP-Ribose) Polymerase Inhibition
2.1. PARP Inhibition and the PI3K/AKT/mTOR Pathway
2.2. PARP Inhibitors Combined with CDK1 Inhibitors
2.3. PARP Inhibitors Combined with Histone Deacetylase Inhibitors
2.4. PARP Inhibitors Combined with EGFR Inhibitors
2.5. ATM Downregulation and PARP Inhibition
2.6. PARP Inhibitors Combined with Androgen Receptor (AR) Inhibitors
3. Clinical Applications of PARP Inhibitors in TNBC
3.1. Olaparib
3.2. Iniparib (BSI-201)
3.3. Niraparib
3.4. Veliparib (ABT-888)
3.5. Talazoparib (BMN-673)
3.6. Rucaparib
3.7. Checkpoint Inhibitors
4. PARP Inhibitor Resistance
4.1. Restoration of HR Repair in PARPi Resistance
4.2. DNA end Resection in PARPi Resistance
4.3. Formation of RAD51-ssDNA Filament and D-Loop in PARPi Resistance
5. Reversion Mutations in PARPi Resistance
5.1. Protection of the DNA Replication Fork in PARPi Resistance
5.2. Epigenetic Modification, Restoration of PARylation, and Pharmacological Alteration in PARPi Resistance
6. Clinical Implications of PARPi Resistance
7. CRISPR/Cas9 in Reverse Mutation
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Rummel, S.K.; Lovejoy, L.; Shriver, C.D.; Ellsworth, R.E. Contribution of germline mutations in cancer predisposition genes to tumor etiology in young women diagnosed with invasive breast cancer. Breast Cancer Res. Treat. 2017, 164, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.D.; Yadav, D.K. TNBC: Potential Targeting of Multiple Receptors for a Therapeutic Breakthrough, Nanomedicine, and Immunotherapy. Biomedicines 2021, 9, 876. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.A.; Oza, G.; Sharma, A.; Arriaga, L.G.; Hernández Hernández, J.M.; Rotello, V.M.; Ramirez, J.T. Triple-Negative Breast Cancer: A Review of Conventional and Advanced Therapeutic Strategies. Int. J. Environ. Res. Public Health 2020, 17, 2078. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-L.; Chen, G.; Chen, T.-Y.; Kuo, Y.-C.; Su, Y.-K. Effects of Cancer Stem Cells in Triple-Negative Breast Cancer and Brain Metastasis: Challenges and Solutions. Cancers 2020, 12, 2122. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-L.; Kuo, Y.-C.; Ho, Y.-S.; Huang, Y.-H. Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness. Cancers 2019, 11, 1334. [Google Scholar] [CrossRef] [Green Version]
- Nederlof, I.; Horlings, H.; Curtis, C.; Kok, M. A High-Dimensional Window into the Micro-Environment of Triple Negative Breast Cancer. Cancers 2021, 13, 316. [Google Scholar] [CrossRef]
- Keung, M.Y.T.; Wu, Y.; Vadgama, J.V. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435. [Google Scholar] [CrossRef] [Green Version]
- Schettini, F.; Giudici, F.; Bernocchi, O.; Sirico, M.; Corona, S.P.; Giuliano, M.; Locci, M.; Paris, I.; Scambia, G.; De Placido, S.; et al. Poly (ADP-ribose) polymerase inhibitors in solid tumours: Systematic review and meta-analysis. Eur. J. Cancer 2021, 149, 134–152. [Google Scholar] [CrossRef]
- Gourley, C.; Balmaña, J.; Ledermann, J.A.; Serra, V.; Dent, R.; Loibl, S.; Pujade-Lauraine, E.; Boulton, S.J. Moving From Poly (ADP-Ribose) Polymerase Inhibition to Targeting DNA Repair and DNA Damage Response in Cancer Therapy. J. Clin. Oncol. 2019, 37, 2257–2269. [Google Scholar] [CrossRef] [Green Version]
- van Beek, L.; McClay, É.; Patel, S.; Schimpl, M.; Spagnolo, L.; de Oliveira, T.M. PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling. Int. J. Mol. Sci. 2021, 22, 5112. [Google Scholar] [CrossRef]
- Langelier, M.-F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural Basis for DNA Damage-Dependent Poly(ADP-Ribosyl)Ation by Human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascal, J.M. The Comings and Goings of PARP-1 in Response to DNA Damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, W.; Wang, Y. PARP-1 and Its Associated Nucleases in DNA Damage Response. DNA Repair 2019, 81, 102651. [Google Scholar] [CrossRef] [PubMed]
- Van Andel, L.; Zhang, Z.; Lu, S.; Kansra, V.; Agarwal, S.; Hughes, L.; Tibben, M.M.; Gebretensae, A.; Lucas, L.; Hillebrand, M.J.X.; et al. Human mass balance study and metabolite profiling of 14Cniraparib, a novel poly(ADP-Ribose) polymerase (PARP)-1 and PARP-2 inhibitor, in patients with advanced cancer. Investig. New Drugs 2017, 35, 751–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, V.; Paunu, K.; Ahlskog, J.K.; Varnai, R.; Sipeky, C.; Sundvall, M. PARP Inhibitors in Prostate Cancer—The Preclinical Rationale and Current Clinical Development. Genes 2019, 10, 565. [Google Scholar] [CrossRef] [Green Version]
