
Orai1–STIM1 Regulates Increased Ca2+ Mobilization, Leading to Contractile Duchenne Muscular Dystrophy Phenotypes in Patient-Derived Induced Pluripotent Stem Cells


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. iPSC Lines
2.3. Plate Preparation
2.4. Generation of the iPSC Line Stably Expressing Tet-Inducible MyoD1
2.5. Feeder-Free iPSC Culture
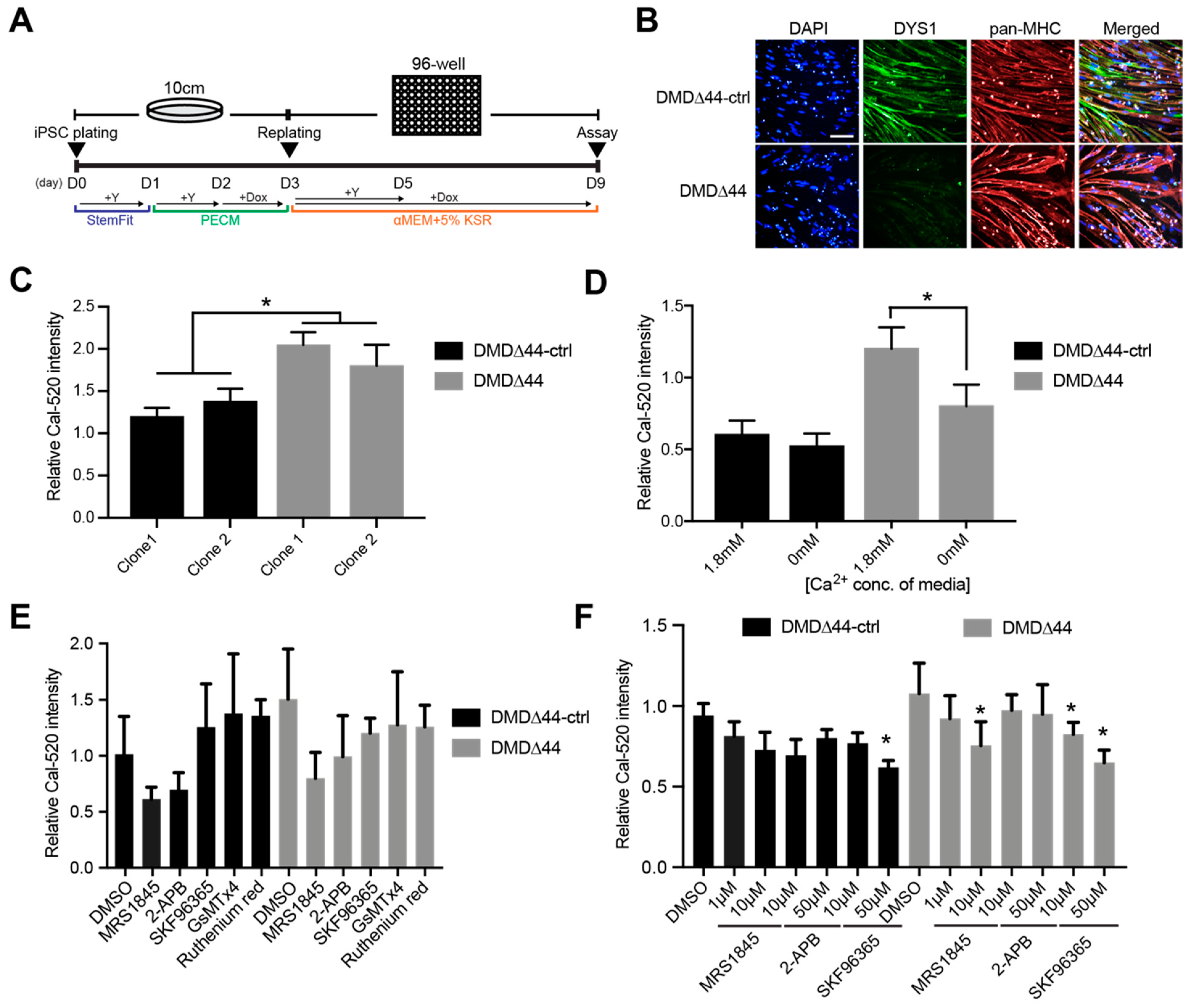
2.6. Skeletal Muscle Differentiation by the Standard Replating Method
2.7. Skeletal Muscle Differentiation by the Modified Replating Method
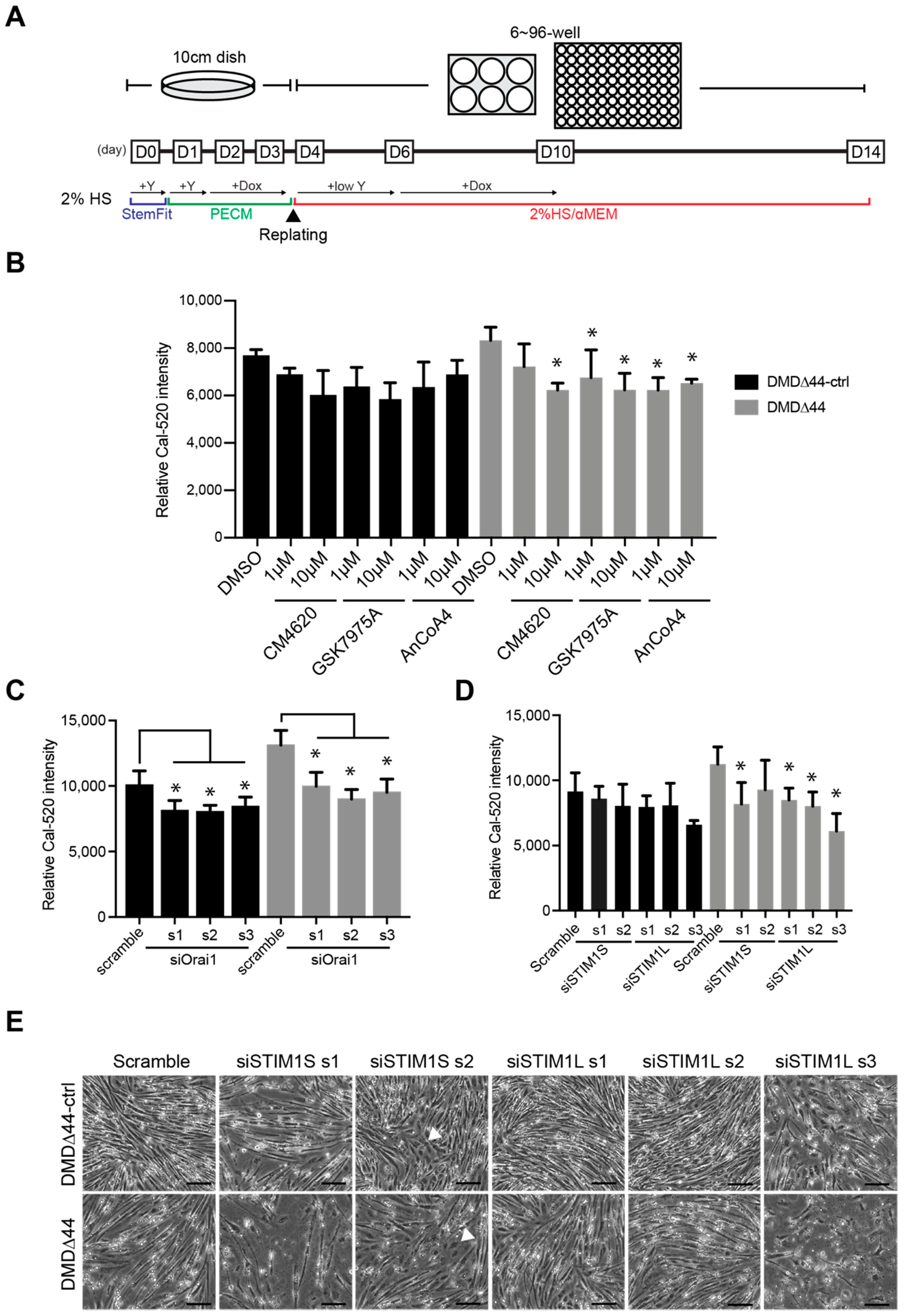
2.8. siRNA Transfection
2.9. Immunocytochemistry
2.10. RT-qPCR Analysis
2.11. Western Blot Analysis
2.12. Ca2+ Mobilization Assay
2.13. Total Ca2+ Content Measurement
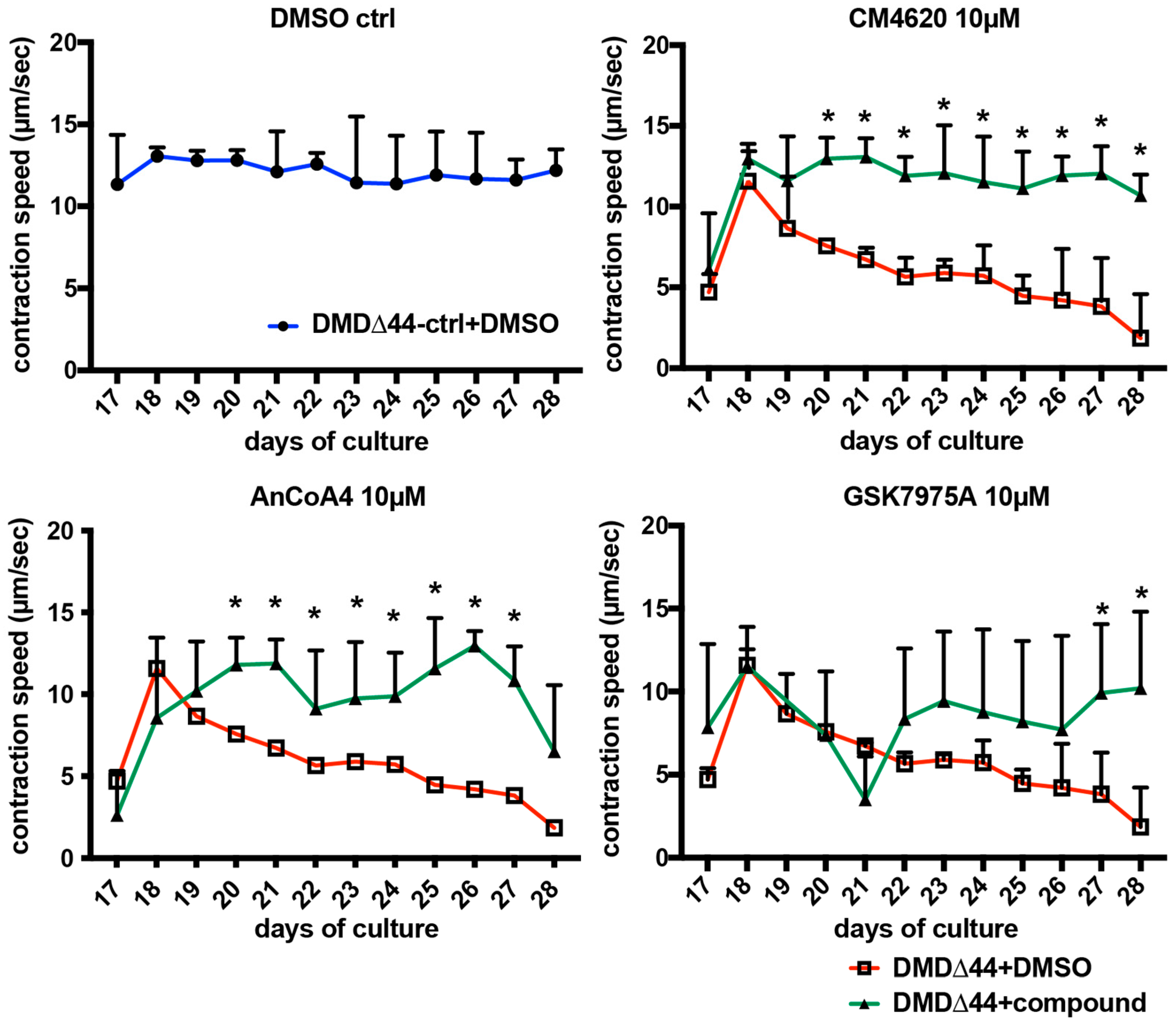
2.14. Electrical Field Stimulation (EFS) and Motion Imaging Assays
2.15. Microscopy
2.16. Statistics Analysis
3. Results
3.1. Ca2+ Overload Condition
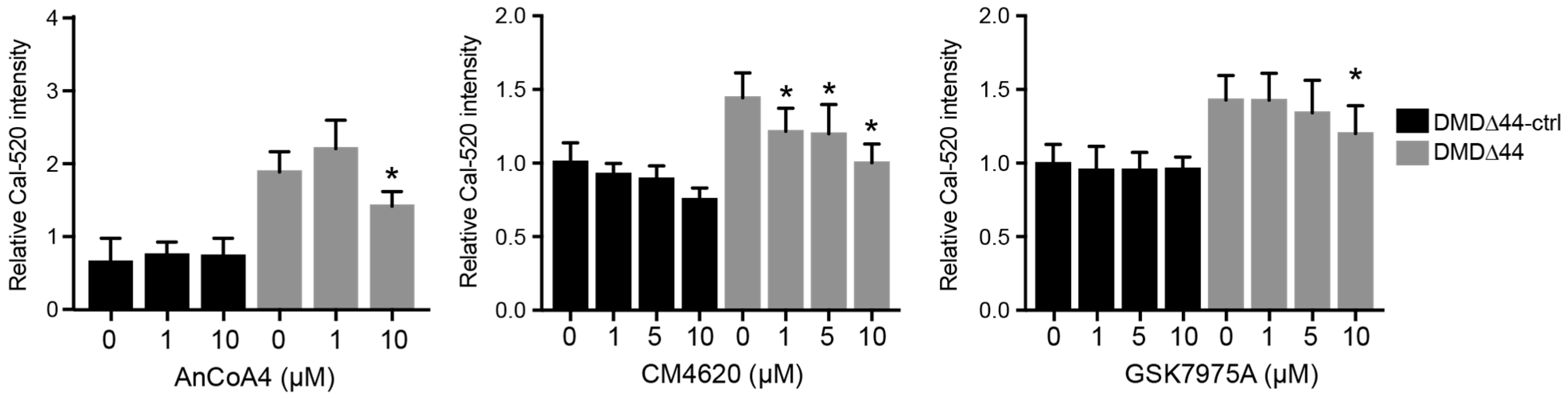
3.2. SOCs
3.3. Orai1 and STIM1 Inhibitors
3.4. Re-Evaluation of the Role of Orai1 and STIM1 in Matured Myotubes
3.5. Effects of Orai1–STIM1 Inhibitors in the Muscle Training Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fairclough, R.J.; Wood, M.J.; Davies, K.E. Therapy for Duchenne muscular dystrophy: Renewed optimism from genetic approaches. Nat. Rev. Genet. 2013, 14, 373–378. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Manzur, A.Y.; Kuntzer, T.; Pike, M.; Swan, A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2008, CD003725. [Google Scholar] [CrossRef]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2016, CD003725. [Google Scholar] [CrossRef] [Green Version]
- Duan, D. Micro-Dystrophin Gene Therapy Goes Systemic in Duchenne Muscular Dystrophy Patients. Hum. Gene Ther. 2018, 29, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Abdel-Hamid, H.Z.; Mah, J.K.; Campbell, C.; Guglieri, M.; Muntoni, F.; Takeshima, Y.; McDonald, C.M.; Kostera-Pruszczyk, A.; Karachunski, P.; et al. Randomized phase 2 trial and open-label extension of domagrozumab in Duchenne muscular dystrophy. Neuromuscul. Disord. 2020, 30, 492–502. [Google Scholar] [CrossRef]
