
Design of α/β-Hybrid Peptide Ligands of α4β1 Integrin Equipped with a Linkable Side Chain for Chemoselective Biofunctionalization of Microstructured Materials

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Methods
2.2. Peptide Synthesis
2.3. Biochemical Characterization
2.3.1. Cell Culture
2.3.2. Cell Adhesion Assays on the Endogenous Ligand FN
2.3.3. Enzymatic Stability
2.4. Preparation of the Biofunctionalized Surfaces 49–Zeolite MLs/50–Zeolite MLs, and of the Negative References BA–Zeolite MLs/49-Plates
2.5. Cell Adhesion on 49–Zeolite MLs/50–Zeolite MLs/BA–Zeolite MLs/49-Plates
2.6. Confocal Microscopy
3. Results
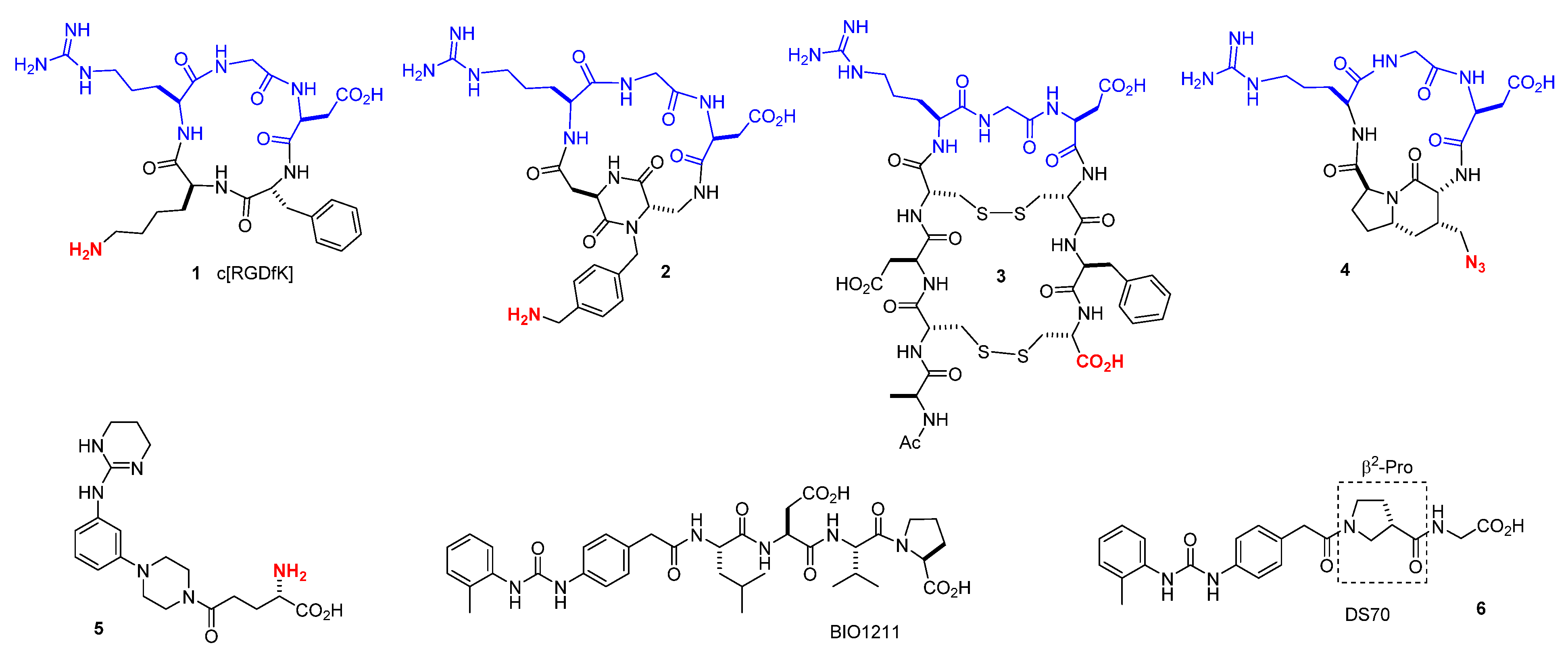
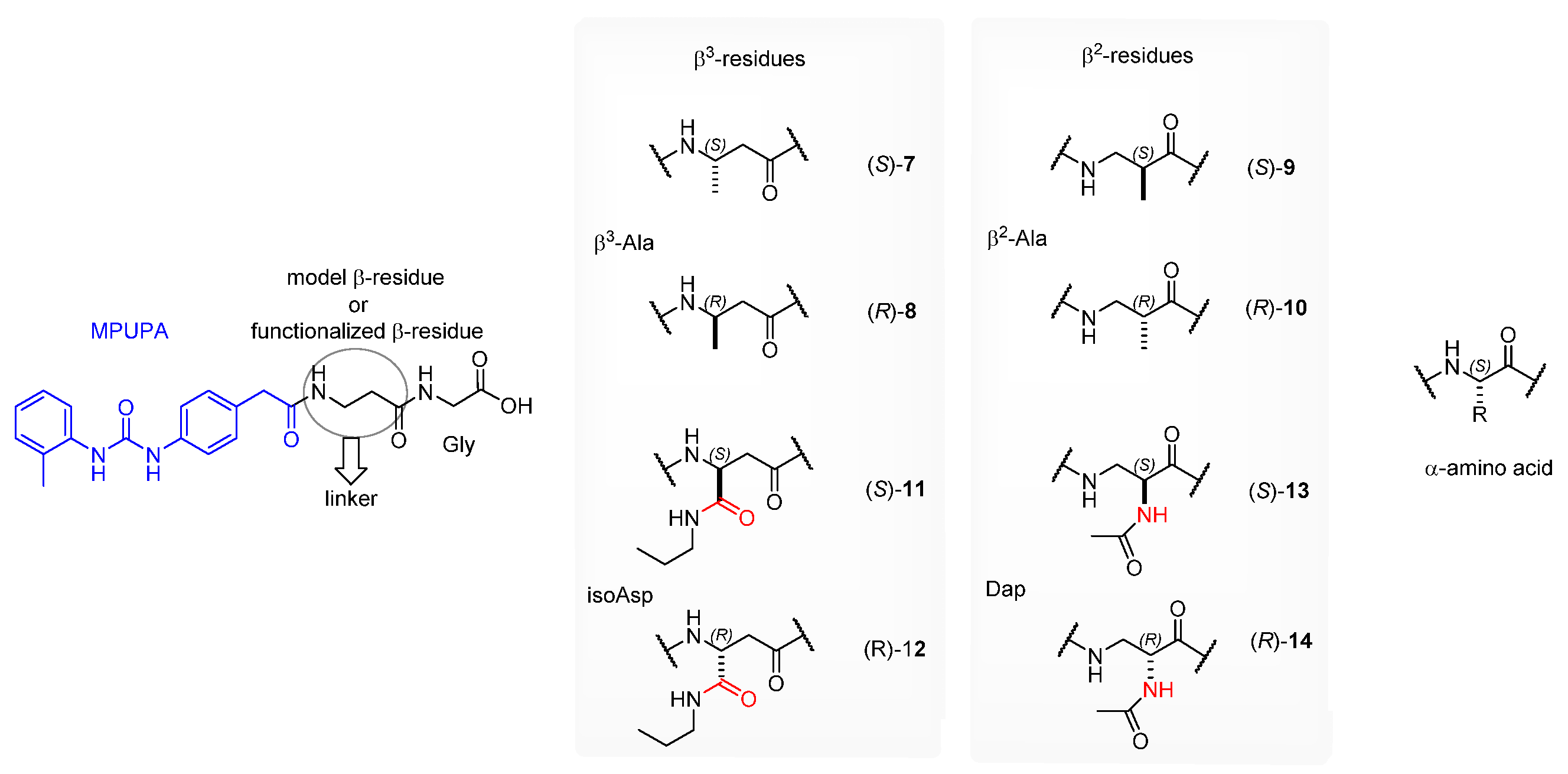
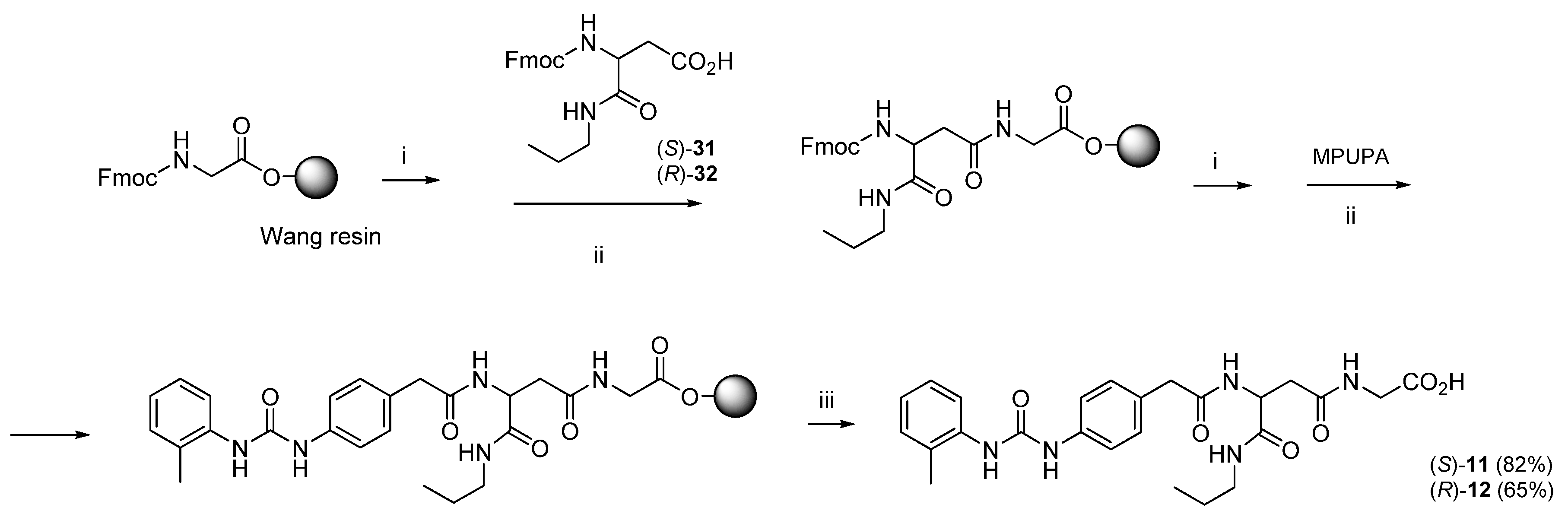
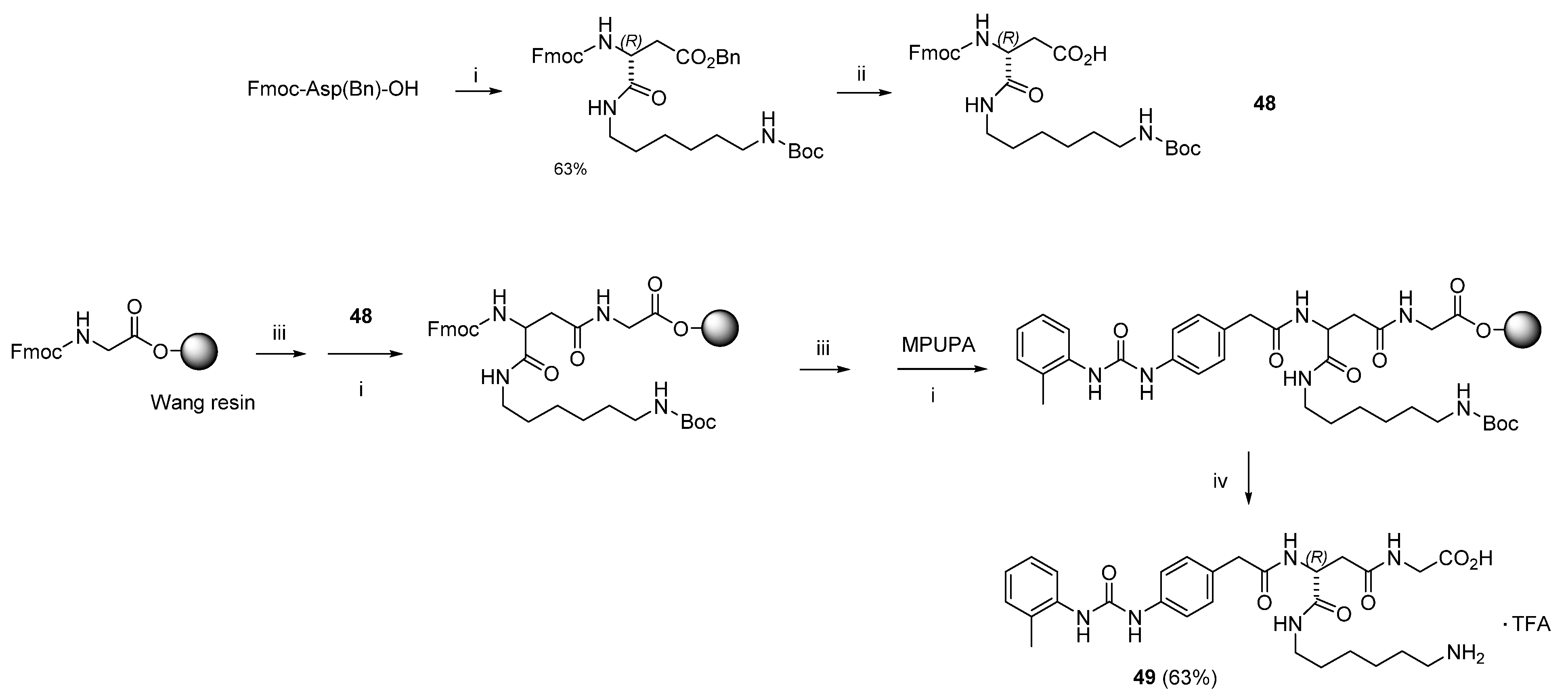
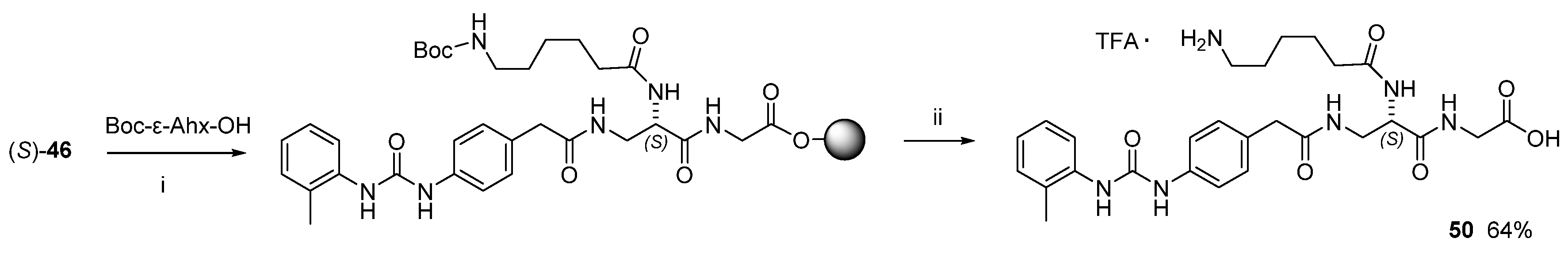
3.1. Peptide Design and Synthesis
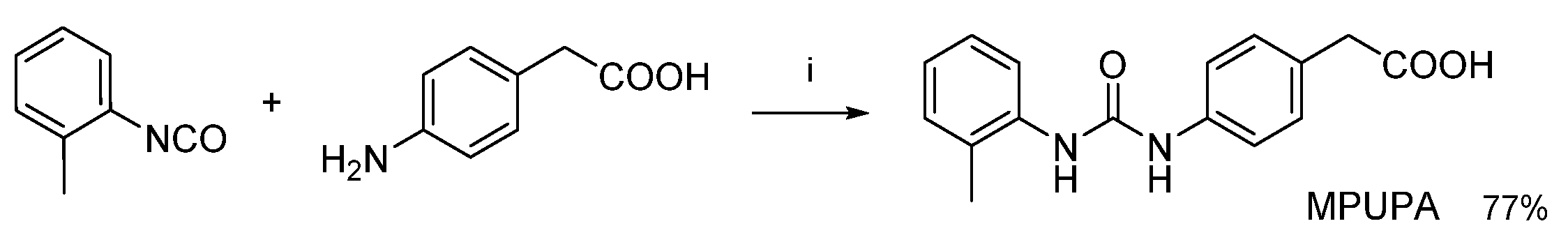
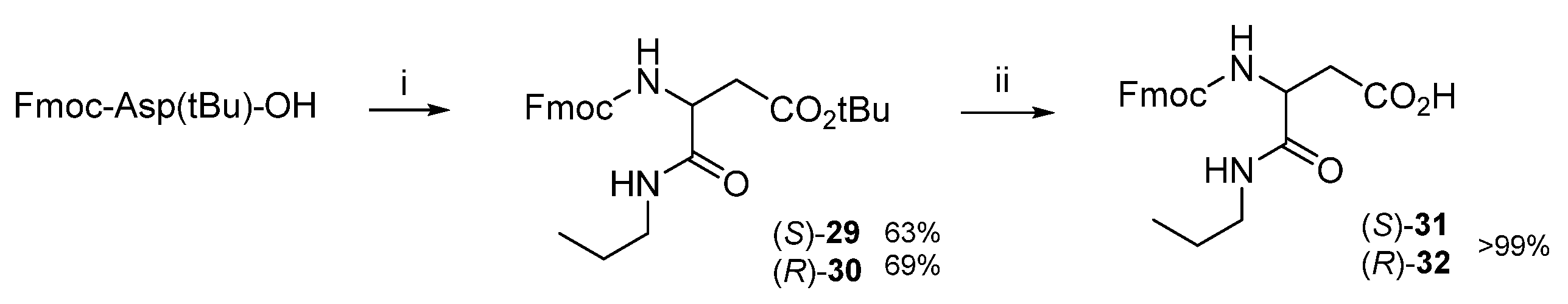
3.1.1. Synthesis of β-Amino Acids
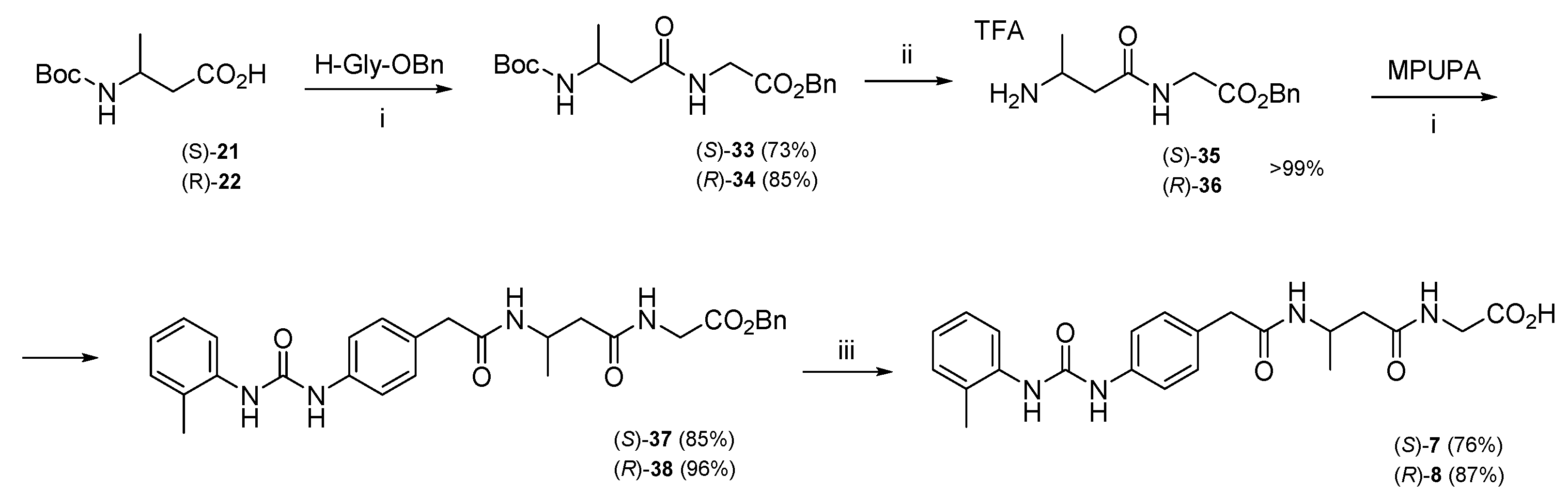
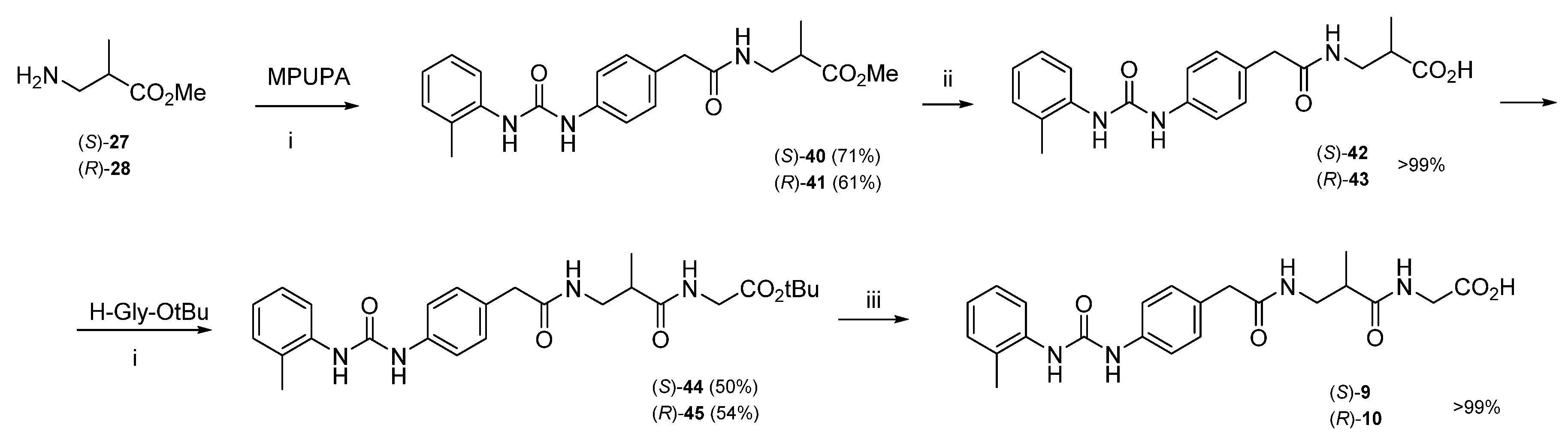
3.1.2. Synthesis of Hybrid Peptides
3.2. Biochemical Cheracterization
3.2.1. Potency and Selectivity of Hybrid Peptides for Human α4β1 Integrin as Measured by Inhibition of Integrin-Mediated Cell Adhesion to Endogenous Ligands
3.2.2. In Vitro Enzymatic Stability of 12, 13
3.3. Design of Linkable α4β1 Integrin Ligands
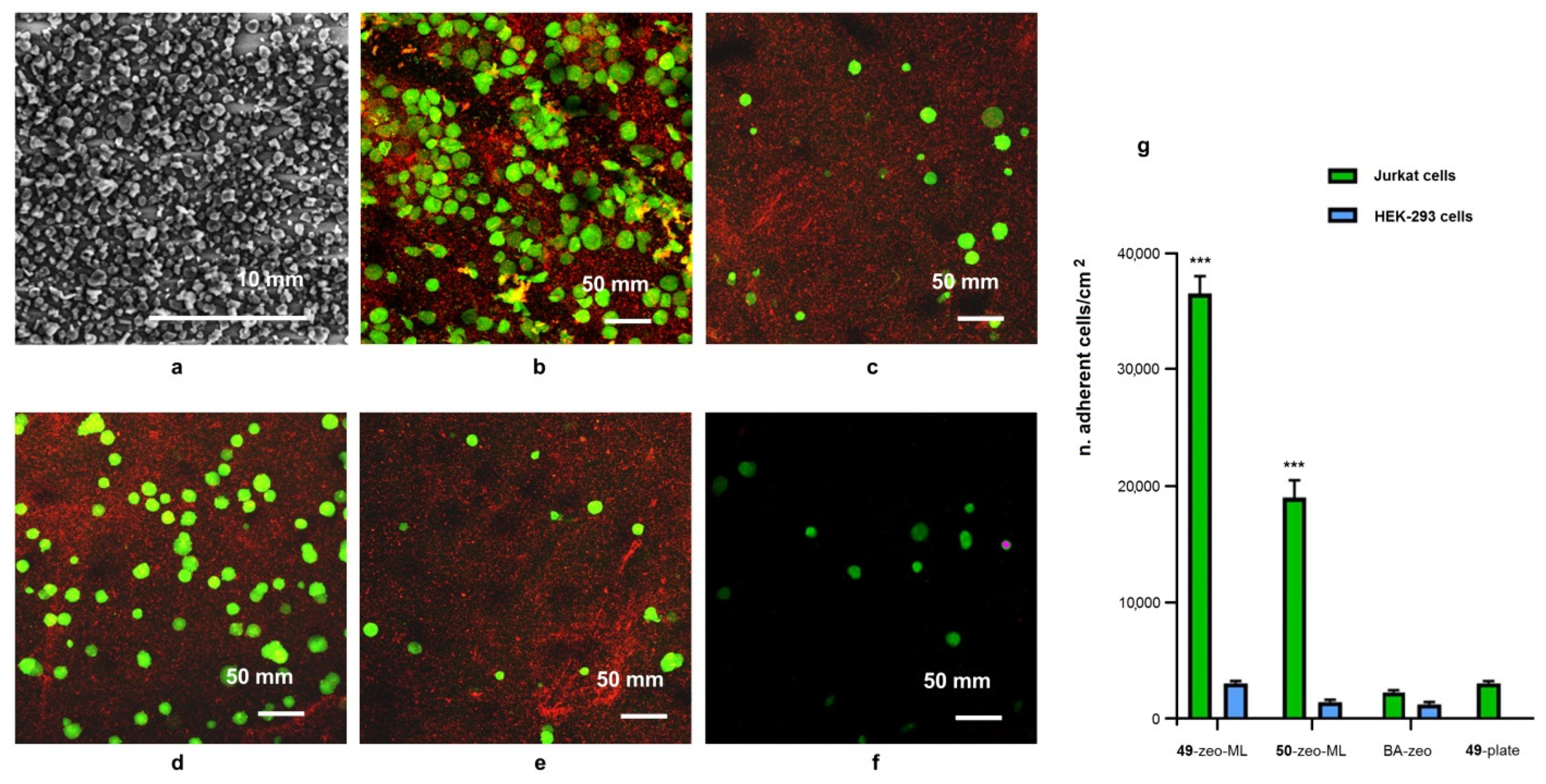
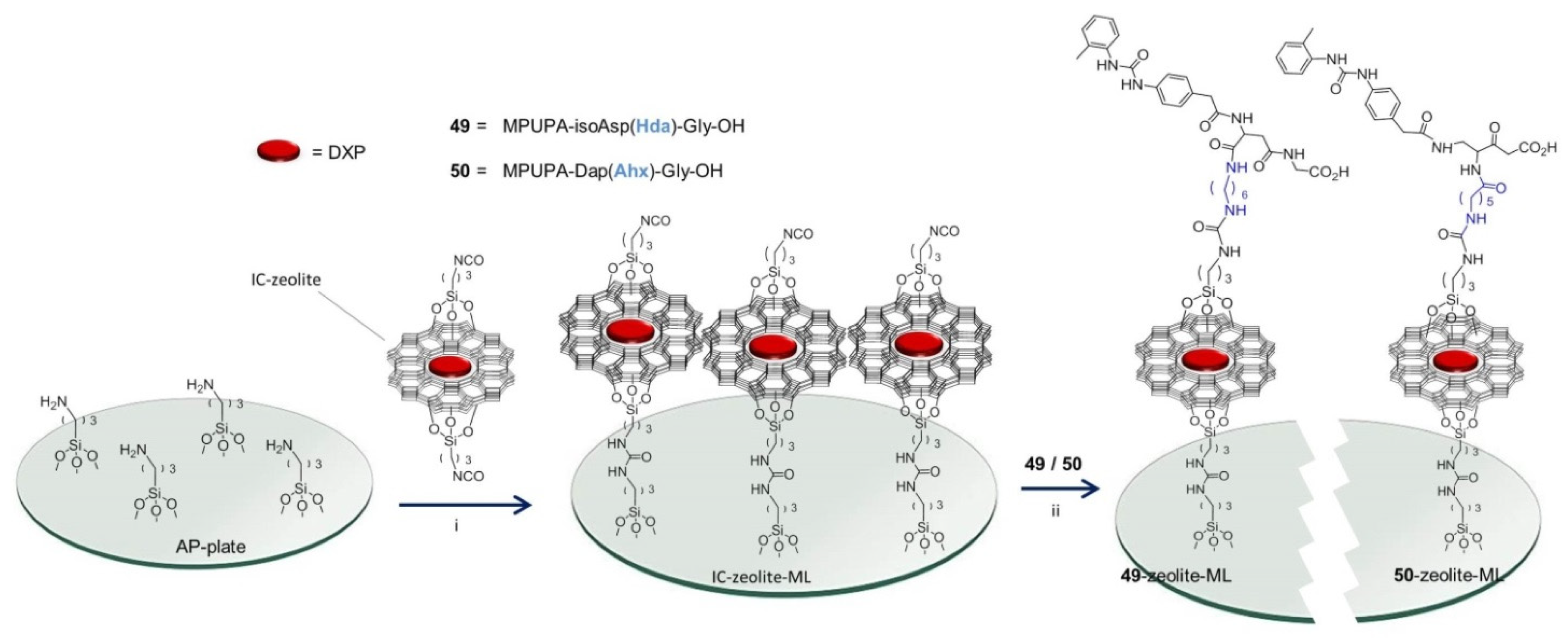
3.4. Preparation of Microstructured Surfaces and Functionalization with Peptides 49, 50
3.5. Cell Adhesion Experiments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [Green Version]
- Ley, K.; Rivera-Nieves, J.; Sandborn, W.J.; Shattil, S. Integrin-based therapeutics: Biological basis, clinical use and new drugs. Nat. Rev. Drug Discov. 2016, 15, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Ruoslahti, E.; Pierschbacher, M.D. Arg-Gly-Asp: A versatile cell recognition signal. Cell 1986, 44, 517–518. [Google Scholar] [CrossRef]
- Gentilucci, L.; Tolomelli, A.; Squassabia, F. Peptides and peptidomimetics in medicine, surgery and biotechnology. Curr. Med. Chem. 2006, 13, 2449–2466. [Google Scholar] [CrossRef] [PubMed]
