
Current Understanding of the Physiopathology, Diagnosis and Therapeutic Approach to Alzheimer’s Disease

 , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
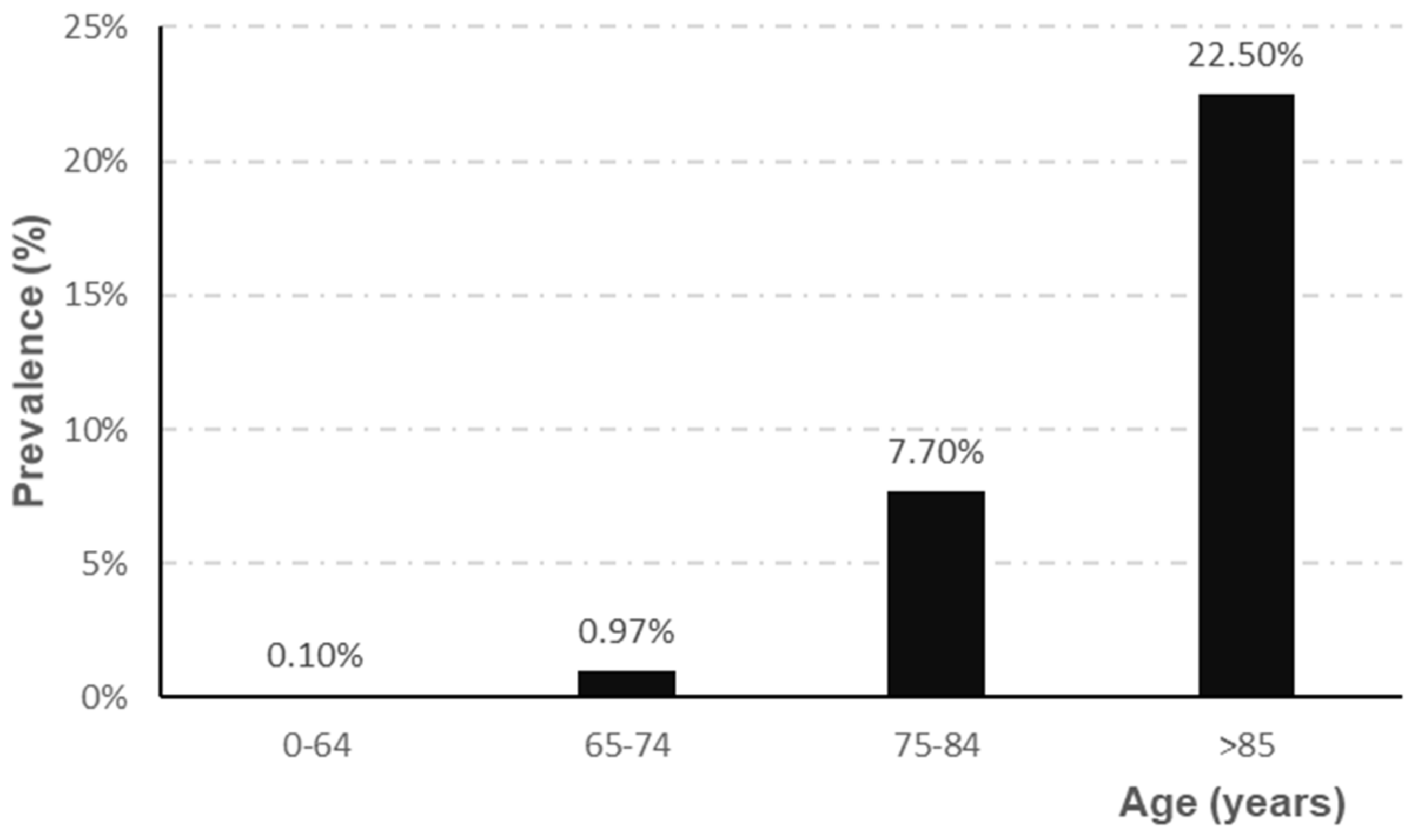
2.1. Etiology of Alzheimer’s Disease
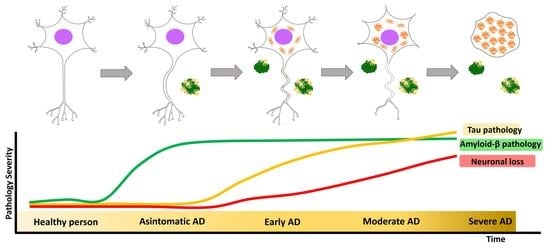
2.2. Clinical Stages of Alzheimer’s Disease
2.3. Neuropathological Features
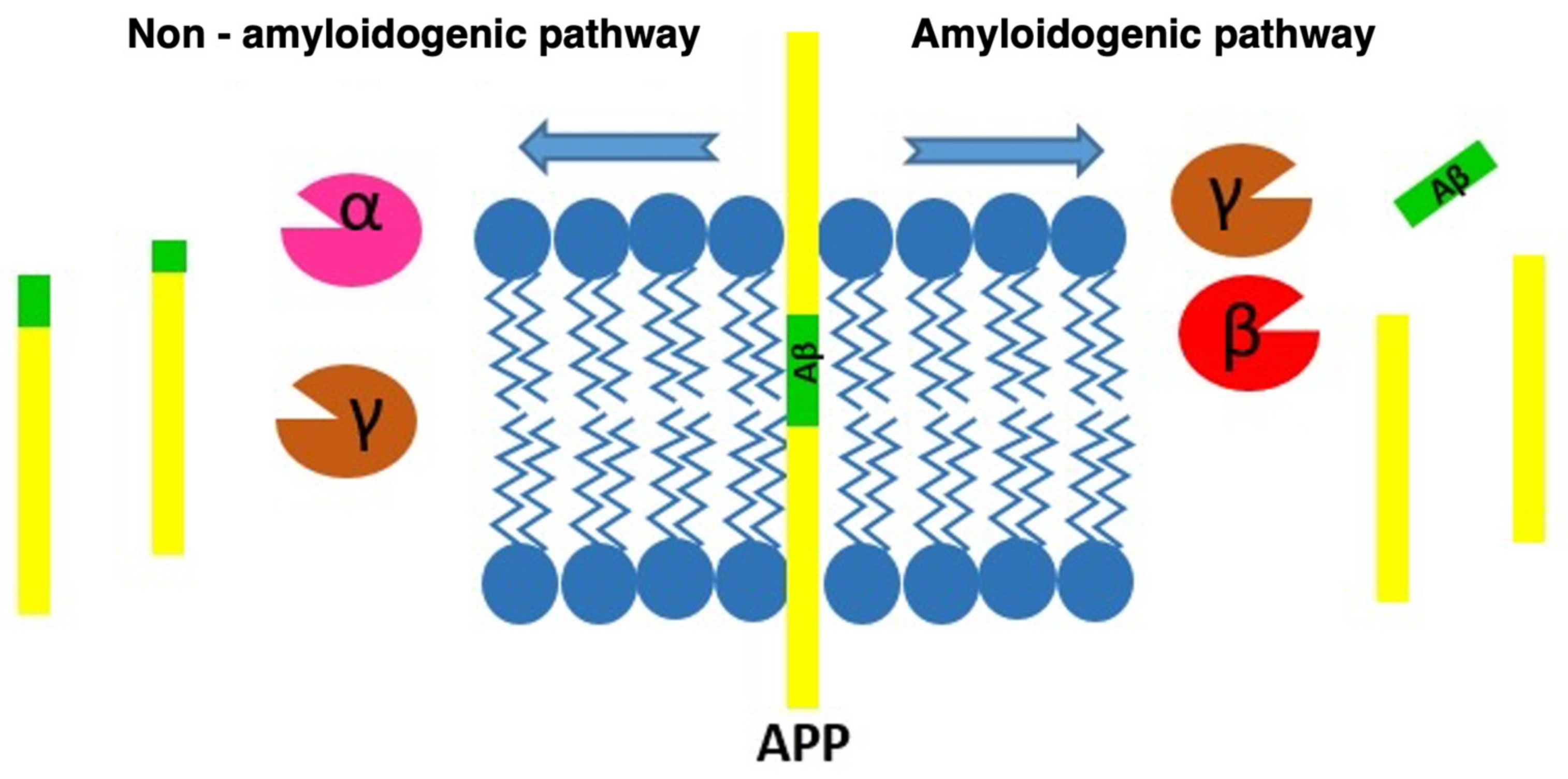
2.3.1. Amyloid-β Pathology (Aβ Pathology)
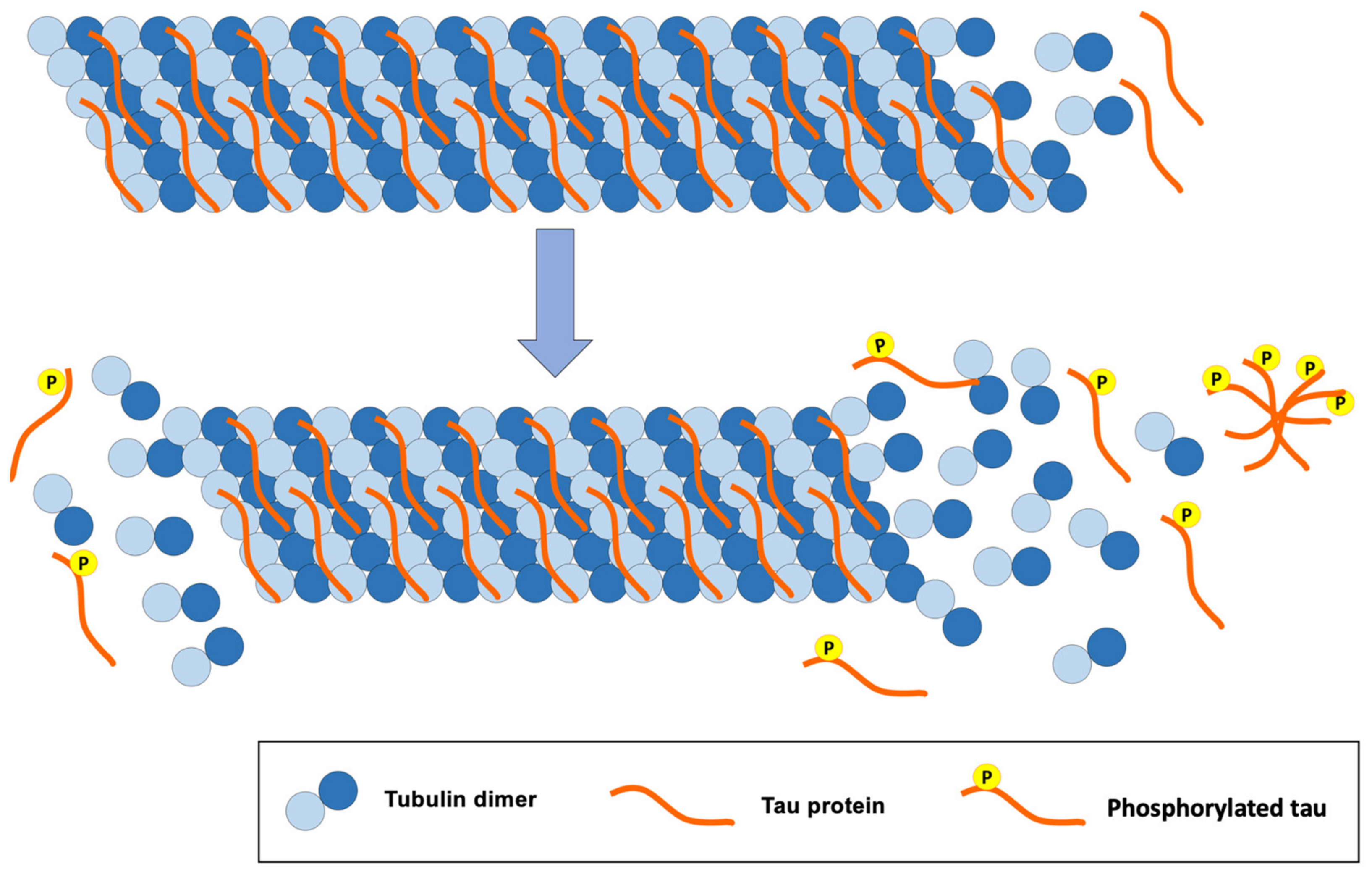
2.3.2. Tau Pathology
2.4. Diagnosis of AD
