
Targeting Inflammasome Activation in COVID-19: Delivery of RNA Interference-Based Therapeutic Molecules

 , and
, and
Abstract
:1. Introduction
2. Immune Dysregulation in COVID-19
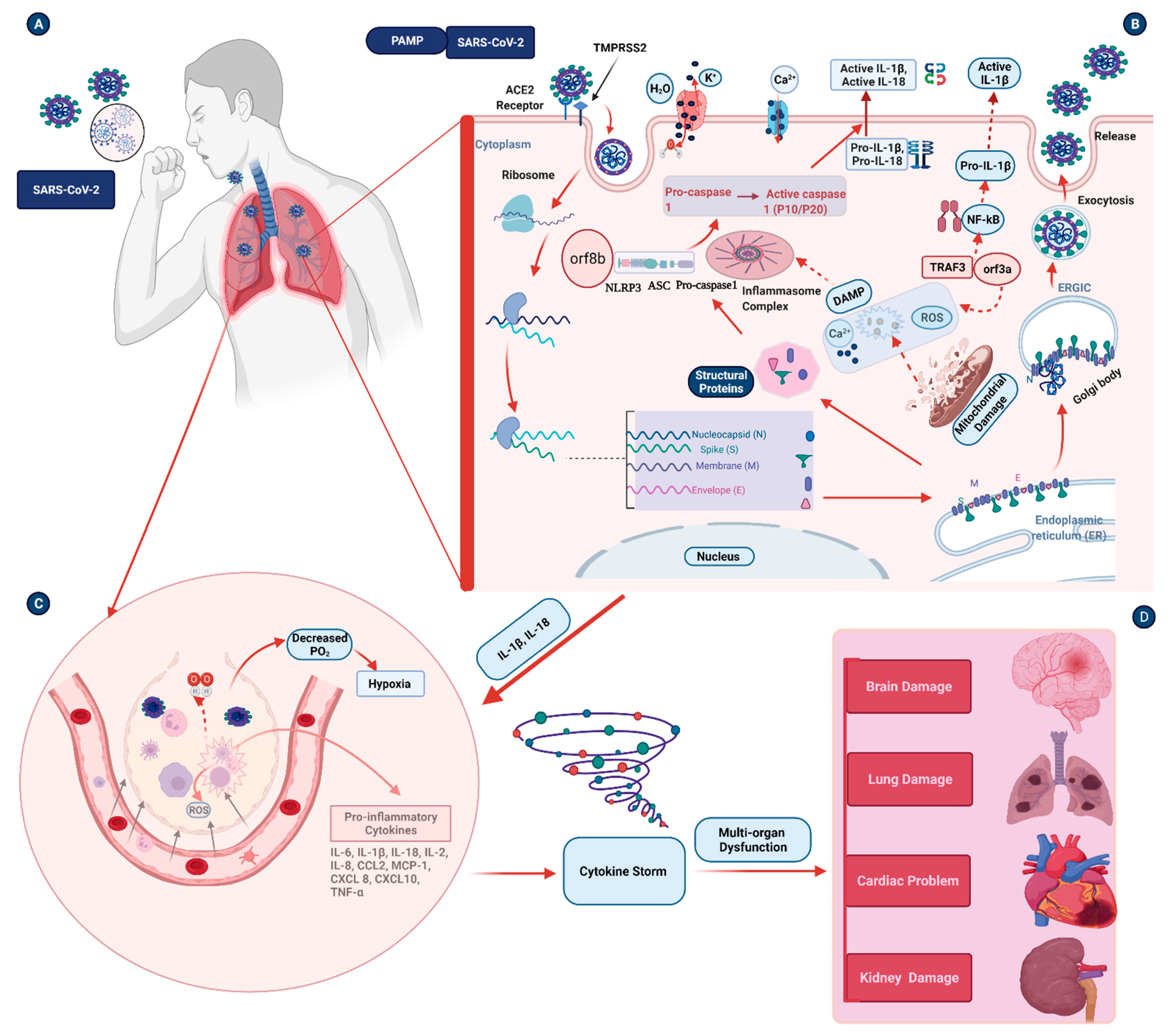
3. Inflammasome Activation in Coronavirus Infections
4. Pathogenesis Triggered by the Inflammasome during COVID-19 Infection
4.1. Inflammation and Pulmonary Damage
4.2. Cardiovascular Damage
4.3. Neurological Damage
5. Potential Therapeutic Agents and Vaccine Strategies for COVID-19
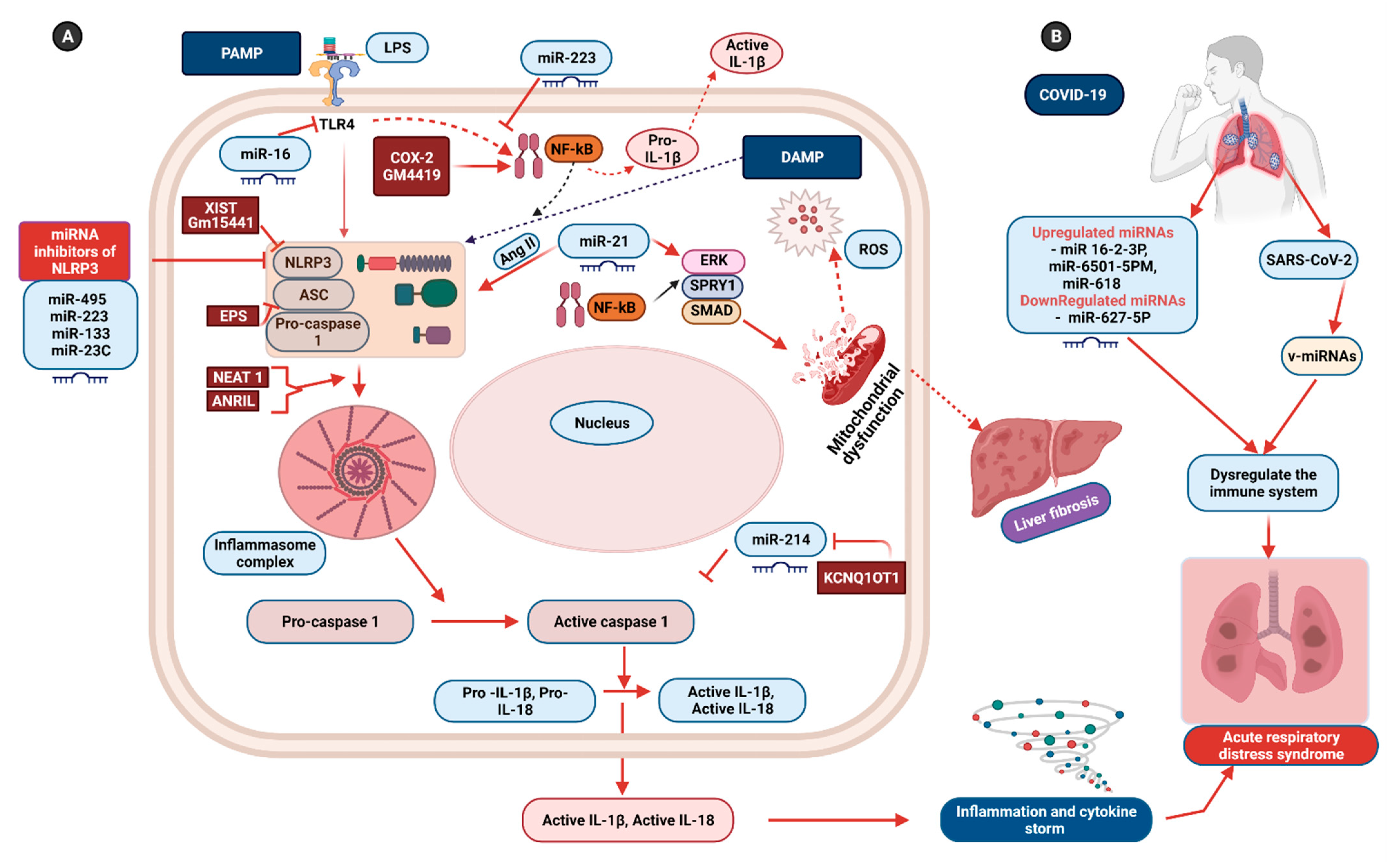
6. Non-Coding RNAs: Potential Therapeutics for Inflammasome-Induced COVID-19 Pathogenesis
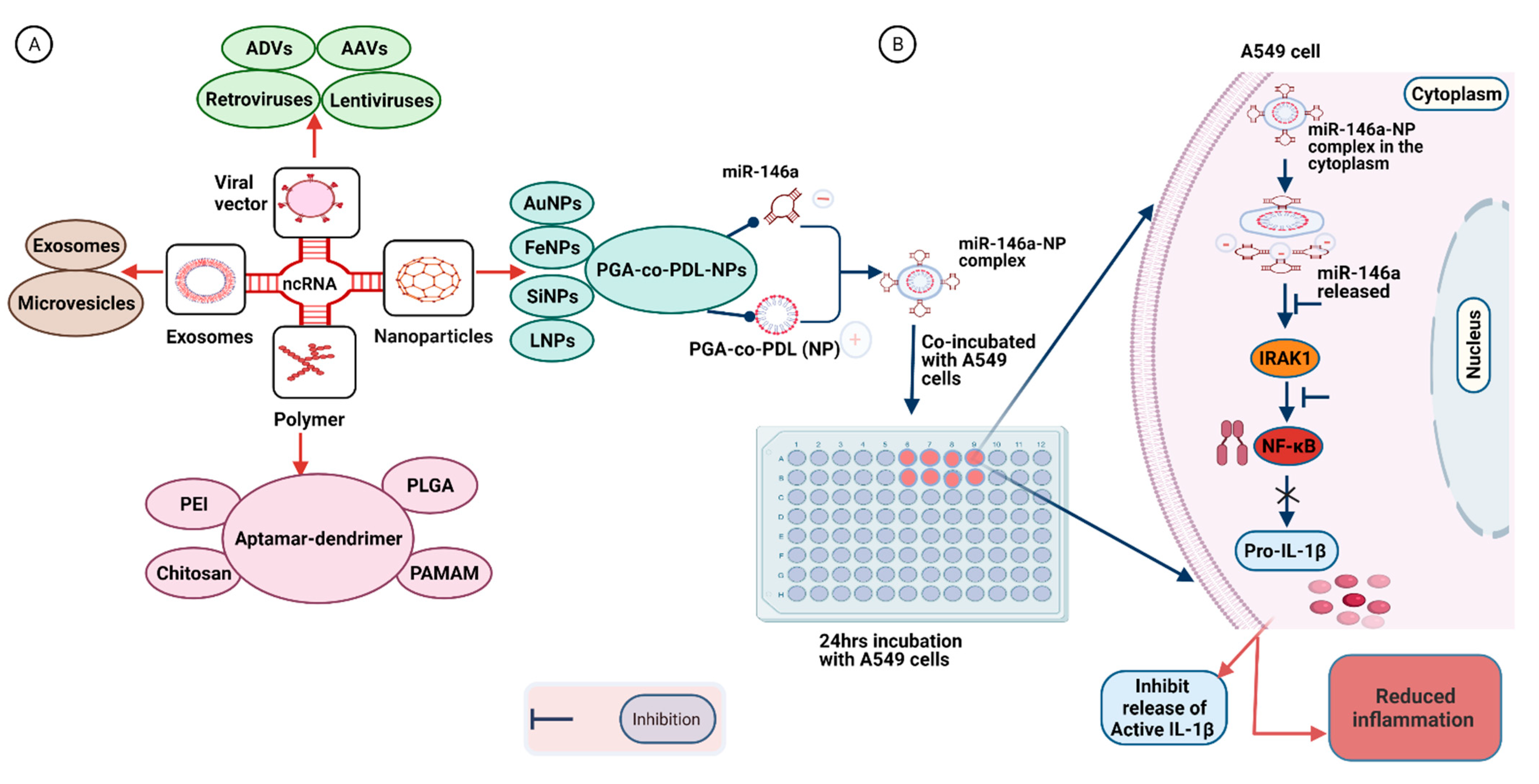
7. Delivery Strategies for Synthetic RNA-Based Therapeutic Molecules
7.1. Virus-Based Vectors
7.2. Polymer-Based Vehicles
7.3. Nanoparticle-Based Vectors
7.4. Exosome-Based Vectors
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johns Hopkins Centers for Civic Impact. Coronavirus Resource Center. 2021. Available online: https://coronavirus.jhu.edu/map.html (accessed on 5 November 2021).
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.S.; de Sá, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasome activation in COVID-19 patients. medRxiv 2020. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.C.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. The inflammasome in times of COVID-19. Front. Immunol. 2020, 11, 583373. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus orf3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef]
- Shi, C.S.; Nabar, N.R.; Huang, N.N.; Kehrl, J.H. SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.Q.; Liu, F.; Zhang, J.Q.; Shao, Q.; Tao, W.Q.; Xiao, R.; Dai, W.; Qian, K.J. Functional crosstalk between long non-coding RNAs and the NLRP3 inflammasome in the regulation of diseases. Mol. Immunol. 2021, 131, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.P.; Hua, K.F. The long non-coding RNAs: Paramount regulators of the NLRP3 inflammasome. Front. Immunol. 2020, 11, 569524. [Google Scholar] [CrossRef]
- Bernier, A.; Sagan, S.M. The diverse roles of microRNAs at the host-virus interface. Viruses 2018, 10, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamani, P.; Oskuee, R.K.; Atkin, S.L.; Navashenaq, J.G.; Sahebkar, A. MicroRNAs as important regulators of the NLRP3 inflammasome. Prog. Biophys. Mol. Biol. 2020, 150, 50–61. [Google Scholar] [CrossRef]
- Zhang, P.; Cao, L.; Zhou, R.; Yang, X.; Wu, M. The lncRNA NEAT1 promotes activation of inflammasomes in macrophages. Nat. Commun. 2019, 10, 1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Qiu, H.; Pei, X.; Fan, Y.G.; Tian, H.Y.; Geng, J. Low-dose sinapic acid abates the pyroptosis of macrophages by downregulation of lncRNA-MALAT1 in rats with diabetic atherosclerosis. J. Cardiovasc. Pharmacol. 2018, 71, 104–112. [Google Scholar] [CrossRef]
- Xue, Z.; Zhang, Z.; Liu, H.; Li, W.; Guo, X.; Zhang, Z.; Liu, Y.; Jia, L.; Li, Y.; Ren, Y.; et al. lincRNA-Cox2 regulates NLRP3 inflammasome and autophagy mediated neuroinflammation. Cell Death Differ. 2019, 26, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Atianand, M.K.; Hu, W.; Satpathy, A.T.; Shen, Y.; Ricci, E.P.; Alvarez-Dominguez, J.R.; Bhatta, A.; Schattgen, S.A.; McGowan, J.D.; Blin, J.; et al. A long noncoding RNA lincRNA-EPS acts as a transcriptional brake to restrain inflammation. Cell 2016, 165, 1672–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocker, C.N.; Kim, D.; Melia, T.; Karri, K.; Velenosi, T.J.; Takahashi, S.; Aibara, D.; Bonzo, J.A.; Levi, M.; Waxman, D.J.; et al. Long non-coding RNA Gm15441 attenuates hepatic inflammasome activation in response to PPARA agonism and fasting. Nat. Commun. 2020, 11, 5847. [Google Scholar] [CrossRef]
- Ma, M.; Pei, Y.; Wang, X.; Feng, J.; Zhang, Y.; Gao, M.Q. LncRNA XIST mediates bovine mammary epithelial cell inflammatory response via NF-κB/NLRP3 inflammasome pathway. Cell Prolif. 2019, 52, e12525. [Google Scholar] [CrossRef] [Green Version]
- Ling, H. Non-coding RNAs: Therapeutic strategies and delivery systems. Adv. Exp. Med. Biol. 2016, 937, 229–237. [Google Scholar] [CrossRef]
- Burnett, J.C.; Rossi, J.J. RNA-based therapeutics: Current progress and future prospects. Chem Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.E. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: From concept to clinic. Mol. Pharm. 2009, 6, 659–668. [Google Scholar] [CrossRef]
- Alabi, C.; Vegas, A.; Anderson, D. Attacking the genome: Emerging siRNA nanocarriers from concept to clinic. Curr. Opin. Pharmacol. 2012, 12, 427–433. [Google Scholar] [CrossRef]
- Smith, D.; Schuller, V.; Engst, C.; Radler, J.; Liedl, T. Nucleic acid nanostructures for biomedical applications. Nanomedicine 2013, 8, 105–121. [Google Scholar] [CrossRef]