- Alday-Parejo, B.; Stupp, R.; Rüegg, C. Are integrins still practicable targets for anti-cancer therapy? Cancers 2019, 11, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelli, G.; Baiula, M.; Caligiana, A.; Galletti, P.; Gentilucci, L.; Artali, R.; Spampinato, S.; Giacomini, D. Could dissecting the molecular framework of β-lactam integrin ligands enhance selectivity? J. Med. Chem. 2019, 62, 10156–10166. [Google Scholar] [CrossRef]
- Anselmi, M.; Borbély, A.; Figueras, E.; Michalek, C.; Kemker, I.; Gentilucci, L.; Sewald, N. Linker hydrophilicity modulates the anticancer activity of RGD–cryptophycin conjugates. Chem. Eur. J. 2021, 27, 1015–1022. [Google Scholar] [CrossRef]
- De Marco, R.; Rampazzo, E.; Zhao, J.; Prodi, L.; Paolillo, M.; Picchetti, P.; Gallo, F.; Calonghi, N.; Gentilucci, L. Integrin-targeting dye-doped PEG-shell/silica-core nanoparticles mimicking the proapoptotic Smac/DIABLO protein. Nanomaterials 2020, 10, 1211. [Google Scholar] [CrossRef]
- Hersel, U.; Dahmen, C.; Kessler, H. RGD modified polymers: Biomaterials for stimulated cell adhesion and beyond. Biomaterials 2003, 24, 4385–4441. [Google Scholar] [CrossRef]
- Zhao, J.; Santino, F.; Giacomini, D.; Gentilucci, L. Integrin-targeting peptides for the design of functional cell-responsive biomaterials. Biomedicines 2020, 8, 307. [Google Scholar] [CrossRef]
- Da Ressurreição, A.S.; Vidu, A.; Civera, M.; Belvisi, L.; Potenza, D.; Manzoni, L.; Ongeri, S.; Gennari, C.; Piarulli, U. Cyclic RGD-peptidomimetics containing bifunctional diketopiperazine scaffolds as new potent integrin ligands. Chem. Eur. J. 2009, 15, 12184–12188. [Google Scholar] [CrossRef] [PubMed]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzoni, L.; Belvisi, L.; Arosio, D.; Civera, M.; Pilkington-Miksa, M.; Potenza, D.; Caprini, A.; Araldi, E.M.V.; Monferini, E.; Mancino, M.; et al. Cyclic RGD-containing functionalized azabicycloalkane peptides as potent integrin antagonists for tumor targeting. ChemMedChem 2009, 4, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Iwama, S.; Kitano, T.; Fukuya, F.; Honda, Y.; Sato, Y.; Notake, M.; Morie, T. Discovery of a potent and selective alpha v beta 3 integrin antagonist with strong inhibitory activity against neointima formation in rat balloon injury model. Bioorg. Med. Chem. Lett. 2004, 14, 2567–2570. [Google Scholar] [CrossRef]