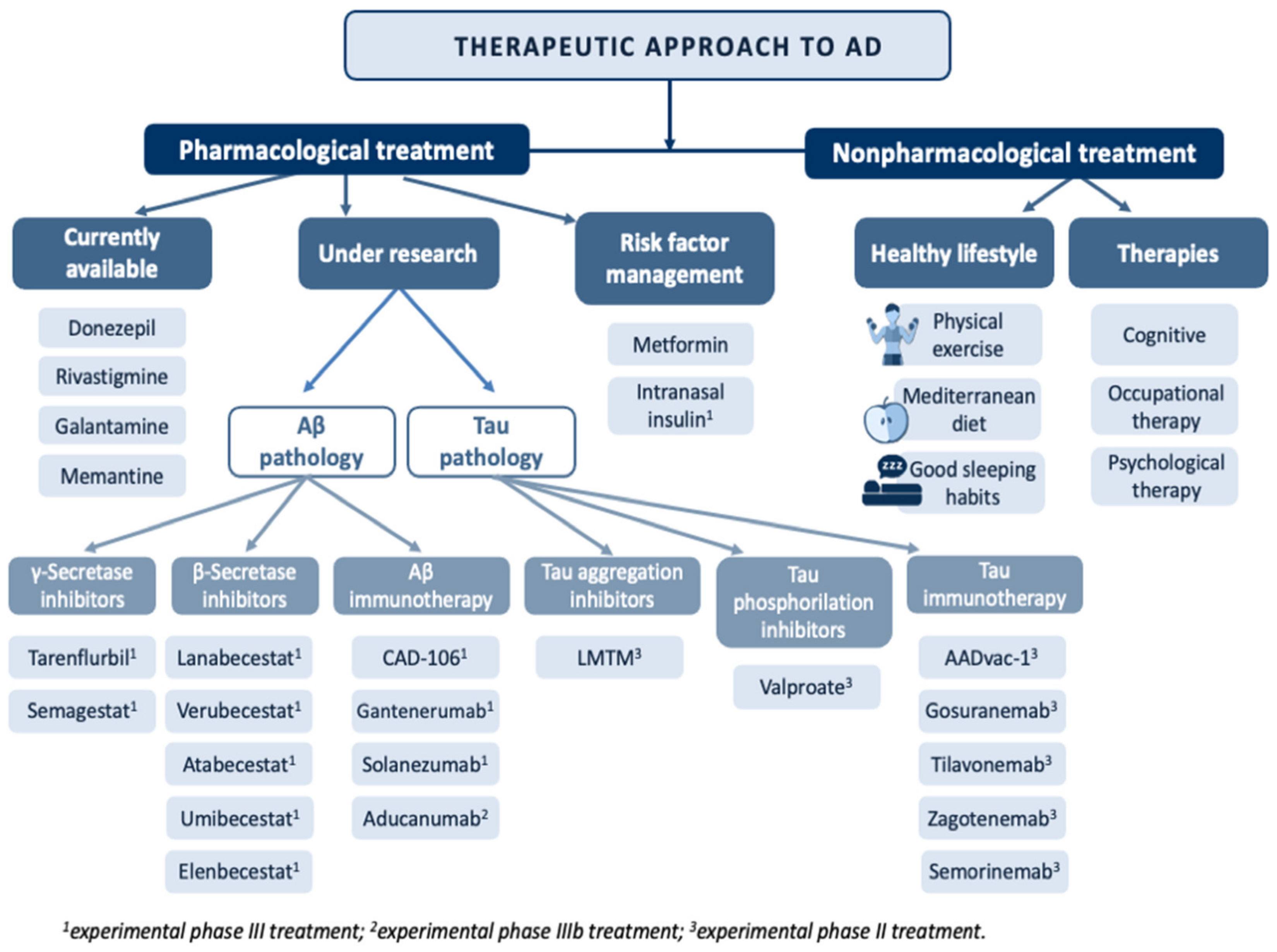
2.5. Alzheimer’s Disease Treatment and Management
2.5.1. Currently Available Pharmacological Treatment
- Donepezil: Approved in 1996 [98], donepezil is the pharmacological treatment of choice for AD. This drug acts by reversibly binding to AChE, inhibiting ACh hydrolysis, resulting in increased bioavailability of this neurotransmitter at neuronal synapses. Its use has been shown to slow cognitive decline and improve behavior in people with AD [97].
- Rivastigmine: This treatment was introduced in 2000 and is indicated for mild and moderate AD. It should be noted that unlike the other AChEIs, rivastigmine is also used to treat dementia associated with Parkinson’s disease [98]. This drug exerts its effect by binding to and inhibiting AChE, increasing ACh levels [97]. The use of rivastigmine in transdermal patches has been shown to be beneficial in AD patients with swallowing problems while decreasing the side effects seen with lower doses in the pill form [99].