- Dzierlega, K.; Yokota, T. Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy. Gene Ther. 2020, 27, 407–416. [Google Scholar] [CrossRef]
- Allen, D.G. Skeletal muscle function: Role of ionic changes in fatigue, damage and disease. Clin. Exp. Pharmacol. Physiol. 2004, 31, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Okinaka, S.; Kumagai, H.; Ebashi, S.; Sugita, H.; Momoi, H.; Toyokura, Y.; Fujie, Y. Serum creatine phosphokinase. Activity in progressive muscular dystrophy and neuromuscular diseases. Arch. Neurol. 1961, 4, 520–525. [Google Scholar] [CrossRef]
- Badalamente, M.A.; Stracher, A. Delay of muscle degeneration and necrosis in mdx mice by calpain inhibition. Muscle Nerve 2000, 23, 106–111. [Google Scholar] [CrossRef]
- Evans, N.P.; Misyak, S.A.; Robertson, J.L.; Bassaganya-Riera, J.; Grange, R.W. Immune-mediated mechanisms potentially regulate the disease time-course of duchenne muscular dystrophy and provide targets for therapeutic intervention. PM&R 2009, 1, 755–768. [Google Scholar]
- Menazza, S.; Blaauw, B.; Tiepolo, T.; Toniolo, L.; Braghetta, P.; Spolaore, B.; Reggiani, C.; Di Lisa, F.; Bonaldo, P.; Canton, M. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum. Mol. Genet. 2010, 19, 4207–4215. [Google Scholar] [CrossRef] [Green Version]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef]
- Sudevan, S.; Takiura, M.; Kubota, Y.; Higashitani, N.; Cooke, M.; Ellwood, R.A.; Etheridge, T.; Szewczyk, N.J.; Higashitani, A. Mitochondrial dysfunction causes Ca2+ overload and ECM degradation-mediated muscle damage in C. elegans. FASEB J. 2019, 33, 9540–9550. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Rebalka, I.A.; Cao, A.; Monaco, C.M.; Varah, N.E.; Edgett, B.A.; Huber, J.S.; Tadi, P.; et al. Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2O2 emission during impaired oxidative phosphorylation. J. Cachexia Sarcopenia Muscle 2019, 10, 643–661. [Google Scholar] [CrossRef] [Green Version]