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [Google Scholar] [CrossRef]
- Rezakhani, L.; Kelishadrokhi, A.F.; Soleimanizadeh, A.; Rahmati, S. Mesenchymal stem cell (MSC)-derived exosomes as a cell-free therapy for patients infected with COVID-19: Real opportunities and range of promises. Chem Phys. Lipids 2021, 234, 105009. [Google Scholar] [CrossRef] [PubMed]
- Abbott, T.R.; Dhamdhere, G.; Liu, Y.; Lin, X.; Goudy, L.; Zeng, L.; Chemparathy, A.; Chmura, S.; Heaton, N.S.; Debs, R.; et al. Development of CRISPR as an antiviral strategy to combat SARS-CoV-2 and influenza. Cell 2020, 181, 865–876.e12. [Google Scholar] [CrossRef] [PubMed]
- Abed, O.S.A. Gene therapy avenues and COVID-19 vaccines. Genes Immun. 2021, 22, 120–124. [Google Scholar] [CrossRef]
- Garcia, L.F. Immune response, inflammation, and the clinical spectrum of COVID-19. Front. Immunol. 2020, 11, 1441. [Google Scholar] [CrossRef]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef]
- Wang, C.; Xie, J.; Zhao, L.; Fei, X.; Zhang, H.; Tan, Y.; Nie, X.; Zhou, L.; Liu, Z.; Ren, Y.; et al. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine 2020, 57, 102833. [Google Scholar] [CrossRef] [PubMed]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef]
- Liu, F.; Han, K.; Blair, R.; Kenst, K.; Qin, Z.; Upcin, B.; Wörsdörfer, P.; Midkiff, C.C.; Mudd, J.; Belyaeva, E.; et al. SARS-CoV-2 infects endothelial cells in vivo and in vitro. Front. Cell. Infect. Microbiol. 2021, 11, 701278. [Google Scholar] [CrossRef]
- Bernard, I.; Limonta, D.; Mahal, L.K.; Hobman, T.C. Endothelium infection and dysregulation by SARS-CoV-2: Evidence and caveats in COVID-19. Viruses 2020, 13, 29. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef]
- Du, F.; Liu, B.; Zhang, S. COVID-19: The role of excessive cytokine release and potential ACE2 down-regulation in promoting hypercoagulable state associated with severe illness. J. Thromb. Thrombolysis 2021, 51, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Asakura, H.; Ogawa, H. COVID-19-associated coagulopathy and disseminated intravascular coagulation. Int. J. Hematol. 2021, 113, 45–57. [Google Scholar] [CrossRef]
- Paniri, A.; Akhavan-Niaki, H. Emerging role of IL-6 and NLRP3 inflammasome as potential therapeutic targets to combat COVID-19: Role of lncRNAs in cytokine storm modulation. Life Sci. 2020, 257, 118114. [Google Scholar] [CrossRef]
- Toldo, S.; Bussani, R.; Nuzzi, V.; Bonaventura, A.; Mauro, A.G.; Cannata, A.; Pillappa, R.; Sinagra, G.; Nana-Sinkam, P.; Sime, P.; et al. Inflammasome formation in the lungs of patients with fatal COVID-19. Inflamm. Res. 2021, 70, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Shah, A. Novel coronavirus-induced NLRP3 inflammasome activation: A potential drug target in the treatment of COVID-19. Front. Immunol. 2020, 11, 1021. [Google Scholar] [CrossRef]
- Chen, I.Y.; Ichinohe, T. Response of host inflammasomes to viral infection. Trends Microbiol. 2015, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Ishii, K.J.; Coban, C.; Akira, S. Innate immune response to viral infection. Cytokine 2008, 43, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Potts, G.K.; Dawson, A.R.; Coon, J.J.; Mehle, A. Phosphorylation at the homotypic interface regulates nucleoprotein oligomerization and assembly of the influenza virus replication machinery. PLoS Pathog. 2015, 11, e1004826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, C.; Fitzgerald, K.A. Molecular mechanisms involved in inflammasome activation. Trends Cell. Biol. 2009, 19, 455–464. [Google Scholar] [CrossRef]
- Halfmann, P.; Hill-Batorski, L.; Kawaoka, Y. The induction of IL-1beta secretion through the NLRP3 inflammasome during Ebola virus infection. J. Infect. Dis. 2018, 218 (Suppl. S5), S504–S507. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Lee, H.K.; Ogura, Y.; Flavell, R.; Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009, 206, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Negash, A.A.; Olson, R.M.; Griffin, S.; Gale, M., Jr. Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PLoS Pathog. 2019, 15, e1007593. [Google Scholar] [CrossRef]
- Leal, V.N.C.; Reis, E.C.; Pontillo, A. Inflammasome in HIV infection: Lights and shadows. Mol. Immunol. 2020, 118, 9–18. [Google Scholar] [CrossRef]
- Darweesh, M.; Kamel, W.; Gavrilin, M.A.; Akusjarvi, G.; Svensson, C. Adenovirus VA RNAI blocks ASC oligomerization and inhibits NLRP3 inflammasome activation. Front. Immunol. 2019, 10, 2791. [Google Scholar] [CrossRef] [Green Version]