- Galantamine: This is a competitive AChE inhibitor approved in 2001 [98]. Its use in mild and moderate AD has been associated with positive evolution of behavior, cognitive performance and development of basic activities of daily living [97]. In addition, it has been observed that galantamine is able to cross the blood–brain barrier more quickly, affecting brain areas such as the hippocampus for an extended period of time (5–7 h) [100].
- Memantine: The latest drug approved for use in moderate and severe AD as monotherapy or in combination with other therapies [101]. It is generally well-tolerated and safe as it exerts its effect without altering neuronal synapses [97]. Memantine treatment has been shown to improve cognitive impairment and general condition in AD patients [101].
2.5.2. Pharmacological Treatment under Investigation
2.5.3. Risk Factor Management
2.5.4. Nonpharmacological Treatment
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lleo, A. Alzheimer’s disease: An ignored condition. Med. Clin. 2018, 150, 432–433. [Google Scholar] [CrossRef]
- Wanleenuwat, P.; Iwanowski, P.; Kozubski, W. Alzheimer’s dementia: Pathogenesis and impact of cardiovascular risk factors on cognitive decline. Postgrad. Med. 2019, 131, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takizawa, C.; Thompson, P.L.; van Walsem, A.; Faure, C.; Maier, W.C. Epidemiological and economic burden of Alzheimer’s disease: A systematic literature review of data across Europe and the United States of America. J. Alzheimer’s Dis. JAD 2015, 43, 1271–1284. [Google Scholar] [CrossRef]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Alvarez-Alvarez, I.; Guillen-Grima, F.; Aguinaga-Ontoso, I. Prevalence and incidence of Alzheimer’s disease in Europe: A meta-analysis. Neurologia 2017, 32, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B. Life expectancy curves reveal major demographic events. Med. Sci. 2017, 33, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 459–509. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.T. New perspectives on Alzheimer’s disease from neuroimaging. Rev. Neurol. 2010, 50 (Suppl. S5), S23–S26. [Google Scholar]
- Garre-Olmo, J. Epidemiology of Alzheimer’s disease and other dementias. Rev. Neurol. 2018, 66, 377–386. [Google Scholar] [PubMed]
- Murman, D.L.; Chen, Q.; Powell, M.C.; Kuo, S.B.; Bradley, C.J.; Colenda, C.C. The incremental direct costs associated with behavioral symptoms in AD. Neurology 2002, 59, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Santana, I.; Farinha, F.; Freitas, S.; Rodrigues, V.; Carvalho, A. The Epidemiology of Dementia and Alzheimer Disease in Portugal: Estimations of Prevalence and Treatment-Costs. Acta Med. Port. 2015, 28, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Shieh, J.C.; Huang, P.T.; Lin, Y.F. Alzheimer’s Disease and Diabetes: Insulin Signaling as the Bridge Linking Two Pathologies. Mol. Neurobiol. 2020, 57, 1966–1977. [Google Scholar] [CrossRef] [PubMed]
- St George-Hyslop, P.H.; Tanzi, R.E.; Polinsky, R.J.; Haines, J.L.; Nee, L.; Watkins, P.C.; Myers, R.H.; Feldman, R.G.; Pollen, D.; Drachman, D.; et al. The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science 1987, 235, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Yamada, M. Risk factors for Alzheimer’s disease. Brain Nerve Shinkei Kenkyu No Shinpo 2010, 62, 679–690. [Google Scholar] [PubMed]
- De Leeuw, F.E.; de Groot, J.C.; Oudkerk, M.; Witteman, J.C.; Hofman, A.; van Gijn, J.; Breteler, M.M. Hypertension and cerebral white matter lesions in a prospective cohort study. Brain J. Neurol. 2002, 125, 765–772. [Google Scholar] [CrossRef]
- Reitz, C.; Tang, M.X.; Manly, J.; Mayeux, R.; Luchsinger, J.A. Hypertension and the risk of mild cognitive impairment. Arch. Neurol. 2007, 64, 1734–1740. [Google Scholar] [CrossRef] [Green Version]
- Pappolla, M.A. Statins, incident Alzheimer disease, change in cognitive function, and neuropathology. Neurology 2008, 71, 2020, author reply 2020–2021. [Google Scholar] [PubMed]
- Pappolla, M.A.; Bryant-Thomas, T.K.; Herbert, D.; Pacheco, J.; Fabra Garcia, M.; Manjon, M.; Girones, X.; Henry, T.L.; Matsubara, E.; Zambon, D.; et al. Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology 2003, 61, 199–205. [Google Scholar] [CrossRef]
- Sensi, S.L. Alzheimer’s Disease, time to turn the tide. Aging 2018, 10, 2537–2538. [Google Scholar] [CrossRef]
- Infante-Garcia, C.; Ramos-Rodriguez, J.J.; Galindo-Gonzalez, L.; Garcia-Alloza, M. Long-term central pathology and cognitive impairment are exacerbated in a mixed model of Alzheimer’s disease and type 2 diabetes. Psychoneuroendocrinology 2016, 65, 15–25. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Infante-Garcia, C.; Galindo-Gonzalez, L.; Garcia-Molina, Y.; Lechuga-Sancho, A.; Garcia-Alloza, M. Increased Spontaneous Central Bleeding and Cognition Impairment in APP/PS1 Mice with Poorly Controlled Diabetes Mellitus. Mol. Neurobiol. 2016, 53, 2685–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Rodriguez, J.J.; Jimenez-Palomares, M.; Murillo-Carretero, M.I.; Infante-Garcia, C.; Berrocoso, E.; Hernandez-Pacho, F.; Lechuga-Sancho, A.M.; Cozar-Castellano, I.; Garcia-Alloza, M. Central vascular disease and exacerbated pathology in a mixed model of type 2 diabetes and Alzheimer’s disease. Psychoneuroendocrinology 2015, 62, 69–79. [Google Scholar] [CrossRef]