- Haubner, R.; Gratias, R.; Diefenbach, B.; Goodman, S.L.; Jonczyk, A.; Kessler, H. Structural and functional aspects of RGD-containing cyclic pentapeptides as highly potent and selective integrin αvβ3 antagonists. J. Am. Chem. Soc. 1996, 118, 7461–7472. [Google Scholar] [CrossRef]
- Dal Corso, A.; Pignataro, L.; Belvisi, L.; Gennari, C. Alpha(v)beta(3) Integrin-targeted peptide/peptidomimetic-drug conjugates: In-depth analysis of the linker technology. Curr. Top. Med. Chem. 2016, 16, 1–16. [Google Scholar] [CrossRef]
- Greco, A.; Maggini, L.; De Cola, L.; De Marco, R.; Gentilucci, L. Diagnostic implementation of fast and selective integrin-mediated adhesion of cancer cells on functionalized zeolite L monolayers. Bioconjug. Chem. 2015, 26, 1873–1878. [Google Scholar] [CrossRef]
- Danhier, F.; Le Breton, A.; Préat, V. RGD-based strategies to target αvβ3 integrin in cancer therapy and diagnosis. Mol. Pharm. 2012, 9, 2961–2973. [Google Scholar] [CrossRef]
- Baiula, M.; Spampinato, S.; Gentilucci, L.; Tolomelli, A. Novel ligands targeting α4β1 integrin: Therapeutic applications and perspectives. Front. Chem. 2019, 7, 489. [Google Scholar] [CrossRef]
- Clerico, M.; Artusi, C.; Liberto, A.; Rolla, S.; Bardina, V.; Barbero, P.; Mercanti, S.F.; Durelli, L. Natalizumab in multiple sclerosis: Long-term management. Int. J. Mol. Sci. 2017, 18, 940. [Google Scholar] [CrossRef]
- Vanderslice, P.; Biediger, R.J.; Woodside, D.G.; Berens, K.L.; Holland, G.W.; Dixon, R.A.F. Development of cell adhesion molecule antagonists as therapeutics for asthma and COPD. Pulm. Pharmacol. Ther. 2004, 17, 1–103. [Google Scholar] [CrossRef]
- Dattoli, S.D.; Baiula, M.; De Marco, R.; Bedini, A.; Anselmi, M.; Gentilucci, L.; Spampinato, S. DS-70, a novel and potent α4 integrin antagonist, is an effective treatment for experimental allergic conjunctivitis in guinea pigs. Br. J. Pharmacol. 2018, 175, 3891–3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baiula, M.; Caligiana, A.; Bedini, A.; Zhao, J.; Santino, F.; Cirillo, M.; Gentilucci, L.; Giacomini, D.; Spampinato, S. Leukocyte integrin antagonists as a novel option to treat dry age-related macular degeneration. Front. Pharmacol. 