- Grskovic, M.; Javaherian, A.; Strulovici, B.; Daley, G.Q. Induced pluripotent stem cells—Opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 2011, 10, 915–929. [Google Scholar] [CrossRef]
- Mareedu, S.; Million, E.D.; Duan, D.; Babu, G.J. Abnormal Calcium Handling in Duchenne Muscular Dystrophy: Mechanisms and Potential Therapies. Front. Physiol. 2021, 12, 647010. [Google Scholar] [CrossRef] [PubMed]
- Shoji, E.; Sakurai, H.; Nishino, T.; Nakahata, T.; Heike, T.; Awaya, T.; Fujii, N.; Manabe, Y.; Matsuo, M.; Sehara-Fujisawa, A. Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci. Rep. 2015, 5, 12831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avila-Medina, J.; Mayoral-Gonzalez, I.; Dominguez-Rodriguez, A.; Gallardo-Castillo, I.; Ribas, J.; Ordoñez, A.; Rosado, J.A.; Smani, T. The Complex Role of Store Operated Calcium Entry Pathways and Related Proteins in the Function of Cardiac, Skeletal and Vascular Smooth Muscle Cells. Front. Physiol. 2018, 9, 257. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelucci, A.; Garcia-Castaneda, M.; Boncompagni, S.; Dirksen, R.T. Role of STIM1/ORAI1-mediated store-operated Ca(2+) entry in skeletal muscle physiology and disease. Cell Calcium 2018, 76, 101–115. [Google Scholar] [CrossRef]
- Wei-LaPierre, L.; Carrell, E.M.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun. 2013, 4, 2805. [Google Scholar] [CrossRef] [Green Version]
- Boncompagni, S.; Michelucci, A.; Pietrangelo, L.; Dirksen, R.T.; Protasi, F. Exercise-dependent formation of new junctions that promote STIM1-Orai1 assembly in skeletal muscle. Sci. Rep. 2017, 7, 14286. [Google Scholar] [CrossRef]
- Zhao, X.; Moloughney, J.G.; Zhang, S.; Komazaki, S.; Weisleder, N. Orai1 mediates exacerbated Ca2+ entry in dystrophic skeletal muscle. PLoS ONE 2012, 7, e49862. [Google Scholar] [CrossRef] [Green Version]