- Ozben, T.; Ozben, S. Neuro-inflammation and anti-inflammatory treatment options for Alzheimer’s disease. Clin. Biochem. 2019, 72, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Joe, E.; Ringman, J.M. Cognitive symptoms of Alzheimer’s disease: Clinical management and prevention. BMJ 2019, 367, l6217. [Google Scholar] [CrossRef] [Green Version]
- Di Resta, C.; Ferrari, M. New molecular approaches to Alzheimer’s disease. Clin. Biochem. 2019, 72, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Tublin, J.M.; Adelstein, J.M.; Del Monte, F.; Combs, C.K.; Wold, L.E. Getting to the Heart of Alzheimer Disease. Circ. Res. 2019, 124, 142–149. [Google Scholar] [CrossRef]
- Shah, S.; Lee, S.F.; Tabuchi, K.; Hao, Y.H.; Yu, C.; LaPlant, Q.; Ball, H.; Dann, C.E., 3rd; Sudhof, T.; Yu, G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell 2005, 122, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s disease. Sub Cell. Biochem. 2012, 65, 329–352. [Google Scholar] [CrossRef]
- Staderini, M.; Martin, M.A.; Bolognesi, M.L.; Menendez, J.C. Imaging of beta-amyloid plaques by near infrared fluorescent tracers: A new frontier for chemical neuroscience. Chem. Soc. Rev. 2015, 44, 1807–1819. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque formation and the intraneuronal accumulation of beta-amyloid in Alzheimer’s disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhao, J.; Zhang, X.; Wang, S.; Viola, K.L.; Chow, F.E.; Zhang, Y.; Lippa, C.; Klein, W.L.; Gong, Y. Amyloid Beta Oligomers Target to Extracellular and Intracellular Neuronal Synaptic Proteins in Alzheimer’s Disease. Front. Neurol. 2019, 10, 1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Wu, J.; Geng, M.; Xiong, J. Role of synaptic activity in the regulation of amyloid beta levels in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1217–1232. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wilde, M.C.; Overk, C.R.; Sijben, J.W.; Masliah, E. Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimer’s Dementia J. Alzheimer’s Ass. 2016, 12, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanc, B.; Cruz, L.; Le, R.; Sanders, J.; Ashe, K.H.; Duff, K.; Stanley, H.E.; Irizarry, M.C.; Hyman, B.T. Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 13990–13995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brendza, R.P.; Bacskai, B.J.; Cirrito, J.R.; Simmons, K.A.; Skoch, J.M.; Klunk, W.E.; Mathis, C.A.; Bales, K.R.; Paul, S.M.; Hyman, B.T.; et al. Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J. Clin. Investig. 2005, 115, 428–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, J.; Lynch, G.; Games, D.; Seubert, P. Alterations in synaptic transmission and long-term potentiation in hippocampal slices from young and aged PDAPP mice. Brain Res. 1999, 840, 23–35. [Google Scholar] [CrossRef]
- Lombardo, J.A.; Stern, E.A.; McLellan, M.E.; Kajdasz, S.T.; Hickey, G.A.; Bacskai, B.J.; Hyman, B.T. Amyloid-beta antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 10879–10883. [Google Scholar] [CrossRef] [Green Version]
- D’Amore, J.D.; Kajdasz, S.T.; McLellan, M.E.; Bacskai, B.J.; Stern, E.A.; Hyman, B.T. In vivo multiphoton imaging of a transgenic mouse model of Alzheimer disease reveals marked thioflavine-S-associated alterations in neurite trajectories. J. Neuropathol. Exp. Neurol. 2003, 62, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Knowles, R.B.; Wyart, C.; Buldyrev, S.V.; Cruz, L.; Urbanc, B.; Hasselmo, M.E.; Stanley, H.E.; Hyman, B.T. Plaque-induced neurite abnormalities: Implications for disruption of neural networks in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1999, 96, 5274–5279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spires, T.L.; Meyer-Luehmann, M.; Stern, E.A.; McLean, P.J.; Skoch, J.; Nguyen, P.T.; Bacskai, B.J.; Hyman, B.T. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 7278–7287. [Google Scholar] [CrossRef] [PubMed]
- Stern, E.A.; Bacskai, B.J.; Hickey, G.A.; Attenello, F.J.; Lombardo, J.A.; Hyman, B.T. Cortical synaptic integration in vivo is disrupted by amyloid-beta plaques. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 4535–4540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.N.; Maass, A.; Harrison, T.M.; Baker, S.L.; Jagust, W.J. Cortical tau deposition follows patterns of entorhinal functional connectivity in aging. eLife 2019, 8, e49132. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, L.; Tammewar, G.; Ayakta, N.; Baker, S.L.; Bejanin, A.; Boxer, A.L.; Gorno-Tempini, M.L.; Janabi, M.; Kramer, J.H.; Lazaris, A.; et al. Local and distant relationships between amyloid, tau and neurodegeneration in Alzheimer’s Disease. NeuroImage Clin. 2018, 17, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Chantran, Y.; Capron, J.; Alamowitch, S.; Aucouturier, P. Anti-Abeta Antibodies and Cerebral Amyloid Angiopathy Complications. Front. Immunol. 2019, 10, 1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Costa, J.F.; Baquero-Toledo, M.; Sastre-Bataller, I.; Mas-Estelles, F.; Vilchez-Padilla, J.J. Inflammatory amyloid angiopathy. Neurologia 2014, 29, 254–256. [Google Scholar] [CrossRef]
- Vinters, H.V. Cerebral amyloid angiopathy. A critical review. Stroke 1987, 18, 311–324. [Google Scholar] [CrossRef] [Green Version]
