
IL-36α and Lipopolysaccharide Cooperatively Induce Autophagy by Triggering Pro-Autophagic Biased Signaling


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Compounds
2.2. Cell Culture
2.3. Immunofluorescence Assay
2.4. Western Blot Assays
2.5. Phospho-Kinase Array Analysis
2.6. Statistical Analysis
3. Results
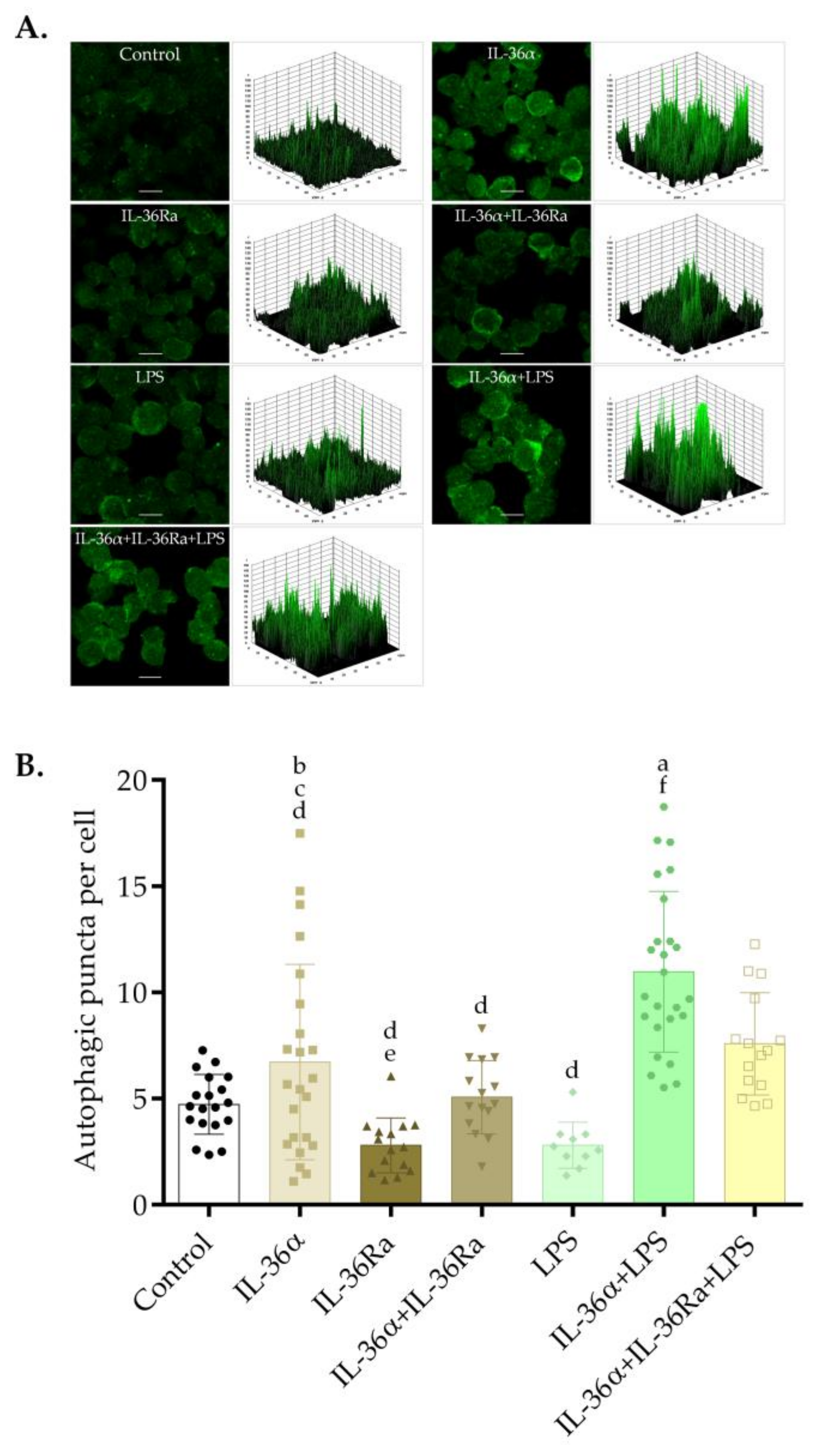
3.1. The Effects of IL-36α and LPS on the Subcellular Localization of LC3B in the THP-1 Cell Line
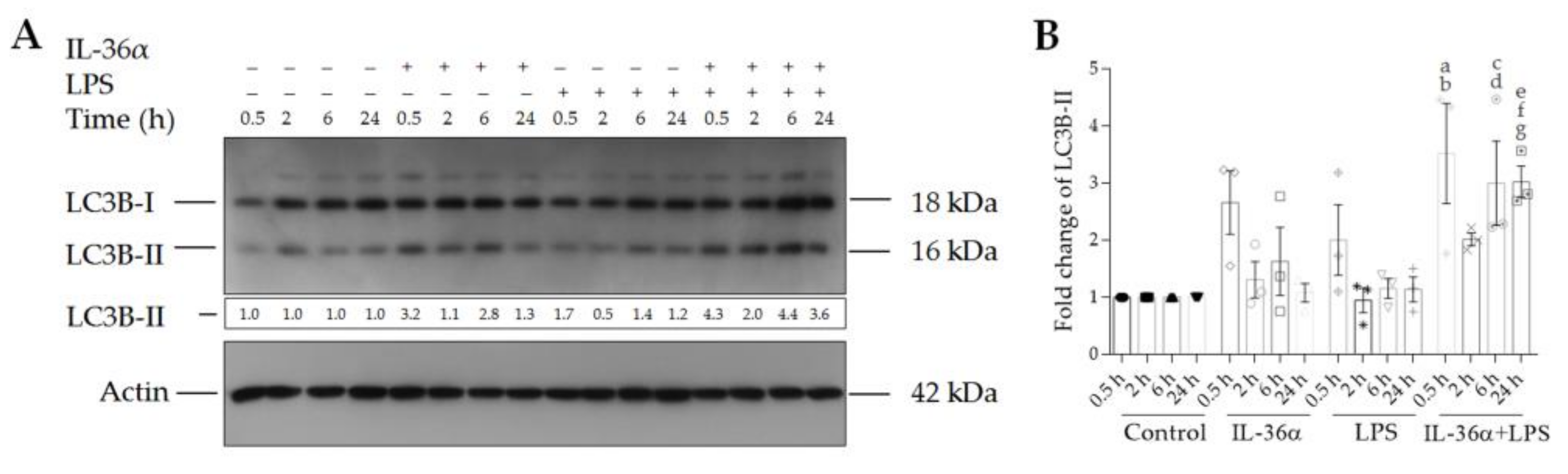
3.2. The Effects of IL-36α and LPS on the Levels of LC3B-I and LC3B-II
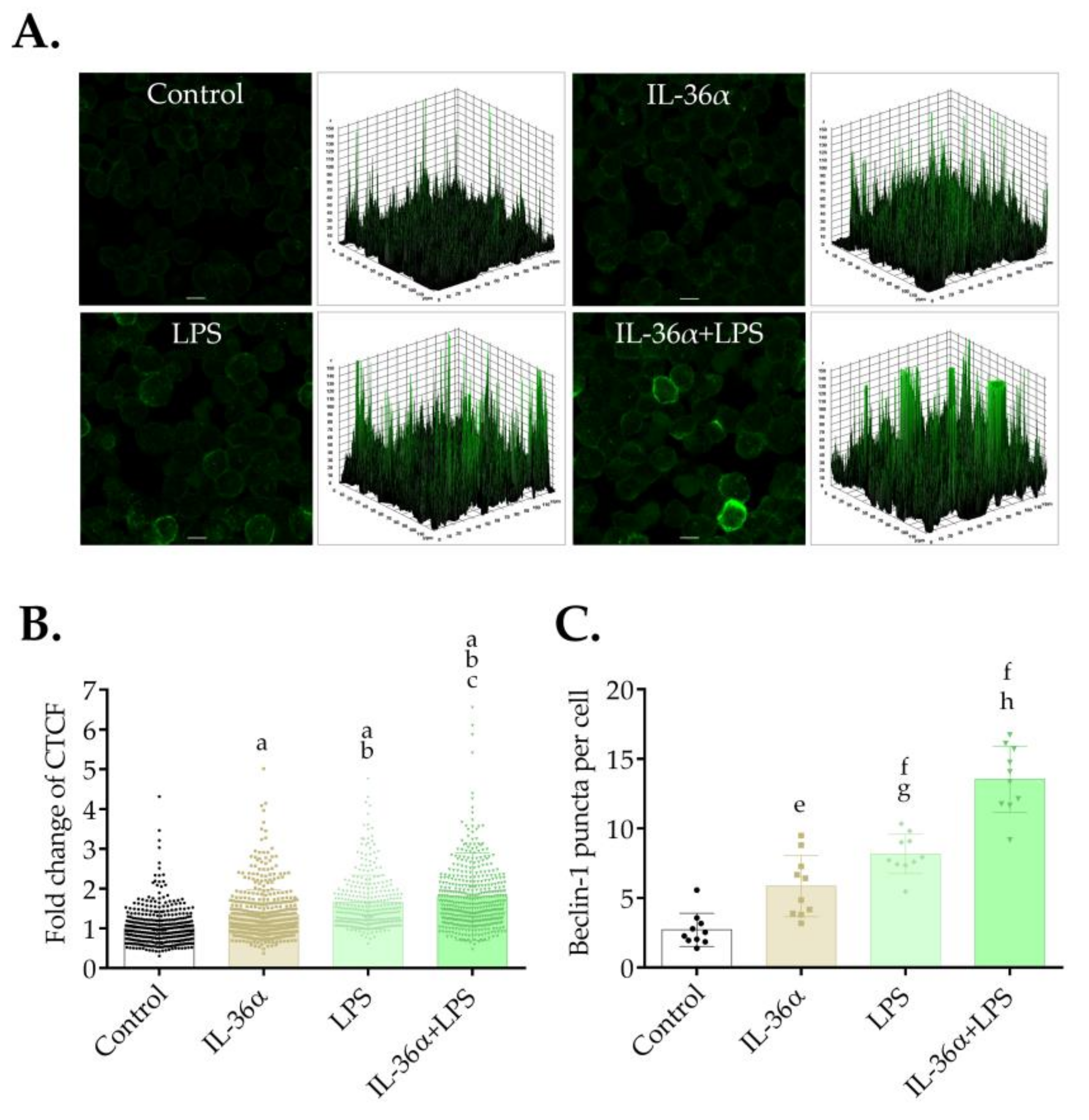
3.3. The Effects of IL-36α and LPS on the Autophagic Flux
3.4. The Effects of IL-36α and LPS on the Level of Beclin-1
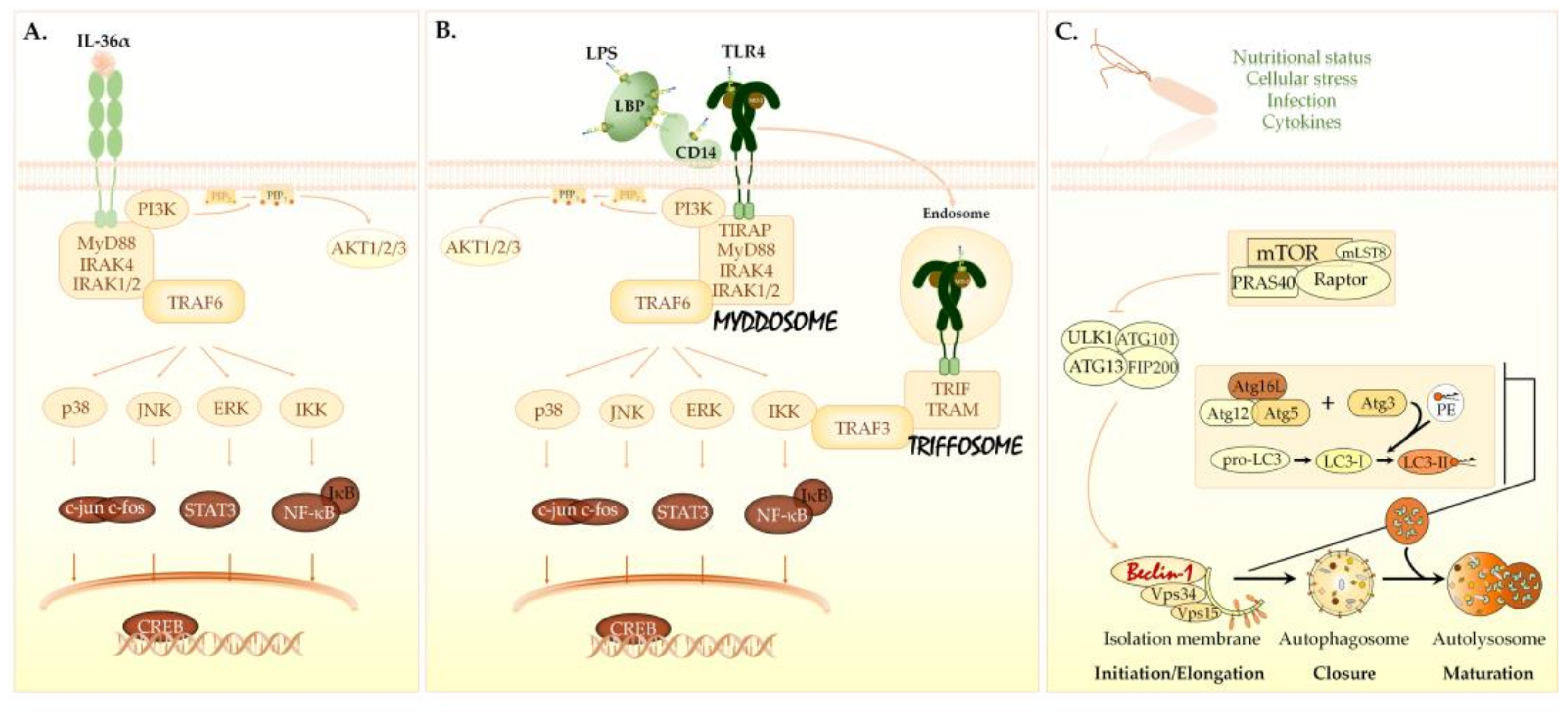
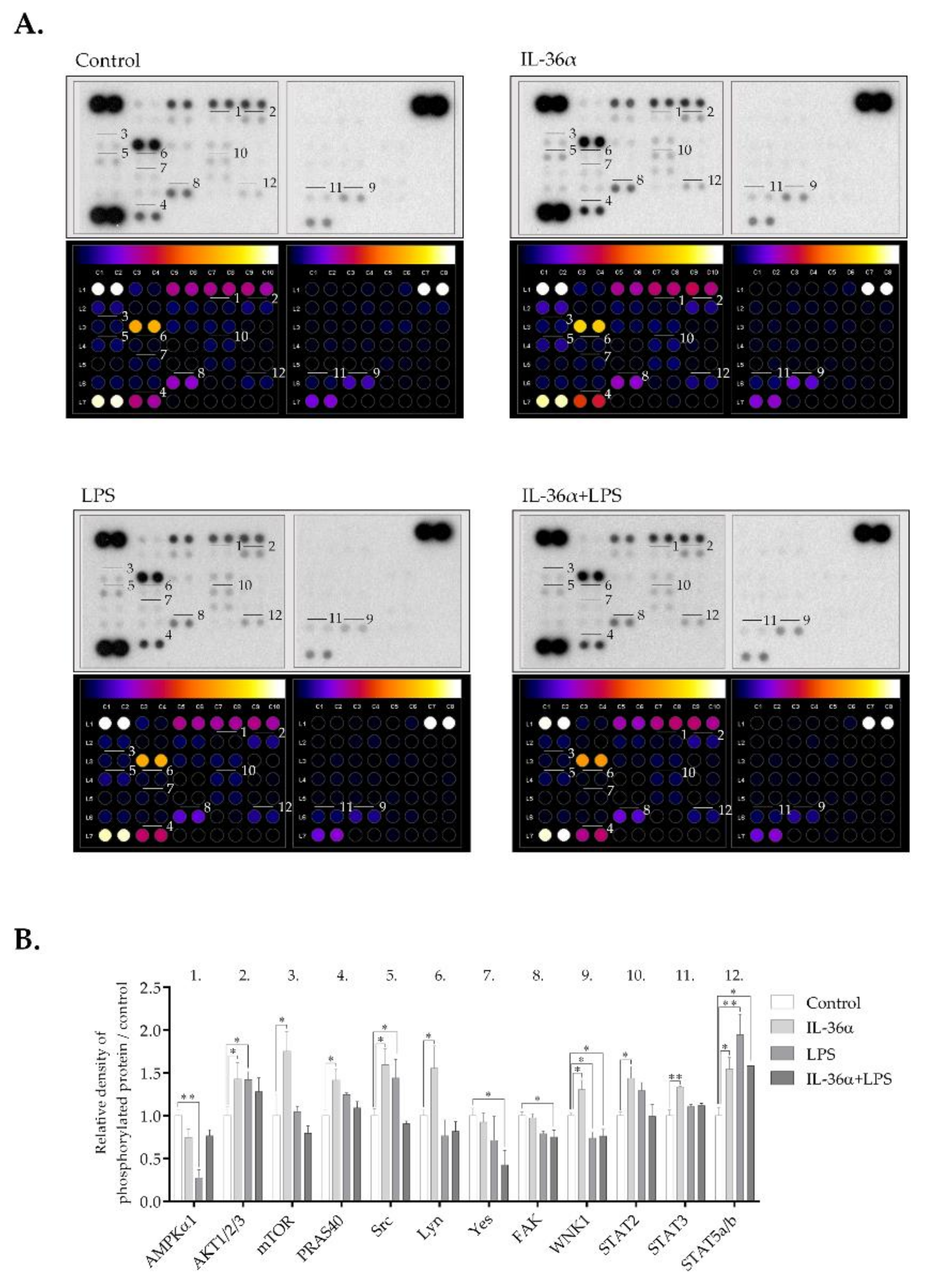
3.5. The Effects of IL-36α and LPS on Cellular Signaling in the THP-1 Cell Line
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dinarello, C.A. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrieta-Coxca, J.M.; Rodríguez-Martínez, S.; Cancino-Diaz, M.E.; Markert, U.R.; Favaro, R.R.; Morales-Prieto, D.M. IL-36 Cytokines: Regulators of Inflammatory Responses and Their Emerging Role in Immunology of Reproduction. Int. J. Mol. Sci. 2019, 20, 1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.E.; Renshaw, B.R.; Ketchem, R.R.; Kubin, M.; Garka, K.E.; Sims, J.E. Four New Members Expand the Interleukin-1 Superfamily. J. Biol. Chem. 2000, 275, 1169–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clancy, D.M.; Henry, C.M.; Sullivan, G.P.; Martin, S.J. Neutrophil Extracellular Traps Can Serve as Platforms for Processing and Activation of IL-1 Family Cytokines. FEBS J. 2017, 284, 1712–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Todorovic, V. Interleukin-36: Structure, Signaling and Function. In Protein Reviews: Volume 21; Atassi, M.Z., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2021; pp. 191–210. ISBN 978-3-030-67814-2. [Google Scholar]