- Nour, A.M.; Reichelt, M.; Ku, C.C.; Ho, M.Y.; Heineman, T.C.; Arvin, A.M. Varicella-zoster virus infection triggers formation of an interleukin-1beta (IL-1beta)-processing inflammasome complex. J. Biol. Chem. 2011, 286, 17921–17933. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.E.; Chikoti, L.; Chandran, B. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J. Virol. 2013, 87, 5005–5018. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Chitre, S.A.; Akinyemi, I.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 viroporin triggers the NLRP3 inflammatory pathway. BioRxiv 2020. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Luis Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Theobald, S.J.; Simonis, A.; Kreer, C.; Zehner, M.; Fischer, J.; Albert, M.-C.; Malin, J.J.; Gräb, J.; Winter, S.; de Silva, U.S.; et al. The SARS-CoV-2 spike protein primes inflammation-mediated interleukin-1- beta secretion in COVID-19 patient-derived macrophages. Res. Sq. 2020, 13, e14150. [Google Scholar] [CrossRef]
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y.; et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyper-inflammation. Nat. Commun. 2021, 12, 4664. [Google Scholar] [CrossRef]
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052. [Google Scholar] [CrossRef]
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahat, R.K.; Panda, S.; Rathore, V.; Swain, S.; Yadav, L.; Sah, S.P. The dynamics of inflammatory markers in coronavirus disease-2019 (COVID-19) patients: A systematic review and meta-analysis. Clin. Epidemiol. Glob. Health 2021, 11, 100727. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodriguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef]
- Zeng, F.; Huang, Y.; Guo, Y.; Yin, M.; Chen, X.; Xiao, L.; Deng, G. Association of inflammatory markers with the severity of COVID-19: A meta-analysis. Int. J. Infect. Dis. 2020, 96, 467–474. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, G.; Kutuzov, M.A.; Ridge, K.M. The inflammasome in lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L627–L633. [Google Scholar] [CrossRef] [Green Version]
- Tal-Singer, R.; Crapo, J.D. COPD at the time of COVID-19: A COPD foundation perspective. Chronic. Obstr. Pulm. Dis. 2020, 7, 73–75. [Google Scholar] [CrossRef] [Green Version]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef] [PubMed]
- Borczuk, A.C.; Salvatore, S.P.; Seshan, S.V.; Patel, S.S.; Bussel, J.B.; Mostyka, M.; Elsoukkary, S.; He, B.; Del Vecchio, C.; Fortarezza, F.; et al. COVID-19 pulmonary pathology: A multi-institutional autopsy cohort from Italy and New York City. Mod. Pathol. 2020, 33, 2156–2168. [Google Scholar] [CrossRef]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.K.Y.; Moriyama, M.; Iwasaki, A. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. J. Immunol. 2020, 205, 307–312. [Google Scholar] [CrossRef]
- Andersson, U.; Ottestad, W.; Tracey, K.J. Extracellular HMGB1: A therapeutic target in severe pulmonary inflammation including COVID-19? Mol. Med. 2020, 26, 42. [Google Scholar] [CrossRef]
- Tseng, C.C.; Fang, W.F.; Leung, S.Y.; Chen, H.C.; Chang, Y.C.; Wang, C.C.; Chang, H.C.; Lin, M.C. Impact of serum biomarkers and clinical factors on intensive care unit mortality and 6-month outcome in relatively healthy patients with severe pneumonia and acute respiratory distress syndrome. Dis. Markers 2014, 2014, 804654. [Google Scholar] [CrossRef] [Green Version]
- Sivakorn, C.; Dechsanga, J.; Jamjumrus, L.; Boonnak, K.; Schultz, M.J.; Dorndorp, A.M.; Phumratanaprapin, W.; Ratanarat, R.; Naorungroj, T.; Wattanawinitchai, P.; et al. High mobility group box 1 and interleukin 6 at intensive care unit admission as biomarkers in critically ill COVID-19 patients. Am. J. Trop. Med. Hyg. 2021, 105, 73–80. [Google Scholar] [CrossRef]
- Abrams, E.M.; t Jong, G.W.; Yang, C.L. Asthma and COVID-19. CMAJ 2020, 192, E551. [Google Scholar] [CrossRef] [Green Version]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet. Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Pottier, N.; Maurin, T.; Chevalier, B.; Puissegur, M.P.; Lebrigand, K.; Robbe-Sermesant, K.; Bertero, T.; Lino Cardenas, C.L.; Courcot, E.; Rios, G.; et al. Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: Implication in epithelial-mesenchymal interactions. PLoS ONE 2009, 4, e6718. [Google Scholar] [CrossRef]
- Pranata, R.; Soeroto, A.Y.; Huang, I.; Lim, M.A.; Santoso, P.; Permana, H.; Lukito, A.A. Effect of chronic obstructive pulmonary disease and smoking on the outcome of COVID-19. Int. J. Tuberc. Lung Dis. 2020, 24, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Henry, B.M. Chronic obstructive pulmonary disease is associated with severe coronavirus disease 2019 (COVID-19). Respir. Med. 2020, 167, 105941. [Google Scholar] [CrossRef]
- Gasse, P.; Riteau, N.; Charron, S.; Girre, S.; Fick, L.; Petrilli, V.; Tschopp, J.; Lagente, V.; Quesniaux, V.F.; Ryffel, B. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am. J. Respir. Crit. Care. Med. 2009, 179, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Hessami, A.; Shamshirian, A.; Heydari, K.; Pourali, F.; Alizadeh-Navaei, R.; Moosazadeh, M.; Abrotan, S.; Shojaie, L.; Sedighi, S.; Shamshirian, D.; et al. Cardiovascular diseases burden in COVID-19: Systematic review and meta-analysis. Am. J. Emerg. Med. 2021, 46, 382–391. [Google Scholar] [CrossRef]