- Levy, E.; Carman, M.D.; Fernandez-Madrid, I.J.; Power, M.D.; Lieberburg, I.; van Duinen, S.G.; Bots, G.T.; Luyendijk, W.; Frangione, B. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248, 1124–1126. [Google Scholar] [CrossRef]
- Van Veluw, S.J.; Reijmer, Y.D.; van der Kouwe, A.J.; Charidimou, A.; Riley, G.A.; Leemans, A.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Histopathology of diffusion imaging abnormalities in cerebral amyloid angiopathy. Neurology 2019, 92, e933–e943. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nature reviews. Neurology 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Belyaev, N.D.; Zhuravin, I.A.; Turner, A.J. The Alzheimer’s amyloid-degrading peptidase, neprilysin: Can we control it? Int. J. Alzheimer’s Dis. 2012, 2012, 383796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, C.; Patte-Mensah, C.; Taleb, O.; Bourguignon, J.J.; Schmitt, M.; Bihel, F.; Maitre, M.; Mensah-Nyagan, A.G. The neuroprotector kynurenic acid increases neuronal cell survival through neprilysin induction. Neuropharmacology 2013, 70, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Folstein, M.F. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: Review and hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef]
- Pivovarova, O.; Hohn, A.; Grune, T.; Pfeiffer, A.F.; Rudovich, N. Insulin-degrading enzyme: New therapeutic target for diabetes and Alzheimer’s disease? Ann. Med. 2016, 48, 614–624. [Google Scholar] [CrossRef]
- Hayrabedyan, S.; Todorova, K.; Spinelli, M.; Barnea, E.R.; Mueller, M. The core sequence of PIF competes for insulin/amyloid beta in insulin degrading enzyme: Potential treatment for Alzheimer’s disease. Oncotarget 2018, 9, 33884–33895. [Google Scholar] [CrossRef] [Green Version]
- Avila, J. Tau phosphorylation and aggregation in Alzheimer’s disease pathology. FEBS Lett. 2006, 580, 2922–2927. [Google Scholar] [CrossRef] [Green Version]
- Matej, R.; Tesar, A.; Rusina, R. Alzheimer’s disease and other neurodegenerative dementias in comorbidity: A clinical and neuropathological overview. Clin. Biochem. 2019, 73, 26–31. [Google Scholar] [CrossRef]
- Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K. Targeting tau protein in Alzheimer’s disease. Drugs Aging 2010, 27, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, A.; West, A.K.; Chuckowree, J.A.; Vickers, J.C.; Dickson, T.C. Does beta-amyloid plaque formation cause structural injury to neuronal processes? Neurotox. Res. 2005, 7, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Jagust, W. Imaging the evolution and pathophysiology of Alzheimer disease. Nature reviews. Neuroscience 2018, 19, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Abeta and Tau Species. Front. Neurosci. 2019, 13, 659. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wes, P.D.; Sayed, F.A.; Bard, F.; Gan, L. Targeting microglia for the treatment of Alzheimer’s Disease. Glia 2016, 64, 1710–1732. [Google Scholar] [CrossRef]
- Morales, I.; Guzman-Martinez, L.; Cerda-Troncoso, C.; Farias, G.A.; Maccioni, R.B. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 2014, 8, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hierro-Bujalance, C.; Bacskai, B.J.; Garcia-Alloza, M. In Vivo Imaging of Microglia with Multiphoton Microscopy. Front. Aging Neurosci. 2018, 10, 218. [Google Scholar] [CrossRef]
- Sankar, S.B.; Infante-Garcia, C.; Weinstock, L.D.; Ramos-Rodriguez, J.J.; Hierro-Bujalance, C.; Fernandez-Ponce, C.; Wood, L.B.; Garcia-Alloza, M. Amyloid beta and diabetic pathology cooperatively stimulate cytokine expression in an Alzheimer’s mouse model. J. Neuroinflamm. 2020, 17, 38. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Lucin, K.M.; Wyss-Coray, T. Immune activation in brain aging and neurodegeneration: Too much or too little? Neuron 2009, 64, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Jagust, W.J. The changing definition of Alzheimer’s disease. Lancet Neurology 2021, 20, 414–415. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2012, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 270–279. [Google Scholar] [CrossRef] [Green Version]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeiro, M.H.; Ardanaz, C.G.; Sola-Sevilla, N.; Dong, J.; Cortes-Erice, M.; Solas, M.; Puerta, E.; Ramírez, M.J. Biomarcadores en la enfermedad de Alzheimer. Adv. Lab. Med. Av. Med. Lab. 2021, 2, 39–50. [Google Scholar] [CrossRef]
- Ortega, R.L.; Dakterzada, F.; Arias, A.; Blasco, E.; Naudi, A.; Garcia, F.P.; Pinol-Ripoll, G. Usefulness of CSF Biomarkers in Predicting the Progression of Amnesic and Nonamnesic Mild Cognitive Impairment to Alzheimer’s Disease. Curr. Aging Sci. 2019, 12, 35–42. [Google Scholar] [CrossRef]
- Humpel, C. Identifying and validating biomarkers for Alzheimer’s disease. Trends Biotechnol. 2011, 29, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauchmann, B.S.; Schneider-Axmann, T.; Perneczky, R. Associations of longitudinal plasma p-tau181 and NfL with tau-PET, Abeta-PET and cognition. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1289–1295. [Google Scholar] [CrossRef]