- Kiviluoto, S.; Decuypere, J.P.; De Smedt, H.; Missiaen, L.; Parys, J.B.; Bultynck, G. STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet. Muscle 2011, 1, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antigny, F.; Sabourin, J.; Saüc, S.; Bernheim, L.; Koenig, S.; Frieden, M. TRPC1 and TRPC4 channels functionally interact with STIM1L to promote myogenesis and maintain fast repetitive Ca2+ release in human myotubes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 806–813. [Google Scholar] [CrossRef]
- Goonasekera, S.A.; Davis, J.; Kwong, J.Q.; Accornero, F.; Wei-LaPierre, L.; Sargent, M.A.; Dirksen, R.T.; Molkentin, J.D. Enhanced Ca2+ influx from STIM1-Orai1 induces muscle pathology in mouse models of muscular dystrophy. Hum. Mol. Genet. 2014, 23, 3706–3715. [Google Scholar] [CrossRef] [Green Version]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Uchimura, T.; Otomo, J.; Sato, M.; Sakurai, H. A human iPS cell myogenic differentiation system permitting high-throughput drug screening. Stem Cell Res. 2017, 25, 98–106. [Google Scholar] [CrossRef]
- Tanaka, A.; Woltjen, K.; Miyake, K.; Hotta, A.; Ikeya, M.; Yamamoto, T.; Nishino, T.; Shoji, E.; Sehara-Fujisawa, A.; Manabe, Y.; et al. Efficient and reproducible myogenic differentiation from human iPS cells: Prospects for modeling Miyoshi Myopathy in vitro. PLoS ONE 2013, 8, e61540. [Google Scholar] [CrossRef]
- Oceguera-Yanez, F.; Kim, S.I.; Matsumoto, T.; Tan, G.W.; Xiang, L.; Hatani, T.; Kondo, T.; Ikeya, M.; Yoshida, Y.; Inoue, H.; et al. Engineering the AAVS1 locus for consistent and scalable transgene expression in human iPSCs and their differentiated derivatives. Methods 2016, 101, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Darbellay, B.; Arnaudeau, S.; Bader, C.R.; Konig, S.; Bernheim, L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 2011, 194, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Michelucci, A.; Boncompagni, S.; Pietrangelo, L.; García-Castañeda, M.; Takano, T.; Malik, S.; Dirksen, R.T.; Protasi, F. Transverse tubule remodeling enhances Orai1-dependent Ca2+ entry in skeletal muscle. eLife 2019, 8, e47576. [Google Scholar] [CrossRef]