- Towne, J.E.; Garka, K.E.; Renshaw, B.R.; Virca, G.D.; Sims, J.E. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 Signal through IL-1Rrp2 and IL-1RAcP to Activate the Pathway Leading to NF-KappaB and MAPKs. J. Biol. Chem. 2004, 279, 13677–13688. [Google Scholar] [CrossRef] [Green Version]
- Boraschi, D.; Tagliabue, A. The Interleukin-1 Receptor Family. Semin. Immunol. 2013, 25, 394–407. [Google Scholar] [CrossRef]
- Yi, G.; Ybe, J.A.; Saha, S.S.; Caviness, G.; Raymond, E.; Ganesan, R.; Mbow, M.L.; Kao, C.C. Structural and Functional Attributes of the Interleukin-36 Receptor. J. Biol. Chem. 2016, 291, 16597–16609. [Google Scholar] [CrossRef] [Green Version]
- Bridgewood, C.; Stacey, M.; Alase, A.; Lagos, D.; Graham, A.; Wittmann, M. IL-36γ Has Proinflammatory Effects on Human Endothelial Cells. Exp. Dermatol. 2017, 26, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.M.; Baliwag, J.; Chen, C.S.; Guzman, A.M.; Stoll, S.W.; Gudjonsson, J.E.; Ward, N.L.; Johnston, A. IL-36 Promotes Myeloid Cell Infiltration, Activation, and Inflammatory Activity in Skin. J. Immunol. 2014, 192, 6053–6061. [Google Scholar] [CrossRef] [Green Version]
- Vigne, S.; Palmer, G.; Martin, P.; Lamacchia, C.; Strebel, D.; Rodriguez, E.; Olleros, M.L.; Vesin, D.; Garcia, I.; Ronchi, F.; et al. IL-36 Signaling Amplifies Th1 Responses by Enhancing Proliferation and Th1 Polarization of Naive CD4+ T Cells. Blood 2012, 120, 3478–3487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, A.; Xing, X.; Guzman, A.M.; Riblett, M.; Loyd, C.M.; Ward, N.L.; Wohn, C.; Prens, E.P.; Wang, F.; Maier, L.E.; et al. IL-1F5, -F6, -F8, and -F9: A Novel IL-1 Family Signaling System That Is Active in Psoriasis and Promotes Keratinocyte Antimicrobial Peptide Expression. J. Immunol. 2011, 186, 2613–2622. [Google Scholar] [CrossRef] [Green Version]