- Singh, K.; Cheung, B.M.; Xu, A. Ultrasensitive detection of blood biomarkers of Alzheimer’s and Parkinson’s diseases: A systematic review. Biomark. Med. 2021, 15, 1693–1708. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, E.H.; La Joie, R.; Strom, A.; Fonseca, C.; Iaccarino, L.; Wolf, A.; Spina, S.; Allen, I.E.; Cobigo, Y.; Heuer, H.; et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer’s disease and frontotemporal lobar degeneration: A retrospective diagnostic performance study. Lancet Neurol. 2021, 20, 739–752. [Google Scholar] [CrossRef]
- Leuzy, A.; Janelidze, S.; Mattsson-Carlgren, N.; Palmqvist, S.; Jacobs, D.; Cicognola, C.; Stomrud, E.; Vanmechelen, E.; Dage, J.L.; Hansson, O. Comparing the Clinical Utility and Diagnostic Performance of CSF P-Tau181, P-Tau217, and P-Tau231 Assays. Neurology 2021, 97, e1681–e1694. [Google Scholar] [CrossRef]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Wiste, H.J.; Weigand, S.D.; Therneau, T.M.; Lowe, V.J.; Knopman, D.S.; Gunter, J.L.; Senjem, M.L.; Jones, D.T.; Kantarci, K.; et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2017, 13, 205–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiqing, N. Magnetic Resonance Imaging in Animal Models of Alzheimer’s Disease Amyloidosis. Int. J. Mol. Sci. 2021, 22, 12768. [Google Scholar] [CrossRef]
- Odusami, M.; Maskeliunas, R.; Damasevicius, R.; Krilavicius, T. Analysis of Features of Alzheimer’s Disease: Detection of Early Stage from Functional Brain Changes in Magnetic Resonance Images Using a Finetuned ResNet18 Network. Diagnostics 2021, 11, 1071. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Kim, M.; Zhu, X.; Kauferb, D.; Xiaofeng, Z.; Wua, G.; Alzheimer’s Disease Neuroimaging Initiative. Long range early diagnosis of Alzheimer’s disease using longitudinal MR imaging data. Med. Image Anal. 2021, 67, 101825. [Google Scholar] [CrossRef]
- Fleisher, A.S.; Chen, K.; Quiroz, Y.T.; Jakimovich, L.J.; Gomez, M.G.; Langois, C.M.; Langbaum, J.B.; Ayutyanont, N.; Roontiva, A.; Thiyyagura, P.; et al. Florbetapir PET analysis of amyloid-beta deposition in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: A cross-sectional study. Lancet Neurol. 2012, 11, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, M.; Shimada, H.; Suhara, T.; Shinotoh, H.; Ji, B.; Maeda, J.; Zhang, M.R.; Trojanowski, J.Q.; Lee, V.M.; Ono, M.; et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 2013, 79, 1094–1108. [Google Scholar] [CrossRef] [Green Version]
- Karas, G.B.; Scheltens, P.; Rombouts, S.A.; Visser, P.J.; van Schijndel, R.A.; Fox, N.C.; Barkhof, F. Global and local gray matter loss in mild cognitive impairment and Alzheimer’s disease. NeuroImage 2004, 23, 708–716. [Google Scholar] [CrossRef]
- Sapkota, S.; Huan, T.; Tran, T.; Zheng, J.; Camicioli, R.; Li, L.; Dixon, R.A. Alzheimer’s Biomarkers From Multiple Modalities Selectively Discriminate Clinical Status: Relative Importance of Salivary Metabolomics Panels, Genetic, Lifestyle, Cognitive, Functional Health and Demographic Risk Markers. Front. Aging Neurosci. 2018, 10, 296. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, S.; Zoltowska, K.M.; Laskowska-Kaszub, K.; Wojda, U. microRNA diagnostic panel for Alzheimer’s disease and epigenetic trade-off between neurodegeneration and cancer. Ageing Res. Rev. 2019, 49, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Boopathi, S.; Poma, A.B.; Garduno-Juarez, R. An Overview of Several Inhibitors for Alzheimer’s Disease: Characterization and Failure. Int. J. Mol. Sci. 2021, 22, 10798. [Google Scholar] [CrossRef] [PubMed]
- Knapskog, A.B.; Engedal, K.; Selbaek, G.; Oksengard, A.R. Alzheimer’s disease—Diagnosis and treatment. Tidsskr. Nor. Laegeforen. Tidsskr. Prakt. Med. Raekke 2021, 141. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandiah, N.; Pai, M.C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, Y.; Zheng, X.; Fang, T.; Yang, X.; Luo, X.; Guo, A.; Newell, K.A.; Huang, X.F.; Yu, Y. Galantamine improves cognition, hippocampal inflammation, and synaptic plasticity impairments induced by lipopolysaccharide in mice. J. Neuroinflamm. 2018, 15, 112. [Google Scholar] [CrossRef] [Green Version]
- Atri, A.; Sheard, S.; Goldfarb, D. A Multidisciplinary Approach for Addressing Challenges in Alzheimer’s Disease. J. Clin. Psychiatry 2019, 80, MS18002WC4C. [Google Scholar] [CrossRef]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Hane, F.T.; Robinson, M.; Lee, B.Y.; Bai, O.; Leonenko, Z.; Albert, M.S. Recent Progress in Alzheimer’s Disease Research, Part 3: Diagnosis and Treatment. J. Alzheimer’s Dis. JAD 2017, 57, 645–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saretz, S.; Basset, G.; Useini, L.; Laube, M.; Pietzsch, J.; Drača, D.; Maksimović-Ivanić, D.; Trambauer, J.; Steiner, H.; Hey-Hawkins, E. Modulation of γ-Secretase Activity by a Carborane-Based Flurbiprofen Analogue. Molecules 2021, 26, 2843. [Google Scholar] [CrossRef]
- Imbimbo, B.P.; Watling, M. Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2019, 28, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Ocampo, A.; Lopera, F. Amyloid-beta immunotherapy: The hope for Alzheimer disease? Colomb. Med. 2016, 47, 203–212. [Google Scholar] [CrossRef]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2021, 17, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Aducanumab: First Approval. Drugs 2021, 81, 1437–1443. [Google Scholar] [CrossRef]