- Dolhnikoff, M.; Ferreira Ferranti, J.; de Almeida Monteiro, R.A.; Duarte-Neto, A.N.; Soares Gomes-Gouvêa, M.; Viu Degaspare, N.; Figueiredo Delgado, A.; Montanari Fiorita, C.; Nunes Leal, G.; Rodrigues, R.M.; et al. SARS-CoV-2 in cardiac tissue of a child with COVID-19-related multisystem inflammatory syndrome. Lancet. Child. Adolesc. Health 2020, 4, 790–794. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Hoel, H.; Heggelund, L.; Reikvam, D.H.; Stiksrud, B.; Ueland, T.; Michelsen, A.E.; Otterdal, K.; Muller, K.E.; Lind, A.; Muller, F.; et al. Elevated markers of gut leakage and inflammasome activation in COVID-19 patients with cardiac involvement. J. Intern. Med. 2021, 289, 523–531. [Google Scholar] [CrossRef]
- Unudurthi, S.D.; Luthra, P.; Bose, R.J.C.; McCarthy, J.R.; Kontaridis, M.I. Cardiac inflammation in COVID-19: Lessons from heart failure. Life Sci. 2020, 260, 118482. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Samstad, E.O.; Niyonzima, N.; Nymo, S.; Aune, M.H.; Ryan, L.; Bakke, S.S.; Lappegard, K.T.; Brekke, O.L.; Lambris, J.D.; Damas, J.K.; et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J. Immunol. 2014, 192, 2837–2845. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Lu, Y.; Cao, Z.; Ma, Q.; Pi, H.; Fang, Y.; Yu, Z.; Hu, H.; Zhou, Z. Cadmium induces NLRP3 inflammasome-dependent pyroptosis in vascular endothelial cells. Toxicol. Lett. 2016, 246, 7–16. [Google Scholar] [CrossRef]
- Zhaolin, Z.; Guohua, L.; Shiyuan, W.; Zuo, W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2019, 52, e12563. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W.; et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2014, 109, 415. [Google Scholar] [CrossRef]
- Grzegorowska, O.; Lorkowski, J. Possible correlations between atherosclerosis, acute coronary syndromes and COVID-19. J. Clin. Med. 2020, 9, 3746. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Dodsworth, M.; Thum, T. Inflammatory cells and their non-coding RNAs as targets for treating myocardial infarction. Basic Res. Cardiol. 2018, 114, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.U.; Hanif, M.; Ali, M.J.; Haider, M.A.; Kherani, D.; Memon, G.M.; Karim, A.H.; Sattar, A. Neurological manifestations of COVID-19 (SARS-CoV-2): A review. Front. Neurol. 2020, 11, 518. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.H.; Beghi, E.; Helbok, R.; Moro, E.; Sampson, J.; Altamirano, V.; Mainali, S.; Bassetti, C.; Suarez, J.I.; McNett, M.; et al. Global incidence of neurological manifestations among patients hospitalized with COVID-19-A report for the GCS-Neuro COVID consortium and the ENERGY Consortium. JAMA Netw. Open 2021, 4, e2112131. [Google Scholar] [CrossRef] [PubMed]
- Bohmwald, K.; Galvez, N.M.S.; Rios, M.; Kalergis, A.M. Neurologic alterations due to respiratory virus infections. Front. Cell. Neurosci. 2018, 12, 386. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, T.; Harii, N.; Goto, J.; Harada, D.; Sugawara, H.; Takamino, J.; Ueno, M.; Sakata, H.; Kondo, K.; Myose, N.; et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int. J. Infect. Dis. 2020, 94, 55–58. [Google Scholar] [CrossRef]
- Liu, T.; Wang, X.; Guo, F.; Sun, X.; Yuan, K.; Wang, Q.; Lan, C. Lysophosphatidylcholine induces apoptosis and inflammatory damage in brain microvascular endothelial cells via GPR4-mediated NLRP3 inflammasome activation. Toxicol. In Vitro 2021, 77, 105227. [Google Scholar] [CrossRef]
- Ito, M.; Shichita, T.; Okada, M.; Komine, R.; Noguchi, Y.; Yoshimura, A.; Morita, R. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 2015, 6, 7360. [Google Scholar] [CrossRef]
- WHO. Coronavirus Disease (COVID-19) Pandemic. 2020. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 11 September 2021).
- Isales, C.M.; Kolhe, R.; Sharma, A.; Lee, T.J.; Yusufu, I.; Sahay, B.; Fulzele, S. COVID-19 virulence in aged patients might be impacted by the host cellular microRNAs abundance/profile. Aging. Dis. 2020, 11, 509–522. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the treatment of COVID-19—Final report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Recovery Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in hospitalized patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef] [PubMed]
- WHO Solidarity Trial Consortium. Repurposed antiviral drugs for COVID-19—interim WHO solidarity trial results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Therapeutics and COVID-19: Living Guideline—World Health Organization (WHO). Available online: https://app.magicapp.org/#/guideline/nBkO1E/rec/jOp0R7 (accessed on 15 November 2021).