- Qu, Q.; Zhai, Z.; Xu, J.; Li, S.; Chen, C.; Lu, B. IL36 Cooperates with Anti-CTLA-4 MAbs to Facilitate Antitumor Immune Responses. Front. Immunol. 2020, 11, 634. [Google Scholar] [CrossRef]
- Harusato, A.; Abo, H.; Ngo, V.L.; Yi, S.W.; Mitsutake, K.; Osuka, S.; Kohlmeier, J.E.; Li, J.D.; Gewirtz, A.T.; Nusrat, A.; et al. IL-36γ Signaling Controls the Induced Regulatory T Cell–Th9 Cell Balance via NFκB Activation and STAT Transcription Factors. Mucosal. Immunol. 2017, 10, 1455–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, V.L.; Kuczma, M.; Maxim, E.; Denning, T.L. IL-36 Cytokines and Gut Immunity. Immunology 2021, 163, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Bassoy, E.Y.; Towne, J.E.; Gabay, C. Regulation and Function of Interleukin-36 Cytokines. Immunol. Rev. 2018, 281, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Gresnigt, M.S.; Rösler, B.; Jacobs, C.W.M.; Becker, K.L.; Joosten, L.A.B.; van der Meer, J.W.M.; Netea, M.G.; Dinarello, C.A.; van de Veerdonk, F.L. The IL-36 Receptor Pathway Regulates Aspergillus Fumigatus-Induced Th1 and Th17 Responses. Eur. J. Immunol. 2013, 43, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Mazgaeen, L.; Gurung, P. Recent Advances in Lipopolysaccharide Recognition Systems. IJMS 2020, 21, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steimle, A.; Autenrieth, I.B.; Frick, J.-S. Structure and Function: Lipid A Modifications in Commensals and Pathogens. Int. J. Med. Microbiol. 2016, 306, 290–301. [Google Scholar] [CrossRef] [Green Version]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieser, K.J.; Kagan, J.C. Multi-Receptor Detection of Individual Bacterial Products by the Innate Immune System. Nat. Rev. Immunol. 