- Michelucci, A.; Boncompagni, S.; Pietrangelo, L.; Takano, T.; Protasi, F.; Dirksen, R.T. Pre-assembled Ca2+ entry units and constitutively active Ca2+ entry in skeletal muscle of calsequestrin-1 knockout mice. J. Gen. Physiol. 2020, 152, e202012617. [Google Scholar] [CrossRef] [PubMed]
- Uchimura, T.; Asano, T.; Nakata, T.; Hotta, A.; Sakurai, H. A muscle fatigue-like contractile decline was recapitulated using skeletal myotubes from Duchenne muscular dystrophy patient-derived iPSCs. Cell Rep. Med. 2021, 2, 100298. [Google Scholar] [CrossRef]
- Kurebayashi, N.; Ogawa, Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J. Physiol. 2001, 533, 185–199. [Google Scholar] [CrossRef]
- Gnanasambandam, R.; Ghatak, C.; Yasmann, A.; Nishizawa, K.; Sachs, F.; Ladokhin, A.S.; Sukharev, S.I.; Suchyna, T.M. GsMTx4: Mechanism of Inhibiting Mechanosensitive Ion Channels. Biophys. J. 2017, 112, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Tripathy, A.; Pasek, D.A.; Meissner, G. Ruthenium red modifies the cardiac and skeletal muscle Ca2+ release channels (ryanodine receptors) by multiple mechanisms. J. Biol. Chem. 1999, 274, 32680–32691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bootman, M.D.; Collins, T.J.; Mackenzie, L.; Roderick, H.L.; Berridge, M.J.; Peppiatt, C.M. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 2002, 16, 1145–1150. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G. The STIM1-ORAI1 microdomain. Cell Calcium 2015, 58, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Sadaghiani, A.M.; Lee, S.M.; Odegaard, J.I.; Leveson-Gower, D.B.; McPherson, O.M.; Novick, P.; Kim, M.R.; Koehler, A.N.; Negrin, R.; Dolmetsch, R.E.; et al. Identification of Orai1 channel inhibitors by using minimal functional domains to screen small molecule microarrays. Chem. Biol. 2014, 21, 1278–1292. [Google Scholar] [CrossRef]