- Kraus, W.L. PARPs and ADP-Ribosylation: 50 Years … and counting. Mol. Cell 2015, 58, 902–910. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.S.; Chang, P. Insights into the Biogenesis, Function, and Regulation of ADP-Ribosylation. Nat. Chem. Biol. 2018, 14, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Langelier, M.-F.; Ruhl, D.D.; Planck, J.L.; Kraus, W.L.; Pascal, J.M. The Zn3 Domain of Human Poly(ADP-Ribose) Polymerase-1 (PARP-1) Functions in Both DNA-Dependent Poly(ADP-Ribose) Synthesis Activity and Chromatin Compaction. J. Biol. Chem. 2010, 285, 18877–18887. [Google Scholar] [CrossRef] [Green Version]
- Barkauskaite, E.; Jankevicius, G.; Ahel, I. Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation. Mol. Cell 2015, 58, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Hoch, N.C.; Polo, L.M. ADP-Ribosylation: From Molecular Mechanisms to Human Disease. Genet. Mol. Biol. 2020, 43, e20190075. [Google Scholar] [CrossRef] [Green Version]
- Durkacz, B.W.; Omidiji, O.; Gray, D.A.; Shall, S. (ADP-Ribose)n Participates in DNA Excision Repair. Nature 1980, 283, 593–596. [Google Scholar] [CrossRef]
- Rudolph, J.; Mahadevan, J.; Luger, K. Probing the Conformational Changes Associated with DNA Binding to PARP1. Biochemistry 2020, 59, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Bilokapic, S.; Suskiewicz, M.J.; Ahel, I.; Halic, M. Bridging of DNA Breaks Activates PARP2–HPF1 to Modify Chromatin. Nature 2020, 585, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Eisemann, T.; Pascal, J.M. Poly(ADP-Ribose) Polymerase Enzymes and the Maintenance of Genome Integrity. Cell. Mol. Life Sci. 2020, 77, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Zandarashvili, L.; Langelier, M.-F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural Basis for Allosteric PARP-1 Retention on DNA Breaks. Science 2020, 368, eaax6367. [Google Scholar] [CrossRef]
- Sukhanova, M.V.; Hamon, L.; Kutuzov, M.M.; Joshi, V.; Abrakhi, S.; Dobra, I.; Curmi, P.A.; Pastre, D.; Lavrik, O.I. A Single-Molecule Atomic Force Microscopy Study of PARP1 and PARP2 Recognition of Base Excision Repair DNA Intermediates. J. Mol. Biol. 2019, 431, 2655–2673. [Google Scholar] [CrossRef]
- Andreidesz, K.; Koszegi, B.; Kovacs, D.; Bagone Vantus, V.; Gallyas, F.; Kovacs, K. Effect of Oxaliplatin, Olaparib and LY294002 in Combination on Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 2056. [Google Scholar] [CrossRef]
- Dirix, L.; Swaisland, H.; Verheul, H.M.; Rottey, S.; Leunen, K.; Jerusalem, G.; Rolfo, C.; Nielsen, D.; Molife, L.R.; Kristeleit, R.; et al. Effect of itraconazole and rifampin on the pharmacokinetics of olaparib in patients with advanced solid tumors: Results of two phase I open-label studies. Clin. Ther. 2016, 38, 2286–2299. [Google Scholar] [CrossRef]
- Ensminger, M.; Löbrich, M. One end to rule them all: Non-homologous end-joining and homologous recombination at DNA double-strand breaks. Br. J. Radiol. 2020, 93, 20191054. [Google Scholar] [CrossRef] [PubMed]
- Plummer, R.; Swaisland, H.; Leunen, K.; Van Herpen, C.M.L.; Jerusalem, G.; De Greve, J.; Lolkema, M.P.; Soetekouw, P.; Mau-Sørensen, M.; Nielsen, D.; et al. Olaparib tablet formulation: Effect of food on the pharmacokinetics after oral dosing in patients with advanced solid tumours. Cancer Chemother. Pharmacol. 2015, 76, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafa, N.M.; Chiu, Y.L.; Rosen, L.S.; Bessudo, A.; Kovacs, X.; Giranda, V.L. A phase 1 study to evaluate effect of food on veliparib pharmacokinetics and relative bioavailability in subjects with solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 583–591. [Google Scholar] [CrossRef]
- Diana, A.; Carlino, F.; Franzese, E.; Oikonomidou, O.; Criscitiello, C.; De Vita, F.; Ciardiello, F.; Orditura, M. Early Triple Negative Breast Cancer: Conventional Treatment and Emerging Therapeutic Landscapes. Cancers 2020, 12, 819. [Google Scholar] [CrossRef] [Green Version]
- Min, A.; Kim, K.; Jeong, K.; Choi, S.; Kim, S.; Suh, K.J.; Lee, K.-H.; Kim, S.; Im, S.-A. Homologous repair deficiency score for identifying breast cancers with defective DNA damage response. Sci. Rep. 2020, 10, 12506. [Google Scholar] [CrossRef]