2021, 11, 617836. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.R.; Hyduk, S.J.; Cybulsky, M.I. Detecting rapid and transient upregulation of leukocyte integrin affinity induced by chemokines and chemoattractants. J. Immunol. Methods 2003, 273, 43–52. [Google Scholar] [CrossRef]
- Yednock, T.A.; Cannon, C.; Vandevert, C.; Goldbach, E.G.; Shaw, G.; Ellis, D.K.; Liaw, C.; Fritz, L.C.; Tanner, L.I. Alpha 4 beta 1 integrin-dependent cell adhesion is regulated by a low affinity receptor pool that is conformationally responsive to ligand. J. Biol. Chem. 1995, 270, 28740–28750. [Google Scholar] [CrossRef]
- Johansson, M.W.; Kelly, E.A.; Busse, W.W.; Jarjour, N.N.; Mosher, D.F. Up-regulation and activation of eosinophil integrins in blood and airway after segmental lung antigen challenge. J. Immunol. 2008, 180, 7622–7635. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.R.; Hyduk, S.J.; Cybulsky, M.I. Chemoattractants induce a rapid and transient upregulation of monocyte alpha4 integrin affinity for vascular cell adhesion molecule 1 which mediates arrest: An early step in the process of emigration. J. Exp. Med. 2001, 193, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, A.; Rosa, M.D.; Bixler, S.; Lobb, R.; Burkly, L.C. Vascular cell adhesion molecule (VCAM)-Ig fusion protein defines distinct affinity states of the very late antigen-4 (VLA-4) receptor. Cell Adhes. Commun. 1995, 3, 131–142. [Google Scholar] [CrossRef]
- Chigaev, A.; Blenc, A.M.; Braaten, J.V.; Kumaraswamy, N.; Kepley, C.L.; Andrews, R.P.; Oliver, J.M.; Edwards, B.S.; Prossnitz, E.R.; Larson, R.S.; et al. Alpha4beta1 integrin affinity changes govern cell adhesion. J. Biol. Chem. 2001, 276, 48670–48682. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.W.; Busse, W.W.; Mosher, D.F. Beta 1 Integrin Activation as a Marker for Asthma. U.S. Patent 20080274482, 6 November 2018. [Google Scholar]