- Bittar, A.; Sengupta, U.; Kayed, R. Prospects for strain-specific immunotherapy in Alzheimer’s disease and tauopathies. NPJ Vaccines 2018, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.W.; Shao, E.; Mucke, L. Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 2021, 371, abb8255. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nature reviews. Neurology 2018, 14, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Haggarty, S.J. Tauopathies: Deciphering Disease Mechanisms to Develop Effective Therapies. Int. J. Mol. Sci. 2020, 21, 8948. [Google Scholar] [CrossRef]
- Soeda, Y.; Takashima, A. New Insights into Drug Discovery Targeting Tau Protein. Front. Mol. Neurosci. 2020, 13, 590896. [Google Scholar] [CrossRef] [PubMed]
- Bendlin, B.B. Antidiabetic therapies and Alzheimer disease. Dialogues Clin. Neurosci. 2019, 21, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, W.H.; Braga, C.F.; Lós, D.B.; Araújo, S.M.R.; França, M.R.; Duarte-Silva, E.; Rodrigues, G.B.; Rocha, S.W.S.; Peixoto, C.A. Metformin prevents p-tau and amyloid plaque deposition and memory impairment in diabetic mice. Exp. Brain Res. 2021, 239, 2821–2839. [Google Scholar] [CrossRef]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef]
- De Toledo Ferraz Alves, T.C.; Ferreira, L.K.; Wajngarten, M.; Busatto, G.F. Cardiac disorders as risk factors for Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2010, 20, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Mayeux, R. Cardiovascular risk factors and Alzheimer’s disease. Curr. Atheroscler. Rep. 2004, 6, 261–266. [Google Scholar] [CrossRef]
- Speh, A.; Wang, R.; Winblad, B.; Kramberger, M.G.; Backman, L.; Qiu, C.; Laukka, E.J. The Relationship Between Cardiovascular Health and Rate of Cognitive Decline in Young-Old and Old-Old Adults: A Population-Based Study. J. Alzheimer’s Dis. JAD 2021, 84, 1523–1537. [Google Scholar] [CrossRef]
- The SPRINT MIND Investigators for the SPRINT Research Group; Williamson, J.D.; Pajewski, N.M.; Auchus, A.P.; Bryan, R.N.; Chelune, G.; Cheung, A.K.; Cleveland, M.L.; Coker, L.H.; Crowe, M.G.; et al. Effect of Intensive vs Standard Blood Pressure Control on Probable Dementia: A Randomized Clinical Trial. JAMA 2019, 321, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, I.M.; Gaussoin, S.A.; Pomponio, R.; Dolui, S.; Erus, G.; Wright, C.B.; Launer, L.J.; Detre, J.A.; Wolk, D.A.; Davatzikos, C.; et al. Association of Intensive vs Standard Blood Pressure Control with Magnetic Resonance Imaging Biomarkers of Alzheimer Disease: Secondary Analysis of the SPRINT MIND Randomized Trial. JAMA Neurol. 2021, 78, 568–577. [Google Scholar] [CrossRef]
- Nordestgaard, L.T.; Christoffersen, M.; Afzal, S.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Frikke-Schmidt, R. Triglycerides as a Shared Risk Factor between Dementia and Atherosclerotic Cardiovascular Disease: A Study of 125 727 Individuals. Clin. Chem. 2021, 67, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Matilla-Mora, R.; Martínez-Piédrola, R.M.; Fernández Huete, J. Effectiveness of occupational therapy and other non-pharmacological therapies in cognitive impairment and Alzheimer’s disease. Rev. Esp. Geriatr. Gerontol. 2016, 51, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Zucchella, C.; Sinforiani, E.; Tamburin, S.; Federico, A.; Mantovani, E.; Bernini, S.; Casale, R.; Bartolo, M. The Multidisciplinary Approach to Alzheimer’s Disease and Dementia. A Narrative Review of Non-Pharmacological Treatment. Front. Neurol. 2018, 9, 1058. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Pei, J.; Zhan, Y.J.; Cai, Y.W. Overview of Meta-Analyses of Five Non-pharmacological Interventions for Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 594432. [Google Scholar] [CrossRef] [PubMed]
- Plascencia-Villa, G.; Perry, G. Preventive and Therapeutic Strategies in Alzheimer’s Disease: Focus on Oxidative Stress, Redox Metals, and Ferroptosis. Antioxid. Redox Signal 2021, 34, 591–610. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Morales, V.; González-Acedo, A.; Melguizo-Rodríguez, L.; Pardo-Moreno, T.; Costela-Ruiz, V.J.; Montiel-Troya, M.; Ramos-Rodríguez, J.J. Current Understanding of the Physiopathology, Diagnosis and Therapeutic Approach to Alzheimer’s Disease. Biomedicines 2021, 9, 1910. https://doi.org/10.3390/biomedicines9121910
García-Morales V, González-Acedo A, Melguizo-Rodríguez L, Pardo-Moreno T, Costela-Ruiz VJ, Montiel-Troya M, Ramos-Rodríguez JJ. Current Understanding of the Physiopathology, Diagnosis and Therapeutic Approach to Alzheimer’s Disease. Biomedicines. 2021; 9(12):1910. https://doi.org/10.3390/biomedicines9121910
Chicago/Turabian StyleGarcía-Morales, Victoria, Anabel González-Acedo, Lucía Melguizo-Rodríguez, Teresa Pardo-Moreno, Víctor Javier Costela-Ruiz, María Montiel-Troya, and Juan José Ramos-Rodríguez. 2021. "Current Understanding of the Physiopathology, Diagnosis and Therapeutic Approach to Alzheimer’s Disease" Biomedicines 9, no. 12: 1910. https://doi.org/10.3390/biomedicines9121910
APA StyleGarcía-Morales, V., González-Acedo, A., Melguizo-Rodríguez, L., Pardo-Moreno, T., Costela-Ruiz, V. J., Montiel-Troya, M., & Ramos-Rodríguez, J. J. (2021). Current Understanding of the Physiopathology, Diagnosis and Therapeutic Approach to Alzheimer’s Disease. Biomedicines, 9(12), 1910. https://doi.org/10.3390/biomedicines9121910