- O’Connor, L.O.; Rulten, S.L.; Cranston, A.N.; Odedra, R.; Brown, H.; Jaspers, J.E.; Jones, L.; Knights, C.; Evers, B.; Ting, A.; et al. The PARP Inhibitor AZD2461 Provides Insights into the Role of PARP3 Inhibition for Both Synthetic Lethality and Tolerability with Chemotherapy in Preclinical Models. Cancer Res. 2016, 76, 6084–6094. [Google Scholar]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)–Function in Genome Maintenance and Relevance of Inhibitors for Anti-Cancer Therapy. Front. Mol. Biosci. 2020, 7, 21. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Tuli, R.; Shiao, S.L.; Nissen, N.; Tighiouart, M.; Kim, S.; Osipov, A.; Bryant, M.; Ristow, L.; Placencio-Hickok, V.; Hoffman, D.; et al. A Phase 1 Study of Veliparib, a PARP-1/2 Inhibitor, with Gemcitabine and Radiotherapy in Locally Advanced Pancreatic Cancer. EBioMedicine 2019, 40, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Liu, H.; Pearson, T.; Iwase, T.; Fuson, J.; Lalani, A.S.; Eli, L.D.; Diala, I.; Tripathy, D.; Lim, B.; et al. PI3K and MAPK Pathways as Targets for Combination with the Pan-HER Irreversible Inhibitor Neratinib in HER2-Positive Breast Cancer and TNBC by Kinome RNAi Screening. Biomedicines 2021, 9, 740. [Google Scholar] [CrossRef] [PubMed]
- Ryu, W.-J.; Sohn, J.H. Molecular Targets and Promising Therapeutics of Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 1008. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Schafer, J.M.; Pendleton, C.S.; Tang, L.; Johnson, K.C.; Chen, X.; Balko, J.M.; Gomez, H.; Arteaga, C.L.; et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014, 16, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.C.; LaBaff, A.; Wei, Y.; Nie, L.; Xia, W.; Huo, L.; Yamaguchi, H.; Hsu, Y.H.; Hsu, J.L.; Liu, D.; et al. Phosphorylation of EZH2 at T416 by CDK2 contributes to the malignancy of triple negative breast cancers. Am. J. Transl. Res. 2015, 7, 1009–1020. [Google Scholar] [PubMed]
- Langelier, M.-F.; Riccio, A.A.; Pascal, J.M. PARP-2 and PARP-3 Are Selectively Activated by 5′ Phosphorylated DNA Breaks Through an Allosteric Regulatory Mechanism Shared with PARP-1. Nucleic Acids Res. 2014, 42, 7762–7775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Ogden, T.E.H.; Yang, J.-C.; Schimpl, M.; Easton, L.E.; Underwood, E.; Rawlins, P.B.; McCauley, M.M.; Langelier, M.-F.; Pascal, J.M.; Embrey, K.J.; et al. Dynamics of the HD Regulatory Subdomain of PARP-1; Substrate Access and Allostery in PARP Activation and Inhibition. Nucleic Acids Res. 2021, 49, 2266–2288. [Google Scholar] [CrossRef]
- Choi, C.; Cho, W.K.; Park, S.; Shin, S.-W.; Park, W.; Kim, H.; Choi, D.H. Checkpoint Kinase 1 (CHK1) Inhibition Enhances the Sensitivity of Triple-Negative Breast Cancer Cells to Proton Irradiation via Rad51 Downregulation. Int. J. Mol. Sci. 2020, 21, 2691. [Google Scholar] [CrossRef] [Green Version]
- Gorecki, L.; Andrs, M.; Korabecny, J. Clinical Candidates Targeting the ATR–CHK1–WEE1 Axis in Cancer. Cancers 2021, 13, 795. [Google Scholar] [CrossRef]
- Nandini, D.; Jennifer, A.; Pradip, D. Therapeutic Strategies for Metastatic Triple-Negative Breast Cancers: From Negative to Positive. Pharmaceuticals 2021, 14, 455. [Google Scholar] [CrossRef]
- Baldan, F.; Mio, C.; Allegri, L.; Puppin, C.; Russo, D.; Filetti, S.; Damante, G. Synergy between HDAC and PARP Inhibitors on Proliferation of a Human Anaplastic Thyroid Cancer-Derived Cell Line. Int. J. Endocrinol. 2015, 2015, 978371. [Google Scholar] [CrossRef]
- Chu, P.-Y.; Tai, Y.-L.; Shen, T.-L. Grb7, a Critical Mediator of EGFR/ErbB Signaling, in Cancer Development and as a Potential Therapeutic Target. Cells 2019, 8, 435. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Gilardini Montani, M.S.; Prodosmo, A.; Stagni, V.; Merli, D.; Monteonofrio, L.; Gatti, V.; Gentileschi, M.P.; Barila, D.; Soddu, S. ATM-depletion in breast cancer cells confers sensitivity to PARP inhibition. J. Exp. Clin. Cancer Res. 2013, 32, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlaam, B.; Pike, K. Identifying high quality, potent and selective inhibitors of ATM kinase: Discovery of AZD0156. Eur. J. Cancer 2016, 61, S118. [Google Scholar] [CrossRef]
- Sari, M.; Saip, P. Myelodysplastic Syndrome after Olaparib Treatment in Heavily Pretreated Ovarian Carcinoma. Am. J. Ther. 2019, 26, e632–e633. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H. Autocrine TGF-β in Cancer: Review of the Literature and Caveats in Experimental Analysis. Int. J. Mol. Sci. 2021, 22, 977. [Google Scholar] [CrossRef] [PubMed]