- De Marco, R.; Greco, A.; Calonghi, N.; Dattoli, D.S.; Baiula, M.; Spampinato, S.; Picchetti, P.; De Cola, L.; Anselmi, M.; Cipriani, F.; et al. Selective detection of α4β1 integrin (VLA-4)-expressing cells using peptide-functionalized nanostructured materials mimicking endothelial surfaces adjacent to inflammatory sites. Pept. Sci. 2018, 110, e23081. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.c.; Ateeq, H.S.; Hsiung, S.H.; Chong, L.T.; Zimmerman, C.N.; Castro, A.; Lee, W.C.; Hammond, C.E.; Kalkunte, S.; Chen, L.L.; et al. Selective, tight-binding inhibitors of integrin α4β1 that inhibit allergic airway responses. J. Med. Chem. 1999, 42, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Karanam, B.V.; Jayra, A.; Rabe, M.; Wang, Z.; Keohane, C.; Strauss, J.; Vincent, S. Effect of enalapril on the in vitro and in vivo peptidyl cleavage of a potent VLA-4 antagonist. Xenobiotica 2007, 37, 487–502. [Google Scholar] [CrossRef]
- Fisher, A.L.; DePuy, E.; Jayaraj, A.; Raab, C.; Braun, M.; Ellis-Hutchings, M.; Zhang, J.; Rogers, J.D.; Musson, D.G. LC/MS/MS plasma assay for the peptidomimetic VLA4 antagonist I and its major active metabolite II: For treatment of asthma by inhalation. J. Pharm. Biomed. Anal. 2002, 27, 57–71. [Google Scholar] [CrossRef]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: Incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef]
- Seebach, D.; Gardiner, J. β-Peptidic peptidomimetics. Acc. Chem. Res. 2008, 41, 1366–1375. [Google Scholar] [CrossRef]
- Cabrele, C.; Martinek, T.A.; Reiser, O.; Berlicki, Ł. Peptides containing β-amino acid patterns: Challenges and successes in medicinal chemistry. J. Med. Chem. 2014, 57, 9718–9739. [Google Scholar] [CrossRef] [PubMed]
- Dattoli, S.D.; De Marco, R.; Baiula, M.; Spampinato, S.; Greco, A.; Tolomelli, A.; Gentilucci, L. Synthesis and assay of retro-α4β1 integrin-targeting motifs. Eur. J. Med. Chem. 2014, 73, 225–232. [Google Scholar] [CrossRef]
- Tolomelli, A.; Baiula, M.; Viola, A.; Ferrazzano, L.; Gentilucci, L.; Dattoli, S.D.; Spampinato, S.; Juaristi, E.; Escudero, M. Dehydro-β-proline containing α4β1 integrin antagonists: Stereochemical recognition in ligand-receptor interplay. ACS Med. Chem. Lett. 2015, 6, 701–706. [Google Scholar] [CrossRef] [Green Version]
- De Marco, R.; Mazzotti, G.; Dattoli, S.D.; Baiula, M.; Spampinato, S.; Greco, A.; Gentilucci, L. 5-Aminomethyloxazolidine-2,4-dione hybrid α/β-dipeptide scaffolds as inductors of constrained conformations: Applications to the synthesis of integrin antagonists. Biopolymers 2015, 104, 636–649. [Google Scholar] [CrossRef]