- Nalawansha, D.A.; Samarasinghe, K.T.G. Double-barreled CRISPR technology as a novel treatment strategy for COVID-19. ACS Pharmacol. Transl. Sci. 2020, 3, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Berber, B.; Aydin, C.; Kocabas, F.; Guney-Esken, G.; Yilancioglu, K.; Karadag-Alpaslan, M.; Caliseki, M.; Yuce, M.; Demir, S.; Tastan, C. Gene editing and RNAi approaches for COVID-19 diagnostics and therapeutics. Gene Ther. 2021, 28, 290–305. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K. RNA Therapy: Current status and future potential. Chonnam. Med. J. 2020, 56, 87–93. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Mao, T.; Israelow, B.; Lucas, C.; Vogels, C.B.F.; Gomez-Calvo, M.L.; Fedorova, O.; Breban, M.I.; Menasche, B.L.; Dong, H.; Linehan, M.; et al. A stem-loop RNA RIG-I agonist protects against acute and chronic SARS-CoV-2 infection in mice. J. Exp. Med. 2021, 219, e20211818. [Google Scholar] [CrossRef]
- Song, L.; Liu, H.; Gao, S.; Jiang, W.; Huang, W. Cellular microRNAs inhibit replication of the H1N1 influenza A virus in infected cells. J. Virol. 2010, 84, 8849–8860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nersisyan, S.; Shkurnikov, M.; Turchinovich, A.; Knyazev, E.; Tonevitsky, A. Integrative analysis of miRNA and mRNA sequencing data reveals potential regulatory mechanisms of ACE2 and TMPRSS2. PLoS ONE 2020, 15, e0235987. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Long, B.; Liu, F.; Wang, J.X.; Liu, C.Y.; Zhao, B.; Zhou, L.Y.; Sun, T.; Wang, M.; Yu, T.; et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. 2016, 37, 2602–2611. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, M.W.; Moore, K.J. MicroRNA regulation of atherosclerosis. Circ. Res. 2016, 118, 703–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslan, S.; Berkan, O.; Lalem, T.; Özbilüm, N.; Göksel, S.; Korkmaz, O.; Cetin, N.; Yvan, D. Cardiolinc™ network. Long non-coding RNAs in the atherosclerotic plaque. Atherosclerosis 2017, 266, 176–181. [Google Scholar] [CrossRef]
- Gu, W.; Yuan, Y.; Wang, L.; Yang, H.; Li, S.; Tang, Z.; Li, Q. Long non-coding RNA TUG1 promotes airway remodelling by suppressing the miR-145-5p/DUSP6 axis in cigarette smoke-induced COPD. J. Cell Mol. Med. 2019, 23, 7200–7209. [Google Scholar] [CrossRef] [Green Version]
- Austin, P.J.; Tsitsiou, E.; Boardman, C.; Jones, S.W.; Lindsay, M.A.; Adcock, I.M.; Chung, K.F.; Perry, M.M. Transcriptional profiling identifies the long noncoding RNA plasmacytoma variant translocation (PVT1) as a novel regulator of the asthmatic phenotype in human airway smooth muscle. J. Allergy Clin. Immunol. 2017, 139, 780–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKiernan, P.J.; Cunningham, O.; Greene, C.M.; Cryan, S.A. Targeting miRNA-based medicines to cystic fibrosis airway epithelial cells using nanotechnology. Int. J. Nanomed. 2013, 8, 3907–3915. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.X.; Munitz, A.; Rothenberg, M.E. MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J. Immunol. 2009, 182, 4994–5002. [Google Scholar] [CrossRef] [Green Version]
- Oglesby, I.K.; McElvaney, N.G.; Greene, C.M. MicroRNAs in inflammatory lung disease-master regulators or target practice? Respir Res. 2010, 11, 148. [Google Scholar] [CrossRef] [Green Version]
- Ashraf, U.; Zhu, B.; Ye, J.; Wan, S.; Nie, Y.; Chen, Z.; Cui, M.; Wang, C.; Duan, X.; Zhang, H.; et al. MicroRNA-19b-3p modulates Japanese encephalitis virus-mediated inflammation via targeting RNF11. J. Virol. 2016, 90, 4780–4795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.; Xu, M.; He, X.Y. Correlations between microRNA-146a and immunoglobulin and inflammatory factors in Guillain-Barre syndrome. J. Int Med. Res. 2020, 48, 300060520904842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, D.C.; Eldahshan, W.; Rutkowski, E. COVID-19-related stroke. Transl. Stroke Res. 2020, 11, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.K.; Hsieh, S.T.; Chao, C.C.; Chen, Y.C.; Lin, Y.H.; Chang, S.C.; Chang, Y.C. Neuromuscular disorders in severe acute respiratory syndrome. Arch. Neurol. 2004, 61, 1669–1673. [Google Scholar] [CrossRef] [PubMed]
- Zhiguo, W. The guideline of the design and validation of miRNA mimics. Methods Mol. Biol. 2011, 676, 211–223. [Google Scholar] [CrossRef]
- Scherr, M.; Venturini, L.; Battmer, K.; Schaller-Schoenitz, M.; Schaefer, D.; Dallmann, I.; Ganser, A.; Eder, M. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res. 2007, 35, e149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirci, M.D.S.; Adan, A. Computational analysis of microRNA-mediated interactions in SARS-CoV-2 infection. Peer J. 2020, 8, e9369. [Google Scholar] [CrossRef] [PubMed]
- Henke, J.I.; Georgen, D.; Zheng, J.; Song, Y.; Schüttler, C.G.; Fehr, C.; Jünemann, C.; Niepmann, M. MicroRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Siu, G.K.; Mok, B.W.; Sun, J.; Fung, K.S.C.; Lam, J.Y.; Wong, N.K.; Gedefaw, L.; Luo, S.; Lee, T.M.H.; et al. Viral microRNAs encoded by nucleocapsid gene of SARS-CoV-2 are detected during infection, and targeting metabolic pathways in host cells. Cells 2021, 10, 1762. [Google Scholar] [CrossRef]
- Haneklaus, M.; Gerlic, M.; Kurowska-Stolarska, M.; Rainey, A.A.; Pich, D.; McInnes, I.B.; Hammerschmidt, W.; O’Neill, L.A.; Masters, S.L. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1beta production. J. Immunol. 2012, 189, 3795–3799. [Google Scholar] [CrossRef] [Green Version]
- Krause, P.R.; Fleming, T.F.; Longini, M.I.; Peto, R.; Briand, S.; Heymann, D.L.; Beral, V.; Snape, M.D.; Rees, H.; Ropero, A.M.; et al. SARS-CoV-2 variants and vaccines. N. Engl. J. Med. 2021, 385, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Fareh, M.; Zhao, W.; Hu, W.; Casan, J.M.L.; Kumar, A.; Symons, J.; Zerbato, J.M.; Fong, D.; Voskoboinik, I.; Ekert, P.G.; et al. Reprogrammed CRISPR-Cas13b suppresses SARS-CoV-2 replication and circumvents its mutational escape through mismatch tolerance. Nat. Commun. 2021, 12, 4270. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, A.; Ferrara, F. Brief review of the mRNA vaccines COVID-19. Inflammopharmacology 2021, 29, 645–649. [Google Scholar] [CrossRef]
- Boehm, E.; Kronig, I.; Neher, R.A.; Eckerle, I.; Vetter, P.; Kaiser, L. Novel SARS-CoV-2 variants: The pandemics within the pandemic. Clin. Microbiol Infect. 2021, 27, 1109–1117. [Google Scholar] [CrossRef]
- Menni, C.; Klaser, K.; May, A.; Polidori, L.; Capdevila, J.; Louca, P.; Sudre, C.H.; Nguyen, L.H.; Drew, D.A.; Merino, J.; et al. Vaccine side-effects and SARS-CoV-2 infection after vaccination in users of the COVID symptom study app in the UK: A prospective observational study. Lancet Infect. Dis. 2021, 21, 939–949. [Google Scholar] [CrossRef]
- Jeyanathan, M.; Afkhami, S.; Smaill, F.; Miller, M.S.; Lichty, B.D.; Xing, Z. Immunological considerations for COVID-19 vaccine strategies. Nat. Rev. Immunol. 2020, 20, 615–632. [Google Scholar] [CrossRef] [PubMed]
- WHO. COVID-19 Vaccine Tracker and Landscape. Available online: https://www.who.int/publications/m/item/draft-landscape-of-COVID-19-candidate-vaccines (accessed on 11 September 2021).