- Brumec, M.; Sobočan, M.; Takač, I.; Arko, D. Clinical Implications of Androgen-Positive Triple-Negative Breast Cancer. Cancers 2021, 13, 1642. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Jin, J.; Yang, F.; Sun, Z.; Zhang, W.; Shi, Y.; Xu, J.; Guan, X. The correlation between PARP1 and BRCA1 in AR positive triple-negative breast cancer. Int. J. Biol. Sci. 2016, 12, 1500–1510. [Google Scholar] [CrossRef] [Green Version]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.; Gradishar, W.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the treatment of androgen receptor-expressing triple-negative breast cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef]
- Ke, Y.; Zhang, J.; Lv, X.; Zeng, X.; Ba, X. Novel Insights into PARPs in Gene Expression: Regulation of RNA Metabolism. Cell. Mol. Life Sci. 2019, 76, 3283–3299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, E.; Bertucci, F.; Sabatier, R.; Gonçalves, A. Targeting BRCA Deficiency in Breast Cancer: What are the Clinical Evidences and the Next Perspectives? Cancers 2018, 10, 506. [Google Scholar] [CrossRef] [Green Version]
- Clarke, N.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Flechon, A.; Redfern, C.; et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018, 19, 975–986. [Google Scholar] [CrossRef]
- Robson, M.; Tung, N.; Conte, P.; Im, S.-A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, E510. [Google Scholar] [CrossRef] [Green Version]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. Immunother. Cancer 2018, 6, 141. [Google Scholar] [CrossRef]
- Nitecki, R.; Gockley, A.A.; Floyd, J.L.; Coleman, R.L.; Melamed, A.; Rauh-Hain, J.A. The incidence of myelodysplastic syndrome in patients receiving poly-ADP ribose polymerase inhibitors for treatment of solid tumors: A meta-analysis. J. Clin. Oncol. 2020, 38, 3641. [Google Scholar] [CrossRef]
- Hossain, F.; Majumder, S.; David, J.; Miele, L. Precision Medicine and Triple-Negative Breast Cancer: Current Landscape and Future Directions. Cancers 2021, 13, 3739. [Google Scholar] [CrossRef]
- Bergin, A.R.T.; Loi, S. Triple-negative breast cancer: Recent treatment advances. F1000Research 2019, 8, 1342. [Google Scholar] [CrossRef]
- Pierce, A.; McGowan, P.M.; Cotter, M.; Mullooly, M.; O’Donovan, N.; Rani, S.; O’Driscoll, L.; Crown, J.; Duffy, M.J. Comparative antiproliferative effects of iniparib and olaparib on a panel of triple-negative and non-triple-negative breast cancer cell lines. Cancer Biol. Ther. 2013, 14, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [Green Version]
- Diéras, V.; Bonnefoi, H.; Alba, E.; Awada, A.; Coudert, B.; Pivot, X.; Gligorov, J.; Jager, A.; Zambelli, S.; Lindeman, G.J.; et al. Iniparib administered weekly or twice-weekly in combination with gemcitabine/carboplatin in patients with metastatic triple-negative breast cancer: A phase II randomized open-label study with pharmacokinetics. Breast Cancer Res. Treat. 2019, 177, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Ong, M.; Tan, D.S.; Gonzalez, M.A.; de Bono, J.S. Appraising iniparib, the PARP inhibitor that never was—What must we learn? Nat. Rev. Clin. Oncol. 2013, 10, 688–696. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Schwartzberg, L.; Danso, M.A.; Miller, K.D.; Rugo, H.S.; Neubauer, M.; Robert, N.; Hellerstedt, B.; Saleh, M.; Richards, P.; et al. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2014, 32, 3840–3847. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, S.K.; Schelman, W.R.; Wilding, G.; Moreno, V.; Baird, R.D.; Miranda, S.; Hylands, L.; Riisnaes, R.; Forster, M.; Omlin, A.; et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2013, 14, 882–892. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined with Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Appleman, L.J.; Beumer, J.H.; Jiang, Y.; Lin, Y.; Ding, F.; Puhalla, S.; Swartz, L.; Owonikoko, T.K.; Donald Harvey, R.; Stoller, R.; et al. Phase 1 study of veliparib (ABT-888), a poly (ADP-ribose) polymerase inhibitor, with carboplatin and paclitaxel in advanced solid malignancies. Cancer Chemother. Pharmacol. 2019, 84, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Diéras, V.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.-P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. Veliparib with carboplatin and paclitaxel in BRCA-mutated advanced breast cancer (BROCADE3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1269–1282. [Google Scholar] [CrossRef]
- Han, H.S.; Diéras, V.; Robson, M.; Palácová, M.; Marcom, P.K.; Jager, A.; Bondarenko, I.; Citrin, D.; Campone, M.; Telli, M.L.; et al. Veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in patients with BRCA1/2 locally recurrent/metastatic breast cancer: Randomized phase II study. Ann. Oncol. 2018, 29, 154–161. [Google Scholar] [CrossRef]