- De Marco, R.; Tolomelli, A.; Juaristi, E.; Gentilucci, L. Integrin ligands with α/β-hybrid peptide structure: Design, bioactivity, and conformational aspects. Med. Res. Rev. 2016, 36, 389–424. [Google Scholar] [CrossRef] [PubMed]
- Podlech, J.; Seebach, D. The Arndt–Eistert reaction in peptide chemistry: A facile access to homopeptides. Angew. Chem. Int. Ed. 1995, 34, 471–472. [Google Scholar] [CrossRef]
- Caputo, R.; Longobardo, L. Enantiopure β3-amino acids-2,2-d2 via homologation of proteinogenic α-amino acids. Amino Acids 2007, 32, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Lee, W.S.; Yun, H.; Hyun, Y.J.; Seo, C.D.; Lee, C.W.; Lim, H.S. Oligomers of N-substituted β2-homoalanines: Peptoids with backbone chirality. Org. Lett. 2016, 18, 3678–3681. [Google Scholar] [CrossRef] [PubMed]
- Blettner, C.; Bradley, M. Asparagine as a masked dehydroalanine residue in solid phase peptide synthesis. Tetrahedron Lett. 1994, 35, 467–470. [Google Scholar] [CrossRef]
- Alon, R.; Kassner, P.D.; Carr, M.W.; Finger, E.B.; Hemler, M.E.; Springer, T. A The integrin VLA-4 supports tethering and rolling in flow on VCAM-1. J. Cell. Biol. 1995, 128, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.; Mazzotti, G.; Greco, A.; Gentilucci, L. Heterocyclic scaffolds in the design of peptidomimetic integrin ligands: Synthetic strategies, structural aspects, and biological activity. Curr. Top. Med. Chem. 2016, 16, 343–359. [Google Scholar] [CrossRef]
- Devaux, A.; Calzaferri, G.; Miletto, I.; Cao, P.; Belser, P.; Brühwiler, D.; Khorev, O.; Häner, R.; Kunzmann, A. Self-absorption and luminescence quantum yields of dye-zeolite L composites. J. Phys. Chem. C 2013, 117, 23034–23047. [Google Scholar] [CrossRef] [Green Version]
- Kehr, N.S.; Riehemann, K.; El-Gindi, J.; Schäfer, A.; Fuchs, H.; Galla, H.-J.; De Cola, L. Cell adhesion and cellular patterning on a self-assembled monolayer of zeolite L crystals. Adv. Funct. Mater. 2010, 20, 2248–2254. [Google Scholar] [CrossRef]