- Nagy, A.; Alhatlani, B. An overview of current COVID-19 vaccine platforms. Comput. Struct. Biotechnol. J. 2021, 19, 2508–2517. [Google Scholar] [CrossRef]
- Frederiksen, L.S.F.; Zhang, Y.; Foged, C.; Thakur, A. The long road toward COVID-19 herd immunity: Vaccine platform technologies and mass immunization strategies. Front. Immunol. 2020, 11, 1817. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef]
- Mascellino, M.T.; Di Timoteo, F.; De Angelis, M.; Oliva, A. Overview of the main anti-SARS-CoV-2 vaccines: Mechanism of action, efficacy and safety. Infect. Drug Resist. 2021, 14, 3459–3476. [Google Scholar] [CrossRef] [PubMed]
- Sadarangani, M.; Marchant, A.; Kollmann, T.R. Immunological mechanisms of vaccine-induced protection against COVID-19 in humans. Nat. Rev. Immunol. 2021, 21, 475–484. [Google Scholar] [CrossRef]
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. N. Engl. J. Med. 2020, 383, 2427–2438. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Farber, D.L. COVID-19 vaccines: Modes of immune activation and future challenges. Nat. Rev. Immunol. 2021, 21, 195–197. [Google Scholar] [CrossRef]
- Yao, C.; Bora, S.A.; Parimon, T.; Zaman, T.; Friedman, O.A.; Palatinus, J.A.; Surapaneni, N.S.; Matusov, Y.P.; Cerro Chiang, G.; Kassar, A.G.; et al. Cell-type-specific immune dysregulation in severely ill COVID-19 patients. Cell Rep. 2021, 34, 108590. [Google Scholar] [CrossRef]
- Pouwels, K.B.; Pritchard, E.; Matthews, P.C.; Stoesser, N.; Eyre, D.W.; Vihta, K.D.; House, T.; Hay, J.; Bell, J.I.; Newton, J.N.; et al. Effect of delta variant on viral burden and vaccine effectiveness against new SARS-CoV-2 infections in the UK. Nat. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Shin, E.C. The type I interferon response in COVID-19: Implications for treatment. Nat. Rev. Immunol. 2020, 20, 585–586. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.C.R.; Acuna, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long non-coding RNAs in the regulation of gene expression: Physiology and disease. Noncoding RNA 2019, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.T. MicroRNAs: Critical regulators of development, cellular physiology and malignancy. Cell Cycle 2005, 4, 1179–1184. [Google Scholar] [CrossRef] [Green Version]
- Ning, Z.W.; Luo, X.Y.; Wang, G.Z.; Li, Y.; Pan, M.X.; Yang, R.Q.; Ling, X.G.; Huang, S.; Ma, X.X.; Jin, S.Y.; et al. MicroRNA-21 mediates angiotensin II-induced liver fibrosis by activating NLRP3 inflammasome/IL-1beta axis via targeting Smad7 and Spry1. Antioxid. Redox Signal. 2017, 27, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Mao, Y.; Yao, M. NLRP3 inflammasome activation by microRNA-495 promoter methylation may contribute to the progression of acute lung injury. Mol. Ther. Nucleic Acids. 2019, 18, 801–814. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Lu, K.; Ye, T.; Zhang, Z. MicroRNA-223 attenuates LPS-induced inflammation in an acute lung injury model via the NLRP3 inflammasome and TLR4/NF-κB signaling pathway via RHOB. Int. J. Mol. Med. 2019, 43, 1467–1477. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yang, F.; Yu, X.; Wang, B.; Yang, Y.; Zhou, X.; Cheng, R.; Xia, S.; Zhou, X. miR-16 inhibits NLRP3 inflammasome activation by directly targeting TLR4 in acute lung injury. Biomed. Pharmacother. 2019, 112, 108664. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, D.Y.; Heo, H.R.; Choi, S.S.; Hong, S.H.; Kim, W.J. Role of miRNA-181a-2-3p in cadmium-induced inflammatory responses of human bronchial epithelial cells. J. Thorac. Dis. 2019, 11, 3055–3069. [Google Scholar] [CrossRef]
- Wan, L.; Yuan, X.; Liu, M.; Xue, B. miRNA-223-3p regulates NLRP3 to promote apoptosis and inhibit proliferation of hep3B cells. Exp. Ther. Med. 2018, 15, 2429–2435. [Google Scholar] [CrossRef]
- Yu, S.Y.; Dong, B.; Tang, L.; Zhou, S.H. LncRNA MALAT1 sponges miR-133 to promote NLRP3 inflammasome expression in ischemia-reperfusion injured heart. Int. J. Cardiol. 2018, 254, 50. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Bai, X.; Lin, Y.; Li, Z.; Fu, J.; Li, M.; Zhao, T.; Yang, H.; Xu, R.; et al. Melatonin prevents endothelial cell pyroptosis via regulation of long noncoding RNA MEG3/miR-223/NLRP3 axis. J. Pineal Res. 2018, 64, 12449. [Google Scholar] [CrossRef]
- Hu, J.; Wu, H.; Wang, D.; Yang, Z.; Dong, J. LncRNA ANRIL promotes NLRP3 inflammasome activation in uric acid nephropathy through miR-122-5p/BRCC3 axis. Biochimie 2018, 157, 102–110. [Google Scholar] [CrossRef]
- Jin, X.; Jin, H.; Shi, Y.; Guo, Y.; Zhang, H. Long non-coding RNA KCNQ1OT1 promotes cataractogenesis via miR-214 and activation of the caspase-1 pathway. Cell Physiol. Biochem. 2017, 42, 295–305. [Google Scholar] [CrossRef]
- Yi, H.; Peng, R.; Zhang, L.Y.; Sun, Y.; Peng, H.M.; Liu, H.D.; Yu, L.J.; Li, A.L.; Zhang, Y.J.; Jiang, W.H.; et al. LincRNA-Gm4419 knockdown ameliorates NF-kappaB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death Dis. 2017, 8, e2583. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hu, X.; Li, L.; Li, J.H. Differential microRNA expression in the peripheral blood from human patients with COVID-19. J. Clin. Lab. Anal. 2020, 34, e23590. [Google Scholar] [CrossRef]
- Zhang, S.; Amahong, K.; Sun, X.; Lian, X.; Liu, J.; Sun, H.; Lou, Y.; Zhu, F.; Qiu, Y. The miRNA: A small but powerful RNA for COVID-19. Brief. Bioinform. 2021, 22, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, F.K. The proteins of severe acute respiratory syndrome coronavirus-2 (SARS CoV-2 or n-COV19), the cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef]
- Natarelli, L.; Parca, L.; Mazza, T.; Weber, C.; Virgili, F.; Fratantonio, D. MicroRNAs and long non-coding RNAs as potential candidates to target specific motifs of SARS-CoV-2. Res. Sq. 2021, 7, 14. [Google Scholar] [CrossRef]
- Mehta, M.; Chellappan, D.K.; Wich, P.R.; Hansbro, N.G.; Hansbro, P.M.; Dua, K. miRNA nanotherapeutics: Potential and challenges in respiratory disorders. Future Med. Chem. 2020, 12, 987–990. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, S.; Peer, D. RNAi nanomedicines: Challenges and opportunities within the immune system. Nano Technol. 2010, 21, 232001. [Google Scholar] [CrossRef] [PubMed]
- Dua, K.; Hansbro, N.G.; Foster, P.S.; Hansbro, P.M. MicroRNAs as therapeutics for future drug delivery systems in treatment of lung diseases. Drug Deliv. Transl. Res. 2017, 7, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.D.; Sood, V.; Shaw, J.R.; Fang, D.; McClintock, K.; MacLachlan, I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 2005, 23, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Mei, D.; Tan, W.S.D.; Tay, Y.; Mukhopadhyay, A.; Wong, W.S.F. Therapeutic RNA strategies for chronic obstructive pulmonary disease. Trends Pharmacol. Sci. 2020, 41, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Kim, J.H.; Kwon, Y.G.; Kim, E.C.; Kim, N.K.; Choi, H.J.; Yun, C.O. VEGF-specific short hairpin RNA-expressing oncolytic adenovirus elicits potent inhibition of angiogenesis and tumor growth. Mol. Ther. 2007, 15, 295–302. [Google Scholar] [CrossRef]
- Dittgen, T.; Nimmerjahn, A.; Komai, S.; Licznerski, P.; Waters, J.; Margrie, T.W.; Helmchen, F.; Denk, W.; Brecht, M.; Osten, P. Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 18206–18211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlosser, K.; Taha, M.; Deng, Y.; Stewart, D.J. Systemic delivery of MicroRNA mimics with polyethylenimine elevates pulmonary microRNA levels, but lacks pulmonary selectivity. Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Merkel, O.M.; Beyerle, A.; Librizzi, D.; Pfestroff, A.; Behr, T.M.; Sproat, B.; Barth, P.J.; Kissel, T. Nonviral siRNA delivery to the lung: Investigation of PEG-PEI polyplexes and their in vivo performance. Mol. Pharm. 2009, 6, 1246–1260. [Google Scholar] [CrossRef]