- de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litton, J.K.; Hurvitz, S.A.; Mina, L.A.; Rugo, H.S.; Lee, K.-H.; Gonçalves, A.; Diab, S.; Woodward, N.; Goodwin, A.; Yerushalmi, R.; et al. Talazoparib versus chemotherapy in patients with germline BRCA1/2-mutated HER2-negative advanced breast cancer: Final overall survival results from the EMBRACA trial. Ann. Oncol. 2020, 31, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Scoggins, M.E.; Hess, K.R.; Adrada, B.E.; Murthy, R.K.; Damodaran, S.; DeSnyder, S.M.; Brewster, A.M.; Barcenas, C.H.; Valero, V.; et al. Neoadjuvant Talazoparib for Patients with Operable Breast Cancer with a Germline BRCA Pathogenic Variant. J. Clin. Oncol. 2020, 38, 388–394. [Google Scholar] [CrossRef]
- Miller, K.; Tong, Y.; Jones, D.R.; Walsh, T.; Danso, M.A.; Ma, C.X.; Silverman, P.; King, M.-C.; Badve, S.S.; Perkins, S.M. Cisplatin with or without rucaparib after preoperative chemotherapy in patients with triple negative breast cancer: Final efficacy results of Hoosier Oncology Group BRE09-146. J. Clin. Oncol. 2015, 33, 1082. [Google Scholar] [CrossRef]
- Drew, Y.; Ledermann, J.; Hall, G.; Rea, D.; Glasspool, R.; Highley, M.; Jayson, G.; Sludden, J.; Murray, J.; Jamieson, D.; et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br. J. Cancer 2016, 114, 723–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durmus, S.; Sparidans, R.W.; van Esch, A.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral availability and brain accumulation of the PARP inhibitor rucaparib (AG-014699). Pharm. Res. 2015, 32, 37–46. [Google Scholar] [CrossRef]
- Simmons, A.D.; Nguyen, M.; Pintus, E. Polyclonal BRCA2 mutations following carboplatin treatment confer resistance to the PARP inhibitor rucaparib in a patient with mCRPC: A case report. BMC Cancer 2020, 20, 215. [Google Scholar] [CrossRef]
- FDA. Grants Accelerated Approval to Rucaparib for BRCA-Mutated Metastatic Castration-Resistant Prostate Cancer. Available online: https://www.fda.gov/drugs/fda-grants-accelerated-approval-rucaparib-brca-mutated-metastatic-castration-resistantprostate (accessed on 15 July 2021).
- Brill, E.; Yokoyama, T.; Nair, J.; Yu, M.; Ahn, Y.R.; Lee, J.M. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer. Oncotarget 2017, 8, 111026–111040. [Google Scholar] [CrossRef]
- Chen, C.-C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–349. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Adam, A.M.A.; Altalhi, T.A.; El-Megharbel, S.M.; Saad, H.A.; Refat, M.S. Using a Modified Polyamidoamine Fluorescent Dendrimer for Capturing Environment Polluting Metal Ions Zn2+, Cd2+, and Hg2+: Synthesis and Characterizations. Crystals 2021, 11, 92. [Google Scholar] [CrossRef]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-g e ovarian carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R.; Duckett, D.R. Therapeutic Targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell 2019, 36, 545–558.e7. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskaya, R.; Raga Gil, E.; Lawrence, N.; Butler, R.; Clifford, G.; Wilson, M.D.; Jackson, S.P. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat. Commun. 2020, 11, 819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.; Genois, M.-M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, Y.; Du, Y.; Zhou, M.; Hu, Y.; Zhang, S. Emerging roles of N6-methyladenosine (m6A) modification in breast cancer. Cell Biosci. 2020, 10, 136. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Fortunati, E.; Liu, D.-X.; Li, Y. Pleiotropic Roles of ABC Transporters in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 3199. [Google Scholar] [CrossRef] [PubMed]
- Aldrak, N.; Alsaab, S.; Algethami, A.; Bhere, D.; Wakimoto, H.; Shah, K.; Alomary, M.N.; Zaidan, N. Oncolytic Herpes Simplex Virus-Based Therapies for Cancer. Cells 2021, 10, 1541. [Google Scholar] [CrossRef]
- Singh, D.D.; Han, I.; Choi, E.-H.; Yadav, D.K. CRISPR/Cas9 based genome editing for targeted transcriptional control in triple- negative breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 2384–2397. [Google Scholar] [CrossRef]
- Singh, D.D.; Verma, R.; Tripathi, S.K.; Sahu, R.; Trivedi, P.; Yadav, D.K. Breast Cancer Transcriptional Regulatory Network Reprogramming by using the CRISPR/Cas9 system: An Oncogenetics Perspective. Curr. Top. Med. Chem. 2021, 21, 1. [Google Scholar] [CrossRef]