- Waldron, R.T.; Chen, Y.; Pham, H.; Go, A.; Su, H.Y.; Hu, C.; Wen, L.; Husain, S.Z.; Sugar, C.A.; Roos, J.; et al. The Orai Ca2+ channel inhibitor CM4620 targets both parenchymal and immune cells to reduce inflammation in experimental acute pancreatitis. J. Physiol. 2019, 597, 3085–3105. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.Y.; Ehrlich, B.E. Signaling in muscle contraction. Cold Spring Harb. Perspect. Biol. 2015, 7, a006023. [Google Scholar] [CrossRef] [PubMed]
- Gailly, P. New aspects of calcium signaling in skeletal muscle cells: Implications in Duchenne muscular dystrophy. Biochim. Biophys. Acta 2002, 1600, 38–44. [Google Scholar] [CrossRef]
- Tutdibi, O.; Brinkmeier, H.; Rudel, R.; Fohr, K.J. Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J. Physiol. 1999, 515 Pt 3, 859–868. [Google Scholar] [CrossRef]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef]
- Kushnir, A.; Todd, J.J.; Witherspoon, J.W.; Yuan, Q.; Reiken, S.; Lin, H.; Munce, R.H.; Wajsberg, B.; Melville, Z.; Clarke, O.B.; et al. Intracellular calcium leak as a therapeutic target for RYR1-related myopathies. Acta Neuropathol. 2020, 139, 1089–1104. [Google Scholar] [CrossRef]
- Williams, G.S.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Belosludtseva, N.V.; Belosludtsev, K.N. The Effect of Deflazacort Treatment on the Functioning of Skeletal Muscle Mitochondria in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 8763. [Google Scholar] [CrossRef]
- Morgan, A.J.; Jacob, R. Ionomycin enhances Ca2+ influx by stimulating store-regulated cation entry and not by a direct action at the plasma membrane. Biochem. J. 1994, 300 Pt 3, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolić, N.; Görgens, S.W.; Thoresen, G.H.; Aas, V.; Eckel, J.; Eckardt, K. Electrical pulse stimulation of cultured skeletal muscle cells as a model for in vitro exercise—Possibilities and limitations. Acta Physiol. 2017, 220, 310–331. [Google Scholar] [CrossRef] [PubMed]