2017, 17, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 Trafficking and Its Influence on LPS-Induced pro-Inflammatory Signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and Localization of Toll-like Receptor Signalling Complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Boonen, B.; Alpizar, Y.A.; Meseguer, V.M.; Talavera, K. TRP Channels as Sensors of Bacterial Endotoxins. Toxins 2018, 10, 326. [Google Scholar] [CrossRef] [Green Version]
- Schappe, M.S.; Szteyn, K.; Stremska, M.E.; Mendu, S.K.; Downs, T.K.; Seegren, P.V.; Mahoney, M.A.; Dixit, S.; Krupa, J.K.; Stipes, E.J.; et al. Chanzyme TRPM7 Mediates the Ca2+ Influx Essential for Lipopolysaccharide-Induced Toll-Like Receptor 4 Endocytosis and Macrophage Activation. Immunity 2018, 48, 59–74.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheraga, R.G.; Abraham, S.; Niese, K.A.; Southern, B.D.; Grove, L.M.; Hite, R.D.; McDonald, C.; Hamilton, T.A.; Olman, M.A. TRPV4 Mechanosensitive Ion Channel Regulates Lipopolysaccharide-Stimulated Macrophage Phagocytosis. J. Immunol. 2016, 196, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat. Rev. Mol. Cell. Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Mammalian Autophagy: Core Molecular Machinery and Signaling Regulation. Curr. Opin. Cell. Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S. Regulation of Autophagy by MTOR-Dependent and MTOR-Independent Pathways: Autophagy Dysfunction in Neurodegenerative Diseases and Therapeutic Application of Autophagy Enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Guan, K.-L. MTOR: A Pharmacologic Target for Autophagy Regulation. J. Clin. Invest. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Guan, K.-L. MTOR as a Central Hub of Nutrient Signalling and Cell Growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Yang, J.; Carra, S.; Zhu, W.