- Singh, D.D.; Hawkins, R.D.; Lahesmaa, R.; Tripathi, S. CRISPR/Cas9 guided genome and epigenome engineering and its ther- apeutic applications in immune mediated diseases. Semin. Cell Dev. Biol. 2019, 96, 32–43. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Compound Structure | Efficacy | IC50 |
|---|---|---|---|
| Nicotinamide |  | PARP inhibitor and by-product of the PARP reaction; many pharmacological actions other than that of inhibiting PARP | 210 μM |
| 3-aminobenzamide |  | Benzamides are free radical scavengers, among other pharmacological actions | 33 μM |
| PD128763 |  | Cytoprotective agent, chemosensitizer, and radiosensitizer; adverse effect of compound causes hypothermia | 420 nM |
| DPQ |  | A commonly used Warner–Lambert PARP inhibitor compound based on an isoquinoline core | 1 μM |
| NU1025 |  | Potentiators of anticancer agent cytotoxicity | 400 nM |
| 4-ANI |  | PARP in DNA repair and cell death | 180 nM |
| ISO |  | PARP in DNA repair and cell death | 390 nM |
| Olaparib (Lynparza) |  | Use in a BRCA1-positive patient with metastatic triple-negative breast cancer, without the initial use of platinum-based chemotherapy, showed significant rapid near-resolution of large liver metastasis while patient experienced gout-like symptoms | 1 nM |
| Niraparib (Zejula) |  | Niraparib in combination with pembrolizumab in patients with triple-negative breast cancer | 4 nM |
| Talazoparib (Talzenna) |  | Ferm line BRCA-mutant, HER2-negative locally advanced or metastatic breast cancer | 0.6 nM |
| Veliparib (ABT-888) |  | Received orphan drug status for lung cancer | 2 nM |
| INO-1001 |  | Potent enhancer of radiation sensitivity and enhances radiation-induced cell killing by interfering with DNA repair mechanisms, resulting in necrotic cell death | 105 nM |
| E7449 |  | Antitumor activity of E7449; a novel PARP 1/2 and tankyrase 1/2 inhibitor | 1 nM |
| CEP-8983 |  | Increases the sensitivity of chemoresistant tumor cells to temozolomide | 20 nM |
| Pamiparib (BGB-290) |  | Pamiparib has potent PARP trapping, the capability to penetrate the brain, and can be used for the research of various cancers including solid tumors | 0.9 nM |
| Fluzoparib (SHR-3162) |  | Inhibitor of poly-adenosine diphosphate(ADP)ribose polymerase (PARP) 1/2 being developed for the treatment of BRCA1/2-mutant solid tumors. | 1.5 nM |
| Name of the Molecules | Tmax (h) | t (h) | AUC (lgh/ mL) | Cmax (lg/mL) | CL/F (L/h) | Vz/F | References |
|---|---|---|---|---|---|---|---|
| Olaparib capsule formulation 300 mg | 1.49 (0.57–3.05) | 13.02 (8.23) | 55.20 (67.4) | 8.05 (24.3) | 6.36 (3.47) | 112.1 (59.84) | [30] |
| Olaparib tablet formulation 300 mg single dose (fasted) | 1.50 (0.50–5.85) | 12.2 (5.31) | 43.6 (54.3) [AUCt] 43.0 (55.2) [AUC] | 7.00 (35.0) | 7.95 (4.23) | 146 (142) | [32] |
| Olaparib tablet formulation 300 mg single dose (fed) | 4.00 (1.00–12.0) | 12.2 (5.31) | 46.0 (56.6) [AUCt] 45.4 (57.1) [AUC] | 5.48 (40.5) | 7.55 (3.99) | 127 (107) | [32] |
| Veliparib monotherapy 40 mg (10 mg, fasting) | 1.2 ± 0.8 | 5.9 ± 1.3 | 2.23 ± 0.82 [AUCt] 2.43 ± 1.07 [AUC] | 0.36 ± 0.13 | 19.0 ± 7.36 | NA | [34,40] |
| Veliparib monotherapy 40 mg (10 mg, fed) | 1.2 ± 0.7 | 5.8 ± 1.2 | 2.45 ± 0.93 [AUCt] 2.65 ± 1.17 [AUCt] | 0.37 ± 0.12 | 17.3 ± 6.41 | NA | |
| Veliparib monotherapy 40 mg (40 mg, fasting) | 1.3 ± 0.9 | 5.8 ± 1.3 | 2.24 ± 0.98 [AUCt] 2.45 ± 1.24 [AUCt] | 0.34 ± 0.12 | 19.5 ± 7.66 | NA | |
| Veliparib monotherapy 40 mg (40 mg, fed) | 2.5 ± 1.1 | 5.8 ± 1.4 | 2.14 ± 0.80 [AUCt] 2.35 ± 1.06 [AUCt] | 0.28 ± 0.09 | 19.7 ± 7.51 | NA | |
| Veliparib metabolite M8 | 2.4 (3.5–9.8) | – | 0.3–1.9 [AUCint] | 0.011 (0.007–0.014) | NA | NA | [34,40] |
| Niraparib 300 mg/day | 3.1 (2.0–6.1) | a | 14.117 (AUC24)b | 1.921 | NA | NA | [12] |
| Niraparib metabolite: unlabeled M1 plasma | 9.02 | 78.4 | 41.2 (AUCt) | 476 | NA | NA | [15] |
| Name of Drug | Types of Inhibitors | Prior Treatment | Type of Population | Status | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|
| AZD1775 in patent with TNBC LYNPARZATM | PARP Inhibitor, patent with TNBC | Olaparib in combination with AZD6738 mutated (ATM) | Inhibitor of Ataxia-Telangiectasia and WEE1 inhibitor | Phase II | NCT03330847 |
| AZD1775 in patent with TNBC LYNPARZATM | PARP Inhibitor, patent with TNBC | Olaparib with radiation therapy, after chemotherapy | Inhibitor of ataxia-telangiectasia | Phase I | NCT03109080 |