- Busby, M.; Devaux, A.; Blum, C.; Subramaniam, V.; Calzaferri, G.; De Cola, L. Interactions of Perylene Bisimide in the One-Dimensional Channels of Zeolite, L.J. Phys. Chem. C 2011, 115, 5974–5988. [Google Scholar] [CrossRef]
- Taherian, A.; Li, X.; Liu, Y.; Haas, T.A. Differences in integrin expression and signaling within human breast cancer cells. BMC Cancer 2011, 11, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinashi, T. Intracellular signalling controlling integrin activation in lymphocytes. Nat. Rev. Immunol. 2005, 5, 546–559. [Google Scholar] [CrossRef] [PubMed]
- De Fougerolles, A.R. Integrins in Immune and Inflammatory Diseases. In I Domains in Integrins; Gullberg, D., Ed.; Plenum Publisher: Georgetown, TX, USA, 2003. [Google Scholar]
- Zamuner, A.; Brun, P.; Scorzeto, M.; Sica, G.; Castagliuolo, I.; Dettin, M. Smart biomaterials: Surfaces functionalized with proteolytically stable osteoblast-adhesive peptides. Bioact. Mater. 2017, 2, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Spicer, C.D.; Pashuck, E.T.; Stevens, M.M. Achieving controlled biomolecule-biomaterial conjugation. Chem. Rev. 2018, 118, 7702–7743. [Google Scholar] [CrossRef]
- Zabala Ruiz, A.; Li, H.; Calzaferri, G. Organizing supramolecular functional dye-zeolite crystals. Angew. Chem. Int. Ed. 2006, 45, 5282–5287. [Google Scholar] [CrossRef]
- Cavalcanti-Adam, E.A.; Volberg, T.; Micoulet, A.; Kessler, H.; Geiger, B.; Spatz, J.P. Cell spreading and focal adhesion dynamics are regulated by spacing of integrin ligands. Biophys. J. 2007, 92, 2964–2974. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
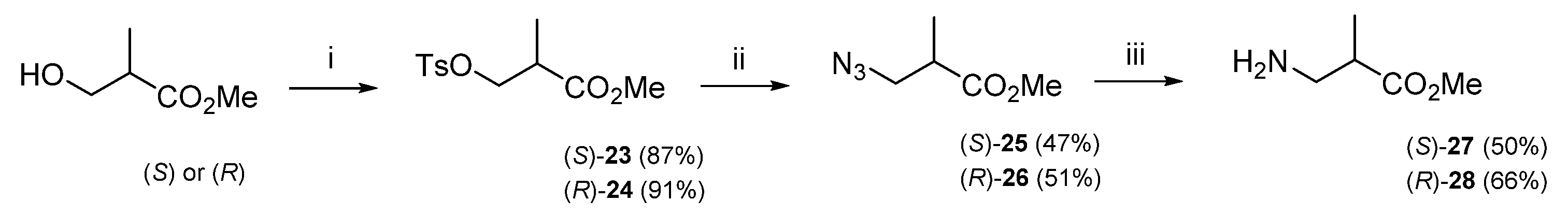{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd β-Residue Type | Structure | IC50 (nM) b |
|---|---|---|
| BIO1211- |  | 5.5 ± 4.0 c |
| DS70 6 β2 |  | 4.3 ± 1.7 c |
| MA97 7 β3 |  | 4060 ± 780 |
| MA99 8 β3 |  | >5000 |
| MA192 9 β2 |  | >5000 |
| MA199 10 β2 |  | >5000 |
| MA62 11 β3 |  | >5000 |
| MA158 12 β3 |  | 9.8 ± 2.1 |
| MA28 13 β2 |  | 236 ± 47 |
| MA29 14 β2 |  | >5000 |
| Material | O(1s) | N(1s) | C(1s) | K(2p) | Si(2p) | Al(2p) | O(1s) |
|---|---|---|---|---|---|---|---|
| Zeolite L | 58.0 ± 1.1 | 0.8 ± 0.2 | 5.8 ± 0.9 | 6.9 ± 1.2 | 21.5 ± 0.2 | 7.0 ± 0.7 | 58.0 ± 1.1 |