- Robin, G.; Berthier, C.; Allard, B. Sarcoplasmic reticulum Ca2+ permeation explored from the lumen side in mdx muscle fibers under voltage control. J. Gen. Physiol. 2012, 139, 209–218. [Google Scholar] [CrossRef]
- Cho, C.H.; Woo, J.S.; Perez, C.F.; Lee, E.H. A focus on extracellular Ca2+ entry into skeletal muscle. Exp. Mol. Med. 2017, 49, e378. [Google Scholar] [CrossRef] [Green Version]
- Harisseh, R.; Chatelier, A.; Magaud, C.; Deliot, N.; Constantin, B. Involvement of TRPV2 and SOCE in calcium influx disorder in DMD primary human myotubes with a specific contribution of alpha1-syntrophin and PLC/PKC in SOCE regulation. Am. J. Physiol.-Cell Physiol. 2013, 304, C881–C894. [Google Scholar] [CrossRef]
- Zhang, I.; Hu, H. Store-Operated Calcium Channels in Physiological and Pathological States of the Nervous System. Front. Cell. Neurosci. 2020, 14, 600758. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Appendino, G.; Nilius, B. Pharmacology of vanilloid transient receptor potential cation channels. Mol. Pharmacol. 2009, 75, 1262–1279. [Google Scholar] [CrossRef]
- Griffiths, E.J. Use of ruthenium red as an inhibitor of mitochondrial Ca2+ uptake in single rat cardiomyocytes. FEBS Lett. 2000, 486, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Saüc, S.; Bulla, M.; Nunes, P.; Orci, L.; Marchetti, A.; Antigny, F.; Bernheim, L.; Cosson, P.; Frieden, M.; Demaurex, N. STIM1L traps and gates Orai1 channels without remodeling the cortical ER. J. Cell Sci. 2015, 128, 1568–1579. [Google Scholar] [CrossRef] [Green Version]
- Horinouchi, T.; Higashi, T.; Higa, T.; Terada, K.; Mai, Y.; Aoyagi, H.; Hatate, C.; Nepal, P.; Horiguchi, M.; Harada, T.; et al. Different binding property of STIM1 and its novel splice variant STIM1L to Orai1, TRPC3, and TRPC6 channels. Biochem. Biophys. Res. Commun. 2012, 428, 252–258. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical | Dose | Activity |