-G.; Kampinga, H.H. The Regulation of the Autophagic Network and Its Implications for Human Disease. Int. J. Biol. Sci. 2013, 9, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, S.R.; Simonsen, A. Membrane Dynamics in Autophagosome Biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The Autophagosome: Origins Unknown, Biogenesis Complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A Ubiquitin-like System Mediates Protein Lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The ATG Conjugation Systems in Autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A Protein Conjugation System Essential for Autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Schulman, B.A. Dynamic Regulation of Macroautophagy by Distinctive Ubiquitin-like Proteins. Nat. Struct. Mol. Biol. 2014, 21, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yoshimori, T. New Insights into Autophagosome–Lysosome Fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in Immunity and Inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Sharma, V.; Verma, S.; Seranova, E.; Sarkar, S.; Kumar, D. Selective Autophagy and Xenophagy in Infection and Disease. Front. Cell Dev. Biol. 2018, 6, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deretic, V. Autophagy: An Emerging Immunological Paradigm. J. Immunol. 2012, 189, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Fang, X.; Wang, X. Autophagy and Inflammation. Clin. Transl. Med. 2017, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Huang, M.; Yao, Y. Autophagy and Proinflammatory Cytokines: Interactions and Clinical Implications. Cytokine Growth Factor Rev. 2018, 43, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of Innate Immune Cell Function by MTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Katholnig, K.; Linke, M.; Pham, H.; Hengstschläger, M.; Weichhart, T. Immune Responses of Macrophages and Dendritic Cells Regulated by MTOR Signalling. Biochem. Soc. Trans. 