| DXP–zeolite | 44.9 ± 3.8 | 2.1 ± 0.3 | 21.1 ± 4.0 | 5.5 ± 0.8 | 18.5 ± 2.0 | 7.9 ± 3.0 | 44.9 ± 3.8 |
| IC–zeolite | 29.5 ± 4.0 | 5.0 ± 1.0 | 32.0 ± 6.0 | 4.0 ± 1.0 | 17.9 ± 3.0 | 11.6 ± 2.0 | 29.5 ± 4.0 |
| IC–zeolite ML | 35.9 ± 1.2 | 5.3 ± 2.0 | 31.0 ± 3.2 | 1.9 ± 1.0 | 17.9 ± 3.1 | 8.0 ± 1.5 | 35.9 ± 1.2 |
| 49–zeolite ML | 31.2 ± 2.0 | 8.3 ± 1.6 | 44.4 ± 4.5 | 0.8 ± 0.4 | 13.5 ± 2.0 | 1.8 ± 0.5 | 31.2 ± 2.0 |
| 50–zeolite ML | 32.2 ± 2.3 | 8.8 ± 2.1 | 43.0 ± 2.0 | 0.5 ± 0.2 | 13.9 ± 1.9 | 1.6 ± 0.3 | 32.2 ± 2.3 |
| BA–zeolite ML | 37.8 ± 3.0 | 4.9 ± 1.1 | 32.2 ± 3.8 | 1.2 ± 0.3 | 17.9 ± 2.5 | 6.0 ± 0.4 | 37.8 ± 3.0 |
| IC-plate 1 | 57.4 ± 3.0 | 0.9 ± 0.4 | 5.0 ± 1.9 | 4.9 ± 1.6 | 19.9 ± 2.0 | 1.9 ± 0.9 | 57.4 ± 3.0 |
| 49-plate 1 | 53.1 ± 4.0 | 2.0 ± 0.6 | 10.1 ± 3.0 | 4.2 ± 1.0 | 18.1 ± 2.8 | 1.6 ± 0.6 | 53.1 ± 4.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anselmi, M.; Baiula, M.; Santino, F.; Zhao, J.; Spampinato, S.; Calonghi, N.; Gentilucci, L. Design of α/β-Hybrid Peptide Ligands of α4β1 Integrin Equipped with a Linkable Side Chain for Chemoselective Biofunctionalization of Microstructured Materials. Biomedicines 2021, 9, 1737. https://doi.org/10.3390/biomedicines9111737
Anselmi M, Baiula M, Santino F, Zhao J, Spampinato S, Calonghi N, Gentilucci L. Design of α/β-Hybrid Peptide Ligands of α4β1 Integrin Equipped with a Linkable Side Chain for Chemoselective Biofunctionalization of Microstructured Materials. Biomedicines. 2021; 9(11):1737. https://doi.org/10.3390/biomedicines9111737
Chicago/Turabian StyleAnselmi, Michele, Monica Baiula, Federica Santino, Junwei Zhao, Santi Spampinato, Natalia Calonghi, and Luca Gentilucci. 2021. "Design of α/β-Hybrid Peptide Ligands of α4β1 Integrin Equipped with a Linkable Side Chain for Chemoselective Biofunctionalization of Microstructured Materials" Biomedicines 9, no. 11: 1737. https://doi.org/10.3390/biomedicines9111737
APA StyleAnselmi, M., Baiula, M., Santino, F., Zhao, J., Spampinato, S., Calonghi, N., & Gentilucci, L. (2021). Design of α/β-Hybrid Peptide Ligands of α4β1 Integrin Equipped with a Linkable Side Chain for Chemoselective Biofunctionalization of Microstructured Materials. Biomedicines, 9(11), 1737. https://doi.org/10.3390/biomedicines9111737