|---|---|---|
| Tetracaine hydrochloride | 50 µM | Voltage-sensitive Ca release inhibitor |
| MRS1845 | 10 µM | Potent SOC blocker |
| 2-APB | 10 µM | Inhibit Ca2+ release from SOC |
| SKF96365 | 10 µM | Pan TRPC inhibitor |
| GsMTx4 | 1 µM | TRPC1/6 inhibitor |
| Ruthenium Red | 10 µM | Pan TRPV and mitochondrial Ca2+ uptake inhibitor |
| Ryanodine | 10 µM | Ca2+ release inhibitor from SR via RyR |
| Nifedipine | 10 µM | L-type Ca channel blocker |
| ML 9 | 10 µM | SOC inhibitor |
| YM 58483 | 10 µM | SOC blocker |
| Disulfiram | 10 µM | Reversibly stimulate Ca-ATPase |
| Ochratoxin A | 10 µM | Stimulate SR ATP-dependent Ca2+ pump |
| Istaroxime | 10 µM | Stimulate SERCA2a |
| 5J 4 | 10 µM | SOC blocker |
| Cyclopiazonic acid | 10 µM | Ca2+-ATPase inhibitor |
| Thapsigargin | 10 µM | SERCA inhibitor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uchimura, T.; Sakurai, H. Orai1–STIM1 Regulates Increased Ca2+ Mobilization, Leading to Contractile Duchenne Muscular Dystrophy Phenotypes in Patient-Derived Induced Pluripotent Stem Cells. Biomedicines 2021, 9, 1589. https://doi.org/10.3390/biomedicines9111589
Uchimura T, Sakurai H. Orai1–STIM1 Regulates Increased Ca2+ Mobilization, Leading to Contractile Duchenne Muscular Dystrophy Phenotypes in Patient-Derived Induced Pluripotent Stem Cells. Biomedicines. 2021; 9(11):1589. https://doi.org/10.3390/biomedicines9111589
Chicago/Turabian StyleUchimura, Tomoya, and Hidetoshi Sakurai. 2021. "Orai1–STIM1 Regulates Increased Ca2+ Mobilization, Leading to Contractile Duchenne Muscular Dystrophy Phenotypes in Patient-Derived Induced Pluripotent Stem Cells" Biomedicines 9, no. 11: 1589. https://doi.org/10.3390/biomedicines9111589
APA StyleUchimura, T., & Sakurai, H. (2021). Orai1–STIM1 Regulates Increased Ca2+ Mobilization, Leading to Contractile Duchenne Muscular Dystrophy Phenotypes in Patient-Derived Induced Pluripotent Stem Cells. Biomedicines, 9(11), 1589. https://doi.org/10.3390/biomedicines9111589