2013, 41, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-S.; Kehrl, J.H. MyD88 and Trif Target Beclin 1 to Trigger Autophagy in Macrophages. J. Biol. Chem. 2008, 283, 33175–33182. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.S.; Kehrl, J.H. TRAF6 and A20 Regulate Lysine 63-Linked Ubiquitination of Beclin-1 to Control TLR4-Induced Autophagy. Sci. Signal. 2010, 3, ra42. [Google Scholar] [CrossRef]
- Harris, J. Autophagy and Cytokines. Cytokine 2011, 56, 140–144. [Google Scholar] [CrossRef]
- Orosz, L.; Papanicolaou, E.G.; Seprényi, G.; Megyeri, K. IL-17A and IL-17F Induce Autophagy in RAW 264.7 Macrophages. Biomed. Pharmacother. 2016, 77, 129–134. [Google Scholar] [CrossRef]
- Gao, Y.; Wen, Q.; Hu, S.; Zhou, X.; Xiong, W.; Du, X.; Zhang, L.; Fu, Y.; Yang, J.; Zhou, C.; et al. IL-36γ Promotes Killing of Mycobacterium Tuberculosis by Macrophages via WNT5A-Induced Noncanonical WNT Signaling. J. Immunol. 2019, 203, 922–935. [Google Scholar] [CrossRef]
- Ge, Y.; Huang, M.; Dong, N.; Yao, Y.-M. Effect of Interleukin-36β on Activating Autophagy of CD4+CD25+ Regulatory T Cells and Its Immune Regulation in Sepsis. J. Infect. Dis. 2020, 222, 1517–1530. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods. 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Dikic, I. Autophagy in Antimicrobial Immunity. Mol. Cell. 2014, 54, 224–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition)1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like Receptors Control Autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento, E.B.M.; Ouwens, D.M. PRAS40: Target or Modulator of MTORC1 Signalling and Insulin Action? Arch. Physiol. Biochem. 2009, 115, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, X.; Shen, X.; Tang, P.; Chen, C.; Zhu, Q.; Li, M.; Xia, R.; Yang, X.; Feng, C.; et al. IL-36β Promotes CD8+ T Cell Activation and Antitumor Immune Responses by Activating MTORC1. Front. Immunol. 