| AZD1775, LYNPARZATM | Patent with TNBC | Olaparib with atezolizumab | Inhibitor of PD-L1 | Phase II | NCT02849496 |
| AZD1775, LYNPARZATM | Patent with TNBC | Oolaparib with paclitaxel and carboplatin | Inhibitor of germline BRCA mutated | Phase II/III | NCT03150576, NCT02789332 |
| AZD1775, LYNPARZATM | Patent with TNBC | Olaparib with AZD2171 orally | Inhibitor of VEGFR tyrosine kinase | Phase I/II | NCT01116648 |
| AZD1775, LYNPARZATM | Patent with TNBC | Olaparib with PI3K inhibitor, BKM120 | Inhibitor of BKM120 | Phase I | NCT01623349 |
| AZD1775, LYNPARZATM | Patent with TNBC | Olaparib with onalespib | Inhibitor of heat shock protein 90 | Phase I | NCT02898207 |
| AZD1775, LYNPARZATM | Patent with TNBC | Olaparib with AZD2014 | mTORC1/2 or Oral AKT inhibitor | Phase I/II | NCT02208375 |
| PARP1/2 inhibitor Veliparib | Patent with TNBC | Veliparib in combination with cyclophosphamide | Inhibitor of EGFR, HER2, BRCA, and tyrosine kinase | Phase II and failed in phase III trials | NCT01306032 |
| PARP1/2 inhibitor Veliparib | Inhibitor of tyrosine kinase, HER2, and BRCA | Veliparib in combination with carboplatin | Patients with TNBC | Completed phase I study | NCT01251874 |
| PARP1/2 inhibitor Veliparib | Inhibitor of EGFR, BRCA, and tyrosine kinase | Veliparib with vinorelbine | Patients with TNBC | Completed phase I | NCT01281150 |
| PARP1/2 inhibitor Veliparib | Inhibitor of EGFR, HER2, BRCA, and tyrosine kinase | Veliparib with cisplatin | Patients with TNBC | Completed phase I | NCT01104259 |
| PARP1/2 inhibitor Veliparib | Inhibitor of EGFR, HER2, BRCA, and tyrosine kinase | Veliparib with pegylation | Patients with TNBC | Completed phase I | NCT01145430 |
| PARP1/2 inhibitor Veliparib | Inhibitor of EGFR, HER2, BRCA, and tyrosine kinase | Veliparib with pegylation | Patients with TNBC | Completed phase I | NCT01145430 |
| PARP1/2 inhibitor Veliparib | Inhibitor of EGFR, HER2, BRCA, and tyrosine kinase | Veliparib with lapatinib | Patients with TNBC | Phase I | NCT02158507 |
| PARP1/2 inhibitor Veliparib | Inhibitor of EGFR, HER2, BRCA, and tyrosine kinase | Veliparib in combination with irinotecan HCl | Patients with TNBC | Phase I I | NCT00576654 |
| PARP1/2 inhibitor Veliparib | Inhibitor of EGFR, HER2, BRCA, and tyrosine kinase | Veliparib with cisplatin | Patients with TNBC | Phase II | NCT02595905 |
| AZD2281 and Ku-0059436 PARP1/2 inhibitor (Selective) | PARP inhibitor; BRCA Mutated | Olaparib alone, or in combination with durvalumab MEDI4736 against PD-L1 | HER2-negative treated mTNBC | Phase-II | NCT00679783 NCT03544125 NCT02484404 NCT03167619 NCT02681562 NCT02484404 |
| PARP1/2 inhibitor Veliparib | Inhibitor of EGFR, HER2, BRCA, and tyrosine kinase | Veliparib plus carboplatin | Patients with TNBC | Phase III | NCT02032277 |
| Iniparib BSI-201 and SAR240550 | Competitive PARP inhibitor; ability to form adducts with many cysteine-containing proteins | Combination with gemcitabine and carboplatin | Patients with TNBC | Phase II | NCT00813956 NCT01045304 NCT01130259 |
| Iniparib BSI-201 and SAR240550 | Competitive PARP inhibitor; ability to form adducts with many cysteine-containing proteins | Combination of iniparib with paclitaxel for TNBC compared to paclitaxel alone | Patients with TNBC | Competed phase II | NCT01204125 |
| Iniparib BSI-201 and SAR240550 | Competitive PARP inhibitor; ability to form adducts with many cysteine-containing proteins | Iniparib with irinotecan | Patients with TNBC | Phase II trial | NCT01173497 |
| Niraparib | ≥1 anti-HER2 treatment; PARP inhibitor | Niraparib plus trastuzumab IV | Metastatic HER2+ breast cancer | Phase Ib/II (recruiting) | NCT03368729 |
| Niraparib | PARP inhibitor | One anthracycline and/or taxane in the (neo-) adjuvant or Niraparib | Advanced/metastatic BRCA1- like | Phase-II, Active, not recruiting | NCT02826512 |
| Niraparib | PARP inhibitor | ≥1 line of therapy Niraparib plus everolimus | Patients with TNBC | Phase I Recruiting | NCT03154281 |
| Niraparib | Germline BRCA mutation-positive (PARP inhibitors) | ≤2 prior cytotoxic regimens and Niraparib versus physician‘s choice | Advanced or metastatic breast cancer | Phase III Active, not yet recruiting | NCT01905592 (BRAVO) |
| Niraparib | Metastatic TNBC inhibitors (PARP inhibitors) | ≤2 lines of cytotoxic therapy, Niraparib plus pembrolizumab | Advanced or metastatic TNBC | Phase I/II Active, not yet recruiting | NCT02657889 (KEYNOTE-162) |