- Chiou, G.Y.; Cherng, J.Y.; Hsu, H.S.; Wang, M.L.; Tsai, C.M.; Lu, K.S.; Chien, Y.; Hung, S.C.; Chen, Y.W.; Wong, C.I.; et al. Cationic polyurethanes-short branch PEI-mediated delivery of Mir145 inhibited epithelial-mesenchymal transdifferentiation and cancer stem-like properties and in lung adenocarcinoma. J. Control. Release 2012, 159, 240–250. [Google Scholar] [CrossRef]
- Cai, C.; Xie, Y.; Wu, L.; Chen, X.; Liu, H.; Zhou, Y.; Zou, H.; Liu, D.; Zhao, Y.; Kong, X.; et al. PLGA-based dual targeted nanoparticles enhance miRNA transfection efficiency in hepatic carcinoma. Sci. Rep. 2017, 7, 46250. [Google Scholar] [CrossRef] [PubMed]
- Santos-Carballal, B.; Aaldering, L.J.; Ritzefeld, M.; Pereira, S.; Sewald, N.; Moerschbacher, B.M.; Götte, M.; Goycoolea, F.M. Physicochemical and biological characterization of chitosan-microRNA nanocomplexes for gene delivery to MCF-7 breast cancer cells. Sci. Rep. 2015, 5, 13567. [Google Scholar] [CrossRef]
- Wu, X.; Tai, Z.; Zhu, Q.; Fan, W.; Ding, B.; Zhang, W.; Zhang, L.; Yao, C.; Wang, X.; Ding, X.; et al. Study on the prostate cancer-targeting mechanism of aptamer-modified nanoparticles and their potential anticancer effect in vivo. Int. J. Nanomed. 2014, 9, 5431–5440. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.K.; Liang, W.; Chan, H.K. Pulmonary delivery of therapeutic siRNA. Adv. Drug Deliv. Rev. 2012, 64, 1–15. [Google Scholar] [CrossRef]
- Mohamed, A.; Kunda, N.K.; Rossa, K.; Hutcheon, G.; Saleem, I.Y. Polymeric nanoparticles for the delivery of miRNA to treat chronic obstructive pulmonary disease (COPD). Eur. J. Pharm. Biopharm. 2019, 136, 1–8. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, X.; Guo, C.; Ren, D.; Zhao, Y.; Xiao, W.; Jiao, W. Aptamer-dendrimer bioconjugates for targeted delivery of miR-34a expressing plasmid and antitumor effects in non-small cell lung cancer cells. PLoS ONE 2015, 10, e0139136. [Google Scholar] [CrossRef]
- Hsu, S.H.; Yu, B.; Wang, X.; Lu, Y.; Schmidt, C.R.; Lee, R.J.; Lee, L.J.; Jacob, S.T.; Ghoshal, K. Cationic lipid nanoparticles for therapeutic delivery of siRNA and miRNA to murine liver tumor. Nanomedicine 2013, 9, 1169–1180. [Google Scholar] [CrossRef] [Green Version]
- McLendon, J.M.; Joshi, S.R.; Sparks, J.; Matar, M.; Fewell, J.G.; Abe, K.; Oka, M.; McMurtry, I.F.; Gerthoffer, W.T. Lipid nanoparticle delivery of a microRNA-145 inhibitor improves experimental pulmonary hypertension. J. Control Release 2015, 210, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, P.T.; Shah, B.P.; Lee, K.B. Combi.ined magnetic nanoparticle-based microRNA and hyperthermia therapy to enhance apoptosis in brain cancer cells. Small 2014, 10, 4106–4112. [Google Scholar] [CrossRef]
- Bertucci, A.; Prasetyanto, E.A.; Septiadi, D.; Manicardi, A.; Brognara, E.; Gambari, R.; Corradini, R.; Cola, L.D. Combined delivery of temozolomide and anti-miR221 PNA using mesoporous silica nanoparticles induces apoptosis in resistant glioma cells. Small 2015, 11, 5687–5695. [Google Scholar] [CrossRef]
- Ghosh, R.; Singh, L.C.; Shohet, J.M.; Gunaratne, P.H. A gold nanoparticle platform for the delivery of functional microRNAs into cancer cells. Biomaterials 2013, 34, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Tian, F.; Hernández, Y.; Bao, C.; Cui, D.; Janssen, K.P.; Ibarra, M.R.; Baptista, P.V.; Stoeger, T.; de la Fuente, J.M. In vivo tumor targeting via nanoparticle-mediated therapeutic siRNA coupled to inflammatory response in lung cancer mouse models. Biomaterials 2013, 34, 7744–7753. [Google Scholar] [CrossRef] [PubMed]
- Bryniarski, K.; Ptak, W.; Jayakumar, A.; Pullmann, K.; Caplan, M.J.; Chairoungdua, A.; Lu, J.; Adams, B.D.; Sikora, E.; Nazimek, K.; et al. Antigen-specific, antibody-coated, exosome-like nanovesicles deliver suppressor T-cell microRNA-150 to effector T cells to inhibit contact sensitivity. J. Allergy Clin. Immunol. 2013, 132, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Charrier, A.; Zhou, Y.; Chen, R.; Yu, B.; Agarwal, K.; Tsukamoto, H.; Lee, L.J.; Paulaitis, M.E.; Brigstock, D.R. Epigenetic regulation of connective tissue growth factor by MicroRNA-214 delivery in exosomes from mouse or human hepatic stellate cells. Hepatology 2014, 59, 1118–1129. [Google Scholar] [CrossRef] [Green Version]
- Munoz, J.L.; Bliss, S.A.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Delivery of functional anti-miR-9 by mesenchymal stem cell-derived exosomes to glioblastoma multiforme cells conferred chemosensitivity. Mol. Ther. Nucleic Acids 2013, 2, e126. [Google Scholar] [CrossRef]
- Nie, H.; Xie, X.; Zhang, D.; Zhou, Y.; Li, B.; Li, F.; Li, F.; Cheng, Y.; Mei, H.; Meng, H.; et al. Use of lung-specific exosomes for miRNA-126 delivery.y in non-small cell lung cancer. Nanoscale 2020, 12, 877–887. [Google Scholar] [CrossRef]
- Pan, Q.; Ramakrishnaiah, V.; Henry, S.; Fouraschen, S.; de Ruiter, P.E.; Kwekkeboom, J.; Tilanus, H.W.; Janssen, H.L.A.; van der Laan, L.J.W. Hepatic cell-to-cell transmission of small silencing RNA can extend the therapeutic reach of RNA interference (RNAi). Gut 2012, 61, 1330–1339. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, D.; Chen, X.; Li, J.; Li, L.; Bian, Z.; Sun, F.; Lu, J.; Yin, Y.; Cai, X.; et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol. Cell 2010, 39, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Ban, E.; Kwon, T.H.; Kim, A. Delivery of therapeutic miRNA using polymer-based formulation. Drug Deliv. Transl. Res. 2019, 9, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.L.; Harper, S.Q. Viral delivery of recombinant short hairpin RNAs. Methods Enzymol. 2005, 392, 145–173. [Google Scholar] [CrossRef]
- Relph, K.L.; Harrington, K.J.; Pandhaa, H. Adenoviral strategies for the gene therapy of cancer. Semin. Oncol. 2005, 32, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Sliva, K.; Schnierle, B.S. Selective gene silencing by viral delivery of short hairpin RNA. Virol. J. 2010, 7, 248. [Google Scholar] [CrossRef] [Green Version]
- Grimm, D.; Pndey, K.; Kay, M.A. Adeno-associated virus vectors for short hairpin RNA expression. Methods Enzymol. 2005, 392, 381–405. [Google Scholar] [CrossRef]
- Matsushita, T.; Elliger, S.; Elliger, C.; Podsakoff, G.; Villarreal, L.; Kurtzman, G.J.; Iwaki, Y.; Colosi, P. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998, 5, 938–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, D.; Kay, M.A. From virus evolution to vector revolution: Use of naturally occurring serotypes of adeno-associated virus (AAV) as novel vectors for human gene therapy. Curr. Gene Ther. 2003, 3, 281–304. [Google Scholar] [CrossRef]
- Brummelkamp, T.R.; Bernards, R.; AGAMI, R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002, 296, 550–553. [Google Scholar] [CrossRef] [Green Version]