2019, 10, 1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallolu Kankanamalage, S.; Lee, A.-Y.; Wichaidit, C.; Lorente-Rodriguez, A.; Shah, A.M.; Stippec, S.; Whitehurst, A.W.; Cobb, M.H. Multistep Regulation of Autophagy by WNK1. Proc. Natl. Acad. Sci. USA 2016, 113, 14342–14347. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The Role of STAT3 in Autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, K.; Lin, L.; Ai, Q.; Wan, J.; Dai, J.; Liu, G.; Tang, L.; Yang, Y.; Ge, P.; Jiang, R.; et al. Lipopolysaccharide-Induced Dephosphorylation of AMPK-Activated Protein Kinase Potentiates Inflammatory Injury via Repression of ULK1-Dependent Autophagy. Front. Immunol. 2018, 9, 1464. [Google Scholar] [CrossRef]
- Rex, J.; Albrecht, U.; Ehlting, C.; Thomas, M.; Zanger, U.M.; Sawodny, O.; Häussinger, D.; Ederer, M.; Feuer, R.; Bode, J.G. Model-Based Characterization of Inflammatory Gene Expression Patterns of Activated Macrophages. PLoS Comput. Biol. 2016, 12, e1005018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, K.; Kitano, H. A Comprehensive Map of the Toll-like Receptor Signaling Network. Mol. Syst. Biol. 2006, 2, 2006.0015. [Google Scholar] [CrossRef]
- Fukao, T.; Koyasu, S. PI3K and Negative Regulation of TLR Signaling. Trends Immunol. 2003, 24, 358–363. [Google Scholar] [CrossRef]
- Aksoy, E.; Taboubi, S.; Torres, D.; Delbauve, S.; Hachani, A.; Whitehead, M.A.; Pearce, W.P.; Berenjeno, I.M.; Nock, G.; Filloux, A.; et al. The P110δ Isoform of the Kinase PI(3)K Controls the Subcellular Compartmentalization of TLR4 Signaling and Protects from Endotoxic Shock. Nat. Immunol. 2012, 13, 1045–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutouja, F.; Brinkmeier, R.; Mastalski, T.; El Magraoui, F.; Platta, H.W. Regulation of the Tumor-Suppressor BECLIN 1 by Distinct Ubiquitination Cascades. Int. J. Mol. Sci. 2017, 18, 2541. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.-D.; Qin, Z.-H. Beclin 1, Bcl-2 and Autophagy. In Autophagy: Biology and Diseases; Qin, Z.-H., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1206, pp. 109–126. ISBN 9789811506017. [Google Scholar]
- Huang, J.; Brumell, J.H. Bacteria-Autophagy Interplay: A Battle for Survival. Nat. Rev. Microbiol. 2014, 12, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-W.; Li, F. Bacterial Interaction with Host Autophagy. Virulence 2019, 10, 352–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Luhaibi, Z.I.I.; Dernovics, Á.; Seprényi, G.; Ayaydin, F.; Boldogkői, Z.; Veréb, Z.; Megyeri, K. IL-36α and Lipopolysaccharide Cooperatively Induce Autophagy by Triggering Pro-Autophagic Biased Signaling. Biomedicines 2021, 9, 1541. https://doi.org/10.3390/biomedicines9111541
Al-Luhaibi ZII, Dernovics Á, Seprényi G, Ayaydin F, Boldogkői Z, Veréb Z, Megyeri K. IL-36α and Lipopolysaccharide Cooperatively Induce Autophagy by Triggering Pro-Autophagic Biased Signaling. Biomedicines. 2021; 9(11):1541. https://doi.org/10.3390/biomedicines9111541
Chicago/Turabian StyleAl-Luhaibi, Zaid I. I., Áron Dernovics, György Seprényi, Ferhan Ayaydin, Zsolt Boldogkői, Zoltán Veréb, and Klára Megyeri. 2021. "IL-36α and Lipopolysaccharide Cooperatively Induce Autophagy by Triggering Pro-Autophagic Biased Signaling" Biomedicines 9, no. 11: 1541. https://doi.org/10.3390/biomedicines9111541
APA StyleAl-Luhaibi, Z. I. I., Dernovics, Á., Seprényi, G., Ayaydin, F., Boldogkői, Z., Veréb, Z., & Megyeri, K. (2021). IL-36α and Lipopolysaccharide Cooperatively Induce Autophagy by Triggering Pro-Autophagic Biased Signaling. Biomedicines, 9(11), 1541. https://doi.org/10.3390/biomedicines9111541