| veliparib | Metastatic TNBC inhibitors (PARP inhibitors) | ≤2 lines of cytotoxic Chemotherapy, Carboplatin, and paclitaxel with or without veliparib | Locally advanced unresectable BRCA associated | Phase III Recruiting | NCT02163694 |
| veliparib | Metastatic TNBC inhibitors (PARP inhibitors) | Veliparib with temozolomide versus veliparib with carboplatin and paclitaxel versus placebo with carboplatin and paclitaxel ≤2 lines of cytotoxic chemotherapy | Metastatic TNBC | Randomized phase II, Ongoing | NCT01506609 |
| veliparib | Metastatic TNBC inhibitors (PARP inhibitors) | Veliparib versus atezolizumab versus veliparib plus atezolizumab | Stage III–IV TNBC | Randomized phase II Ongoing | NCT02849496 |
| veliparib | Metastatic TNBC inhibitors PARP inhibitors) | Cisplatin and placebo versus cisplatin and veliparib ≤1 line of cytotoxic chemotherapy for metastatic disease | Metastatic TNBC and/or BRCA mutation-associated breast cancer | Phase II Active, not recruiting | NCT02595905 |
| veliparib | Metastatic TNBC inhibitors PARP inhibitors) | Temozolomide and veliparib ≥1 chemotherapy regimen | Metastatic TNBC and/or BRCA mutation-associated breast cancer | Phase II, Active, not recruiting | NCT01009788 |
| Talazoparib | Neoadjuvant therapy | None | Primary breast cancer ≥1 cm with a deleterious BRCA mutation | Phase II, Active, not recruiting | NCT02282345 |
| Talazoparib | Advanced TNBC and HR deficiency and advanced HER2-negative breast cancer or other solid tumors with a mutation in HR pathway genes | ≥1 line of therapy | Talazoparib | Phase II, Recruiting | NCT02401347 |
| Talazoparib | Metastatic TNBC inhibitors PARP inhibitors | Platinum-containing regimen with disease progression > 8 weeks | Metastatic breast cancer with BRCA mutation | Phase II Terminated (Primary Analysis and study completed Not stopped | NCT02034916 (ABRAZO) |
| Talazoparib | Metastatic TNBC inhibitors PARP inhibitors | ≤3 chemotherapy-inclusive regimens Talazoparib versus physician‘s choice | Locally advanced and/or metastatic breast cancer with germline BRCA mutations | Phase III Completed | NCT01945775 (EMBRACA) |
| Rucaparib | Metastatic TNBC inhibitors PARP inhibitors | ≤5 prior chemotherapy Rucaparib regimens in the last 5 years | Patients presenting with metastatic breast cancer (MBC) | Phase II, Completed | NCT00664781 |
| Rucaparib | Metastatic TNBC inhibitors PARP inhibitors | ≥1 line of chemotherapy, Rucaparib | Patients with a BRCAness genomic signature | Phase II Completed | NCT02505048 (RUBY) |
| Rucaparib | Stage I–III patients with TNBC or inhibitors PARP inhibitors | Neoadjuvant chemotherapy Cisplatin with rucaparib | ER/PR+, HER2- negative breast cancer with known BRCA1/2 mutations | Phase II Completed | NCT01074970 |
| Rucaparib | TNBC inhibitors | ≥3 prior chemotherapy regimens, Rucaparib | Patients with advanced solid tumors with evidence of germline | Phase I/II Active, not recruiting | NCT01482715 |
| Rucaparib | TNBC inhibitors | ≤5 prior chemotherapy regimens in the last 5 years, Rucaparib | Patients with MBC carriers of a BRCA1/2 | Phase II Completed | NCT00664781 |
| Rucaparib | TNBC inhibitors | ≥1 line of chemotherapy Rucaparib | Patients with a BRCAness genomic signature | Phase II Completed | NCT02505048 (RUBY) |
| Rucaparib | TNBC inhibitors | Neoadjuvant chemotherapy Cisplatin with rucaparib | Advanced solid tumors with evidence of germline or somatic BRCA mutation | Completed | NCT01074970 |
| Rucaparib | TNBC inhibitors | ≥3 prior chemotherapy regimens | Advanced solid tumors with evidence of germline or somatic BRCA mutation | Phase I/II Active, not recruiting | NCT01482715 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, D.D.; Parveen, A.; Yadav, D.K. Role of PARP in TNBC: Mechanism of Inhibition, Clinical Applications, and Resistance. Biomedicines 2021, 9, 1512. https://doi.org/10.3390/biomedicines9111512
Singh DD, Parveen A, Yadav DK. Role of PARP in TNBC: Mechanism of Inhibition, Clinical Applications, and Resistance. Biomedicines. 2021; 9(11):1512. https://doi.org/10.3390/biomedicines9111512
Chicago/Turabian StyleSingh, Desh Deepak, Amna Parveen, and Dharmendra Kumar Yadav. 2021. "Role of PARP in TNBC: Mechanism of Inhibition, Clinical Applications, and Resistance" Biomedicines 9, no. 11: 1512. https://doi.org/10.3390/biomedicines9111512
APA StyleSingh, D. D., Parveen, A., & Yadav, D. K. (2021). Role of PARP in TNBC: Mechanism of Inhibition, Clinical Applications, and Resistance. Biomedicines, 9(11), 1512. https://doi.org/10.3390/biomedicines9111512