- Al Yacoub, N.; Romanowska, M.; Haritonova, N.; Foerster, J. Optimized production and concentration of lentiviral vectors containing large inserts. J. Gene Med. 2007, 9, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Preat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Ansary, R.H.; Awang, M.B.; Rahman, M.M. Biodegradable poly(D, L-lactic-co-glycolic acid)-based micro/nanoparticles for sustained release of protein drugs—A review. Trop. J. Pharm. Res. 2014, 13, 1179–1190. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Kong, L.; Ni, Q.; Lu, Y.; Ding, W.; Liu, G.; Pu, L.; Tang, W.; Kong, L. miR-146a ameliorates liver ischemia/reperfusion injury by suppressing IRAK1 and TRAF6. PLoS ONE 2014, 9, e101530. [Google Scholar] [CrossRef]
- Bolu, B.S.; Sanyal, R.; Sanyal, A. Drug delivery systems from self-assembly of dendron-polymer conjugates. Molecules 2018, 23, 1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.W.L.; Paoletti, C.; Campisia, M.; Osaki, T.; Adriani, G.; Kamm, R.D.; Mattu, C.; Chiono, V. MicroRNA delivery through nanoparticles. J. Control. Release 2019, 313, 80–95. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, L. Lipid nanoparticles for gene delivery. Adv. Genet. 2014, 88, 13–36. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Lee, R.J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 2016, 99, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Pilgaard, L.; Moos, T.; Duroux, M. A comprehensive overview of exosomes as drug delivery vehicles—endogenous nanocarriers for targeted cancer therapy. Biochim. Biophys. Acta 2014, 1846, 75–87. [Google Scholar] [CrossRef]
- Fan, Q.; Yang, L.; Zhang, X.; Peng, X.; Wei, S.; Su, D.; Zhai, Z.; Hua, X.; Li, H. The emerging role of exosome-derived non-coding RNAs in cancer biology. Cancer Lett. 2018, 414, 107–115. [Google Scholar] [CrossRef]
- O’Loughlin, A.J.; Mager, I.; de Jong, O.G.; Varela, M.A.; Schiffelers, R.M.; El Andaloussi, S.; Wood, M.J.A.; Vader, P. Functional delivery of lipid-conjugated siRNA by extracellular vesicles. Mol. Ther. 2017, 25, 1580–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.S.; Roh, T.Y.; Park, J.; Nilsson, J.; Lotvall, L.; Kim, Y.K.; Gho, Y.S. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano 2013, 7, 7698–7710. [Google Scholar] [CrossRef]
- Lundstrom, K. Viral vectors for COVID-19 vaccine development. Viruses 2021, 13, 317. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Wang, Q.; Shan, C.; Yang, C.; Feng, Y.; Wu, J.; Liu, X.; Zhou, Y.; Jiang, R.; Hu, P.; et al. An adenovirus-vectored COVID-19 vaccine confers protection from SARS-CoV-2 challenge in rhesus macaques. Nat. Commun. 2020, 11, 4207. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Potential Therapeutic Ncrna | Disease | Potential Mechanisms |
|---|---|---|
| LNCRNAS 1 | ||
| SHRNA-TUG1 | COPD | Preventing airway remodeling and inflammation by the knockdown of the lncRNA TUG1 [110] |
| SHRNA-PVT1 | Asthma | Regulation of inflammation by the knockdown of lncRNA PVT1 [111] |
| SHRNA-XIST, SHRNA-TLR8-AS1 | Cystic fibrosis | Preventing inflammation by downregulating lncRNAs XIST and TLR8-AS1 [112] |
| HEART-RELATED CIRCULAR RNA (HRCR) | Myocardial infarction | Sponge the pro-inflammatory effects of miR-223 [87,107] |
| MIRNAS 2 | ||
| ANTI-MIR-21 | Asthma | Regulation of inflammation by the knockdown of miR-21 [113] |
| ANTI-MIR-126 | Cystic fibrosis | Regulation of innate immune response [114] |
| ANTI-MIR-155 | Pulmonary fibrosis | Downregulation of NLRP3 inflammasome by silencing miR-155 [72] |
| MIR-200 FAMILY | Cardiac inflammation | Knockdown of ACE2 [106] |
| SYNTHETIC MIRNA MIMICS OF MIR-181B AND MIR-146A | Atherosclerosis | Prevent inflammation [108] |
| SHRNA | Atherosclerosis | Targeting lncRNA ANRIL and MIAT [109] |
| MIR-19B-3P | Encephalitis | Reduction in inflammation [115] |
| MIR-200 | Ischemic stroke | Knockdown of ACE2 [106] |
| ANTI-MIR-146A | GBS | Downregulation of inflammatory response [116] |
| Delivery Vector | Payload | Target | Therapeutic Impact | Limitations |
|---|---|---|---|---|
| Viral Vectors | ||||
| Adenovirus vectors/Adeno-associated viruses | ShRNA (sh-VEGF), ShRNA (sh-Hec1) | Endothelial cells, SF9 tumor cell line | Inhibits tumor growth and angiogenesis, depletion ofHEC1 protein in SF9 cells | Poor vector stability, trigger immune response, cytotoxicity [166] |
| Lentivirus vectors | ShRNA | Cortical neurons | Target gene knock down | Not reported [167] |
| Non Viral Vectors | ||||
| PEI/PEG–PEI polymer/PU-PEI | MiRNA mimics, DsiRNA, MiR-145 | Lungs | Elevates pulmonary miRNA levels, knock-down of target genes, inhibits EMT and tumor growth | Lacks pulmonary selectivity, moderate inflammatory effects [168,169,170] |
| PLGA | MiR-99a | Hepatic carcinoma | Downregulation of target genes, reduction in tumor size | Not reported [171] |
| Chitosan | MiR-145 | MCF-7 breast cancer cells | Downregulation of target mRNA | Not reported [172] |
| PAMAM | MiRNA | Prostate cancer (PCa) cells/xenograft mouse model | Enhanced survival of tumor-bearing mouse | Not reported [173] |
| Poly (ester amine)-alt-PEG | SiRNA | Lungs | Suppressed progression of lung cancer | Not reported [174] |
| PEI/Chitosan | MiR-126 | Cystic fibrosis (CF) | Knockdown of target gene | Not reported [112] |
| PGA-co-PDL | MiR-146a | COPD | Reduced expression of the IRAK1 gene | Not reported [175] |
| Aptamer-dendrimer | MiR-34a | Lung cancer | Reduced cancer cell growth, invasion, induced apoptosis | Not reported [176] |
| Hyaluronic acid coated PEI-PLGA NPs | MiR-542-3p and doxorubicin | Breast cancer | Tumor cell apoptosis | Not reported [107] |
| Lipid-based nanoparticles | MiR-122 mimics, miR-145 | Hepatocellular carcinoma, lungs | Suppression of target genes and tumor xenograft, reduced pulmonary hypertension | Low cytotoxicity [177,178] |
| Iron-based nanoparticles | MiR-let7a | Brain cancer cells | Enhances apoptosis of cancer cells | Not reported [179] |
| Silica-based nanostructures | Anti-miR-221 | Glioma cells | Induction of apoptosis | Not reported [180] |
| Gold nanoparticles | MiRNA mimics, SiRNA | Cancer cell lines, lung cancer | Affected proliferation and target gene expression, reduced cancer cell proliferation and tumor growth | Not reported [181,182] |
| Exosomes | MiR-150, miR-214, anti-miR-9, miR-126, siRNA | Allergic cutaneous sensitivity (mouse), hepatic cells, lung cancer, glioblastoma cells | T-cell regulation, halting of fibrosis, downregulation of MiR-9, supressed the migration and proliferation of cancer cells, enhanced therapeutic efficiency of RNAi drug | Not reported [183,184,185,186,187] |
| THP-1 cell-derived microvesicles | MiR-150 | HMEC-1 cells | Enhanced endothelial cell migration | Not reported [188] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gedefaw, L.; Ullah, S.; Lee, T.M.H.; Yip, S.P.; Huang, C.-L. Targeting Inflammasome Activation in COVID-19: Delivery of RNA Interference-Based Therapeutic Molecules. Biomedicines 2021, 9, 1823. https://doi.org/10.3390/biomedicines9121823
Gedefaw L, Ullah S, Lee TMH, Yip SP, Huang C-L. Targeting Inflammasome Activation in COVID-19: Delivery of RNA Interference-Based Therapeutic Molecules. Biomedicines. 2021; 9(12):1823. https://doi.org/10.3390/biomedicines9121823
Chicago/Turabian StyleGedefaw, Lealem, Sami Ullah, Thomas M. H. Lee, Shea Ping Yip, and Chien-Ling Huang. 2021. "Targeting Inflammasome Activation in COVID-19: Delivery of RNA Interference-Based Therapeutic Molecules" Biomedicines 9, no. 12: 1823. https://doi.org/10.3390/biomedicines9121823
APA StyleGedefaw, L., Ullah, S., Lee, T. M. H., Yip, S. P., & Huang, C. -L. (2021). Targeting Inflammasome Activation in COVID-19: Delivery of RNA Interference-Based Therapeutic Molecules. Biomedicines, 9(12), 1823. https://doi.org/10.3390/biomedicines9121823